
Heat to Hydrogen by Reverse Electrodialysis—Using a Non-Equilibrium Thermodynamics Model to Evaluate Hydrogen Production Concepts Utilising Waste Heat

 ,
,  , and
, and
Abstract
:1. Introduction
2. Theory
2.1. Outline
2.2. Contribution from the Electrodes
2.3. Contribution from the Rinse Solutions
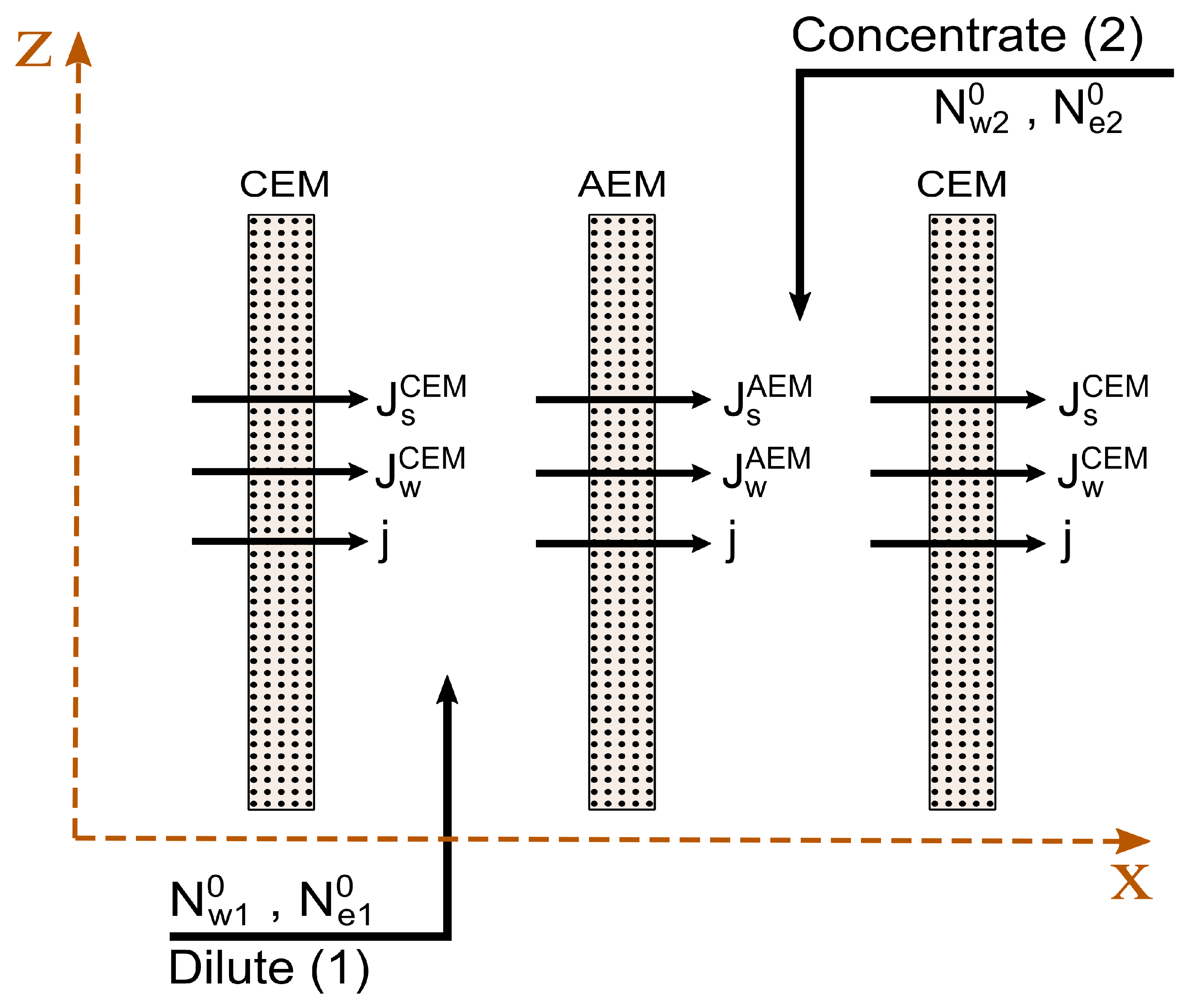
2.4. Contribution from a Unit Cell
2.5. The Total Cell
2.6. Component Fluxes
2.7. System Power
2.8. Mass Balances
2.9. Energy Balances
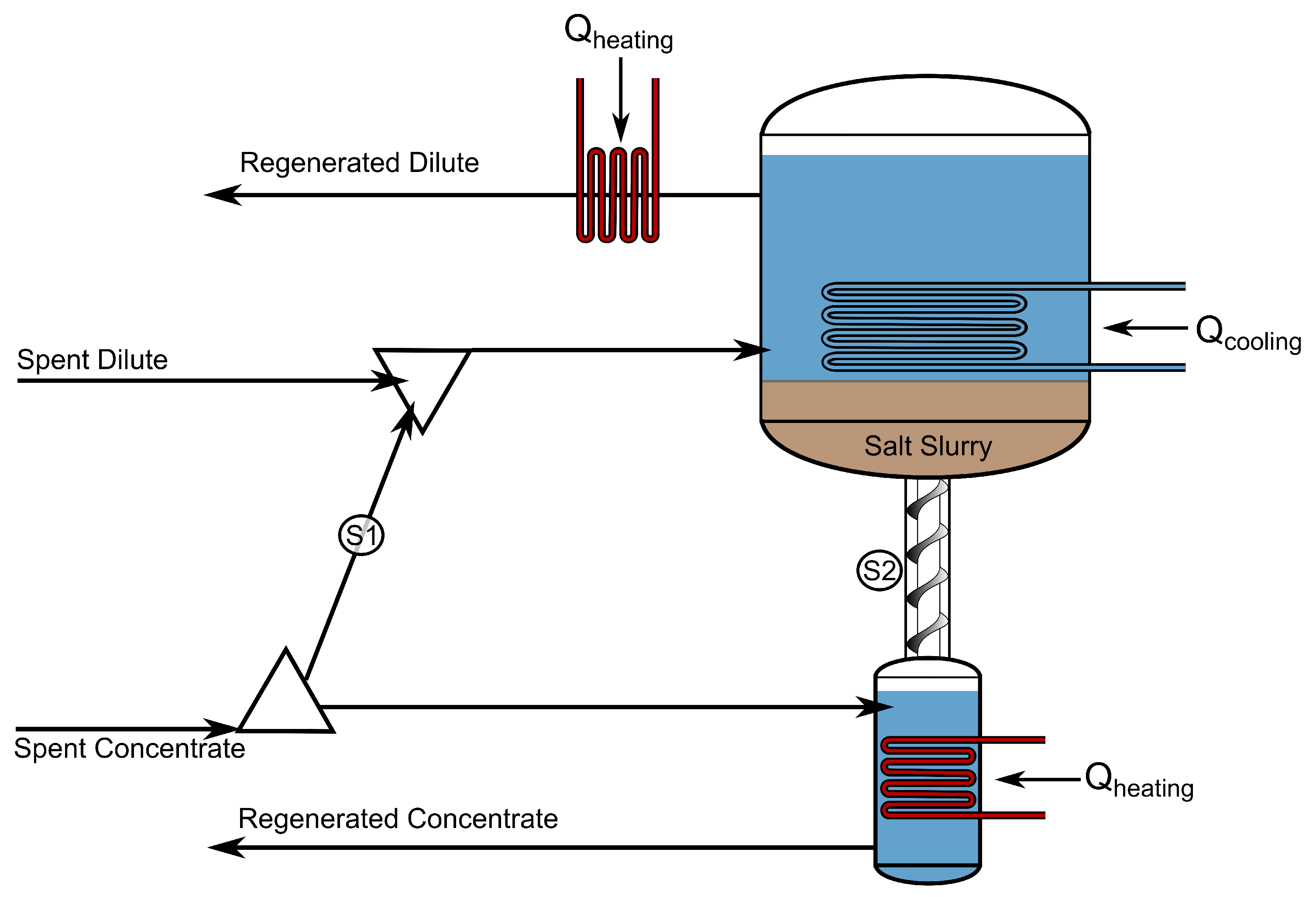
2.10. Solution Regeneration with Evaporation
2.11. Solution Regeneration with Precipitation
3. Computational Procedure
- The co-current flow IVP is solved using initial (inlet) values of molar flow rates of components and temperatures in one concentrate and one dilute channel. Fluxes are calculated by Equation (22) for each numerical step, and the peak power current density is used.
- The counter-current flow BVP is solved using the state variable profiles from the solved IVP as an initial guess.
- The state variable profiles from the solved BVP are used to compute the hydrogen production from the RED stack.
- The difference between the outlet and the inlet state variables is used to compute the mass and energy balances for streams and in order to regenerate the electrolyte solutions.
- concentration polarisation phenomena in bulk solutions are neglected;
- viscous dissipation and pressure drops are neglected;
- parasitic current draws in the feed channels of the cell are unaccounted for;
- electrode rinse solutions are assumed to have constant chemical potentials at both sides of the cell;
- variations in solution thermodynamic and transport properties with temperature are neglected; experimental data at 25 °C is used for solutions at 10 and 40 °C.
- Evaporation case: Evaporation (MED) is here used as the regeneration system. The inlet molality of concentrate is 4.5 mol kg, and 1 mol kg for the dilute. The inlet water mass flow is 0.27 g s and the inlet temperature is 40 °C for both channels.
- Precipitation case: Precipitation is here used as the regeneration system. The inlet molality of concentrate is 5.37 mol kg (solubility at 40 °C), and 4.15 mol kg (solubility at 10 °C) for the dilute. The inlet water mass flow is 0.27 g s and the inlet temperature is 40 °C for both channels.
4. Results and Discussion
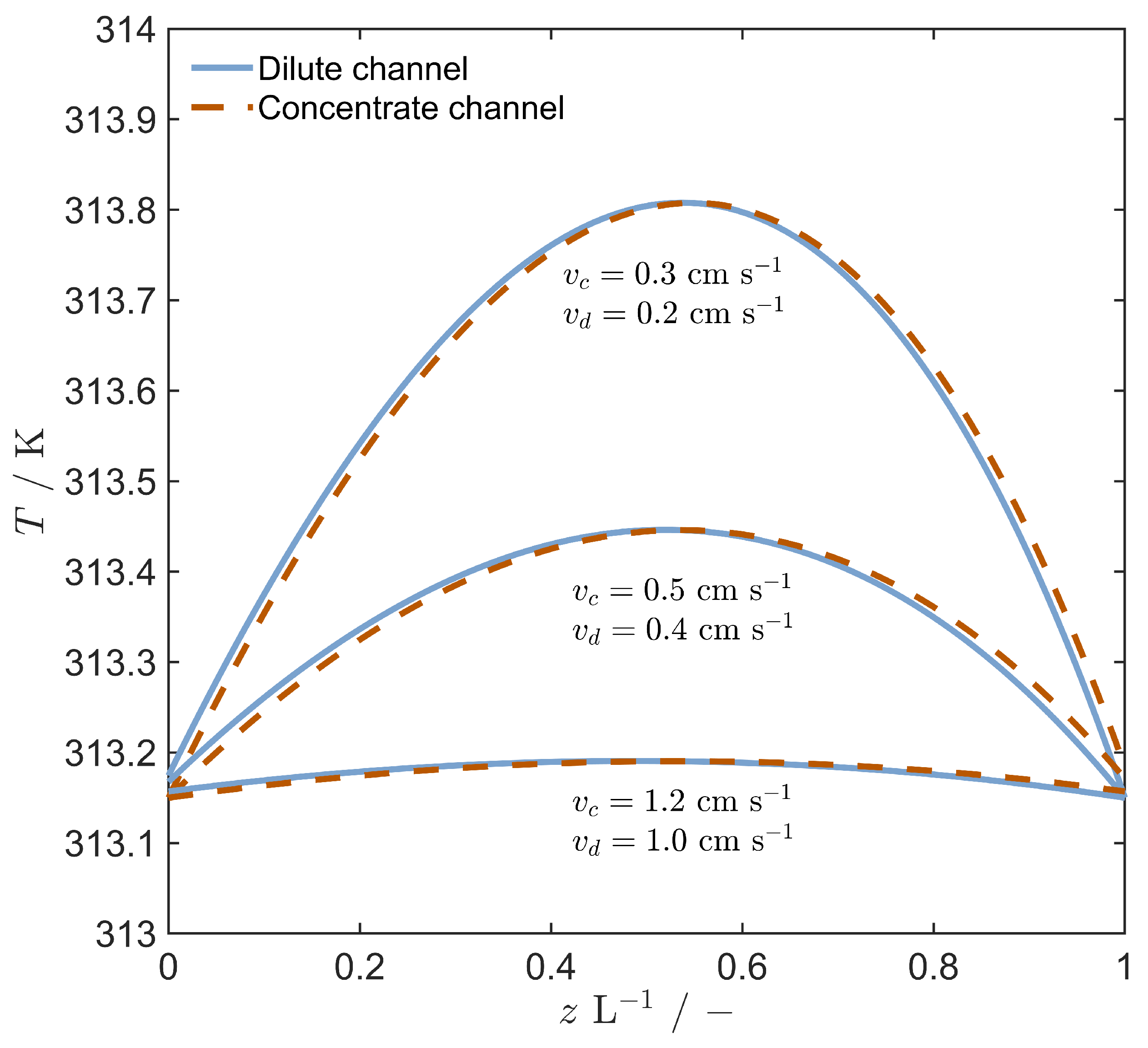
4.1. Model Validation
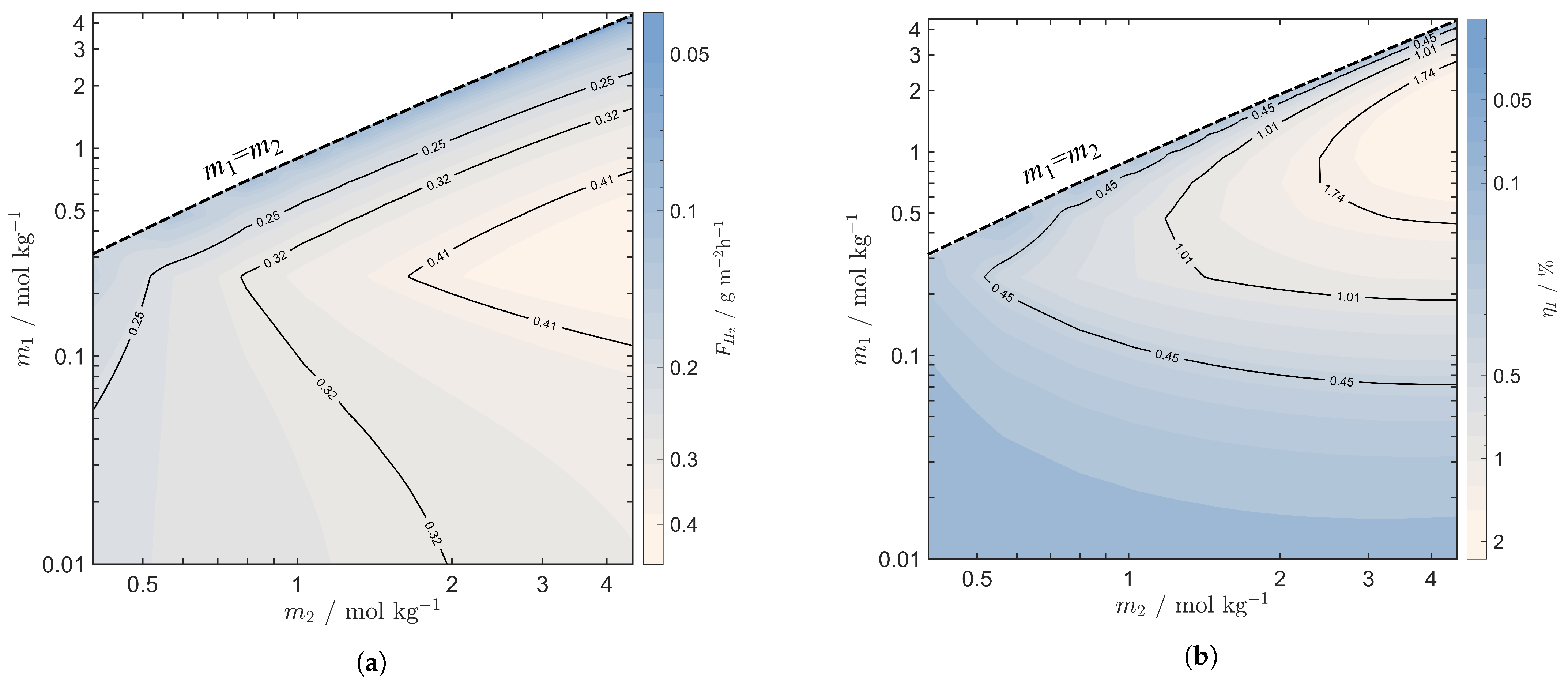
4.2. Precipitation and Evaporation Comparison
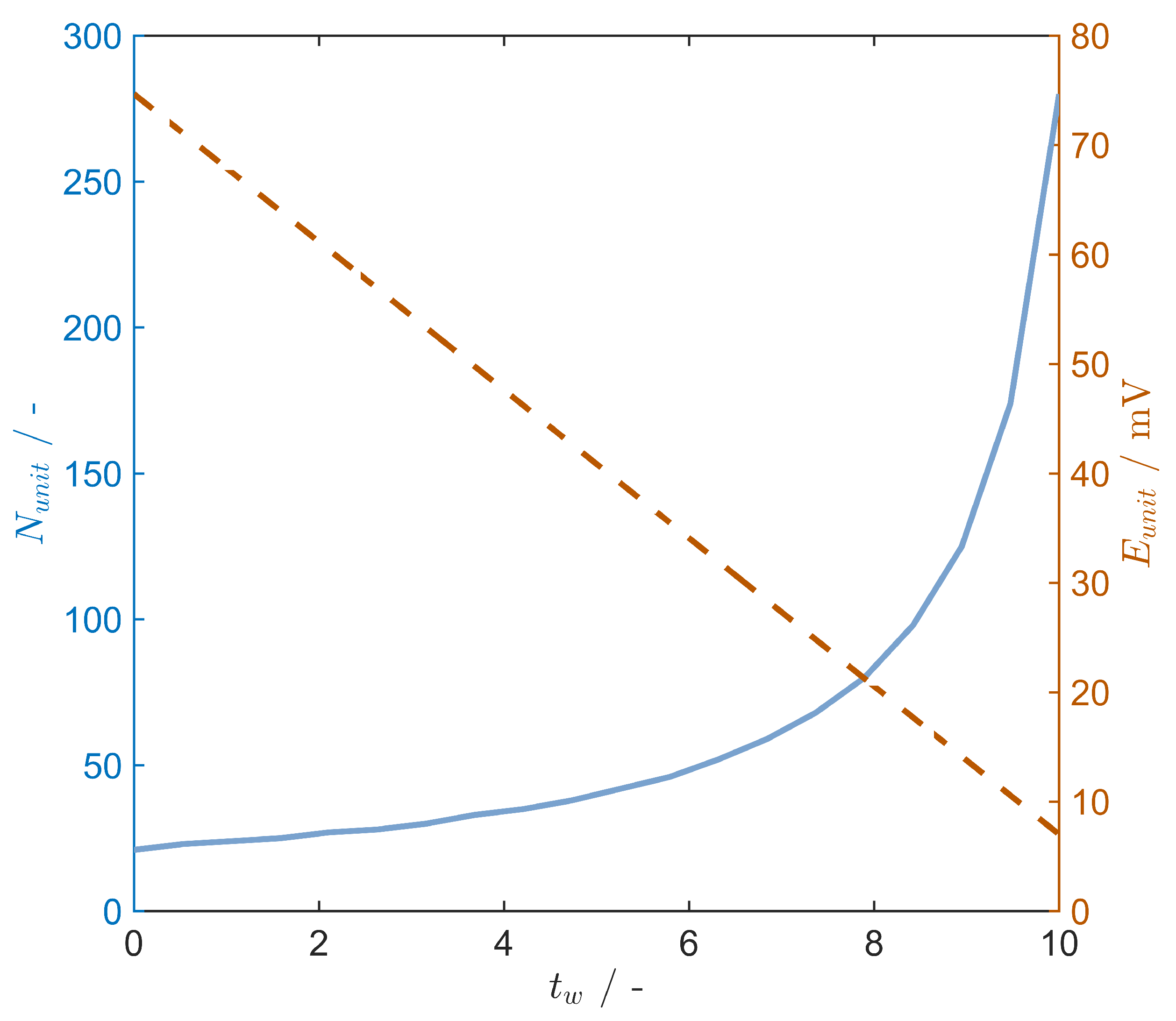
4.3. Stack Performance with Evaporation Regeneration
4.4. Membrane Water Transport
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Post, J.W. Blue Energy: Electricity Production from Salinity Gradients by Reverse Electrodialysis. Ph.D. Thesis, Wageningen University, Wageningen, The Netherlands, 2009. [Google Scholar]
- Krakhella, K.W.; Morales, M.; Bock, R.; Seland, F.; Burheim, O.S.; Einarsrud, K.E. Electrodialytic Energy Storage System: Permselectivity, Stack Measurements and Life-Cycle Analysis. Energies 2020, 13, 1247. [Google Scholar] [CrossRef]
- Scialdone, O.; Guarisco, C.; Grispo, S.; D’Angelo, A.; Galia, A. Investigation of electrode material–Redox couple systems for reverse electrodialysis processes. Part I: Iron redox couples. J. Electroanal. Chem. 2012, 681, 66–75. [Google Scholar] [CrossRef]
- Pattle, R. Production of electric power by mixing fresh and salt water in the hydroelectric pile. Nature 1954, 174, 660. [Google Scholar] [CrossRef]
- Loeb, S. Method and Apparatus for Generating Power Utilizing Pressure-Retarded-Osmosis. U.S. Patent 3,906,250, 19 September 1975. [Google Scholar]
- Loeb, S. Method and Apparatus for Generating Power Utilizing Reverse Electrodialysis. U.S. Patent 4,171,409, 16 October 1979. [Google Scholar]
- Tedesco, M.; Scalici, C.; Vaccari, D.; Cipollina, A.; Tamburini, A.; Micale, G. Performance of the first reverse electrodialysis pilot plant for power production from saline waters and concentrated brines. J. Membr. Sci. 2016, 500, 33–45. [Google Scholar] [CrossRef]
- Papapetrou, M. Reverse Electrodialysis Power Production-Progress in the development of an innovative system. In Proceedings of the 4th International Conference on Ocean Energy, Dublin, Ireland, 17 October 2012. [Google Scholar]
- Tedesco, M.; Cipollina, A.; Tamburini, A.; van Baak, W.; Micale, G. Modelling the Reverse ElectroDialysis process with seawater and concentrated brines. Desalin. Water Treat. 2012, 49, 404–424. [Google Scholar] [CrossRef]
- Tedesco, M.; Cipollina, A.; Tamburini, A.; Micale, G.; Helsen, J.; Papapetrou, M. REAPower: Use of desalination brine for power production through reverse electrodialysis. Desalin. Water Treat. 2015, 53, 3161–3169. [Google Scholar] [CrossRef]
- Tufa, R.A.; Curcio, E.; van Baak, W.; Veerman, J.; Grasman, S.; Fontananova, E.; Di Profio, G. Potential of brackish water and brine for energy generation by salinity gradient power-reverse electrodialysis (SGP-RE). RSC Adv. 2014, 4, 42617–42623. [Google Scholar] [CrossRef]
- Tedesco, M.; Brauns, E.; Cipollina, A.; Micale, G.; Modica, P.; Russo, G.; Helsen, J. Reverse electrodialysis with saline waters and concentrated brines: A laboratory investigation towards technology scale-up. J. Membr. Sci. 2015, 492, 9–20. [Google Scholar] [CrossRef]
- Tedesco, M.; Cipollina, A.; Tamburini, A.; Micale, G. Towards 1 kW power production in a reverse electrodialysis pilot plant with saline waters and concentrated brines. J. Membr. Sci. 2017, 522, 226–236. [Google Scholar] [CrossRef]
- Ni, M.; Leung, M.K.; Leung, D.Y. Energy and exergy analysis of hydrogen production vby a proton exchange membrane (PEM) electrolyzer plant. Energy Convers. Manag. 2008, 49, 2748–2756. [Google Scholar] [CrossRef]
- Kumar, S.S.; Himabindu, V. Hydrogen production by PEM water electrolysis–A review. Mater. Sci. Energy Technol. 2019, 2, 442–454. [Google Scholar]
- Lamb, J.J.; Burheim, O.S.; Pollet, B.G. Hydrogen Fuel Cells and Water Electrolysers. In Micro-Optics and Energy; Springer Nature Switzerland AG: Cham, Switzerland, 2020; pp. 61–71. [Google Scholar]
- Lamb, J.J.; Hillestad, M.; Rytter, E.; Bock, R.; Nordgård, A.S.; Lien, K.M.; Burheim, O.S.; Pollet, B.G. Traditional routes for hydrogen production and carbon conversion. In Hydrogen, Biomass and Bioenergy: Integration Pathways for Renewable Energy Applications; Academic Press: Cambridge, MA, USA, 2020; p. 21. [Google Scholar]
- Lamb, J.J.; Pollet, B.G.; Burheim, O.S. Energy storage. In Energy-Smart Buildings Design: Construction and Monitoring of Buildings for Improved Energy Efficiency; IOP Publishing: Bristol, UK, 2020. [Google Scholar] [CrossRef]
- Rytter, E.; Hillestad, M.; Austbø, B. Thermochemical production of fuels. In Hydrogen, Biomass and Bioenergy: Integration Pathways for Renewable Energy Applications; Academic Press: Cambridge, MA, USA, 2020; p. 89. [Google Scholar]
- Borgschulte, A. The hydrogen grand challenge. Front. Energy Res. 2016, 4, 11. [Google Scholar] [CrossRef]
- Boyano, A.; Blanco-Marigorta, A.; Morosuk, T.; Tsatsaronis, G. Exergoenvironmental analysis of a steam methane reforming process for hydrogen production. Energy 2011, 36, 2202–2214. [Google Scholar] [CrossRef]
- Rand, D.A. A journey on the electrochemical road to sustainability. J. Solid State Electrochem. 2011, 15, 1579–1622. [Google Scholar] [CrossRef]
- Acar, C.; Dincer, I. Comparative assessment of hydrogen production methods from renewable and non-renewable sources. Int. J. Hydrog. Energy 2014, 39, 1–12. [Google Scholar] [CrossRef]
- Hatzell, M.C.; Ivanov, I.; Cusick, R.D.; Zhu, X.; Logan, B.E. Comparison of hydrogen production and electrical power generation for energy capture in closed-loop ammonium bicarbonate reverse electrodialysis systems. Phys. Chem. Chem. Phys. 2014, 16, 1632–1638. [Google Scholar] [CrossRef]
- Tamburini, A.; Cipollina, A.; Papapetrou, M.; Piacentino, A.; Micale, G. Salinity gradient engines. In Sustainable Energy from Salinity Gradients; Woodhead Publishing: Sawston, UK, 2016; pp. 219–256. [Google Scholar]
- Johnson, I.; Choate, W.T.; Davidson, A. Waste Heat Recovery. Technology and Opportunities in U.S. Industry; Technical Report; U.S. Department of Energy: Washington, DC, USA, 2008. [Google Scholar] [CrossRef]
- Miró, L.; Brückner, S.; Cabeza, L.F. Mapping and discussing Industrial Waste Heat (IWH) potentials for different countries. Renew. Sustain. Energy Rev. 2015, 51, 847–855. [Google Scholar] [CrossRef]
- Hammond, G.; Norman, J. Heat recovery opportunities in UK industry. Appl. Energy 2014, 116, 387–397. [Google Scholar] [CrossRef]
- Papapetrou, M.; Kosmadakis, G.; Cipollina, A.; La Commare, U.; Micale, G. Industrial waste heat: Estimation of the technically available resource in the EU per industrial sector, temperature level and country. Appl. Therm. Eng. 2018, 138, 207–216. [Google Scholar] [CrossRef]
- Rattner, A.S.; Garimella, S. Energy harvesting, reuse and upgrade to reduce primary energy usage in the USA. Energy 2011, 36, 6172–6183. [Google Scholar] [CrossRef]
- Horenburg, P. Transforming Waste Heat into Electricity. Steam Expansion Engine Makes Efficient and Flexible Use of Low-Temperature Heat with ORC Technology; Bundesministerium fuer Wirtschaft und Technologie (BMWi): Berlin, Germany, 2011. [Google Scholar]
- Tamburini, A.; Tedesco, M.; Cipollina, A.; Micale, G.; Ciofalo, M.; Papapetrou, M.; Van Baak, W.; Piacentino, A. Reverse electrodialysis heat engine for sustainable power production. Appl. Energy 2017, 206, 1334–1353. [Google Scholar] [CrossRef]
- Zimmermann, P.; Solberg, S.B.B.; Tekinalp, Ö.; Lamb, J.J.; Wilhelmsen, Ø.; Deng, L.; Burheim, O.S. Heat to Hydrogen by RED—Reviewing Membranes and Salts for the RED Heat Engine Concept. Membranes 2022, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Delgado, B.; Giacalone, F.; Catrini, P.; Cipollina, A.; Piacentino, A.; Tamburini, A.; Micale, G. Reverse electrodialysis heat engine with multi-effect distillation: Exergy analysis and perspectives. Energy Convers. Manag. 2019, 194, 140–159. [Google Scholar] [CrossRef]
- Kim, D.H.; Park, B.H.; Kwon, K.; Li, L.; Kim, D. Modeling of power generation with thermolytic reverse electrodialysis for low-grade waste heat recovery. Appl. Energy 2017, 189, 201–210. [Google Scholar] [CrossRef]
- Olkis, C.; Brandani, S.; Santori, G. Adsorption reverse electrodialysis driven by power plant waste heat to generate electricity and provide cooling. Int. J. Energy Res. 2021, 45, 1971–1987. [Google Scholar] [CrossRef]
- Bevacqua, M.; Tamburini, A.; Papapetrou, M.; Cipollina, A.; Micale, G.; Piacentino, A. Reverse electrodialysis with NH4HCO3-water systems for heat-to-power conversion. Energy 2017, 137, 1293–1307. [Google Scholar] [CrossRef]
- Papapetrou, M.; Kosmadakis, G.; Giacalone, F.; Ortega-Delgado, B.; Cipollina, A.; Tamburini, A.; Micale, G. Evaluation of the Economic and Environmental Performance of Low-Temperature Heat to Power Conversion using a Reverse Electrodialysis—Multi-Effect Distillation System. Energies 2019, 12, 3206. [Google Scholar] [CrossRef]
- Krakhella, K.W.; Bock, R.; Burheim, O.S.; Seland, F.; Einarsrud, K.E. Heat to H2: Using waste heat for hydrogen production through reverse electrodialysis. Energies 2019, 12, 3428. [Google Scholar] [CrossRef]
- Raka, Y.D.; Karoliussen, H.; Lien, K.M.; Burheim, O.S. Opportunities and challenges for thermally driven hydrogen production using reverse electrodialysis system. Int. J. Hydrog. Energy 2020, 45, 1212–1225. [Google Scholar] [CrossRef]
- Raka, Y.D.; Bock, R.; Karoliussen, H.; Wilhelmsen, Ø.; Burheim, O.S. The Influence of Concentration and Temperature on the Membrane Resistance of Ion Exchange Membranes and the Levelised Cost of Hydrogen from Reverse Electrodialysis with Ammonium Bicarbonate. Membranes 2021, 11, 135. [Google Scholar] [CrossRef]
- Wu, X.; Gong, Y.; Xu, S.; Yan, Z.; Zhang, X.; Yang, S. Electrical Conductivity of Lithium Chloride, Lithium Bromide, and Lithium Iodide Electrolytes in Methanol, Water, and Their Binary Mixtures. J. Chem. Eng. Data 2019, 64, 4319–4329. [Google Scholar] [CrossRef]
- Zlotorowicz, A.; Strand, R.V.; Burheim, O.S.; Wilhelmsen, Ø.; Kjelstrup, S. The permselectivity and water transference number of ion exchange membranes in reverse electrodialysis. J. Membr. Sci. 2017, 523, 402–408. [Google Scholar] [CrossRef]
- Signe Kjelstrup, D.B. Non-Equilibrium Thermodynamics of Heterogeneous Systems; World Scientific: Singapore, 2008. [Google Scholar]
- Kristiansen, K.R.; Barragán, V.M.; Kjelstrup, S. Thermoelectric Power of Ion Exchange Membrane Cells Relevant to Reverse Electrodialysis Plants. Phys. Rev. Appl. 2019, 11, 044037. [Google Scholar] [CrossRef]
- Førland, T.F.K.S.; Kjelstrup, S. Irreversible Thermodynamics: Theory and Applications; Wiley: Chichester, UK, 1988. [Google Scholar]
- Bedeaux, D.; Kjelstrup, S. Local Properties of a Formation Cell as Described by Nonequilibrium Thermodynamics. J. Non-Equilib. Thermodyn. 2000, 25, 161–178. [Google Scholar] [CrossRef]
- Friedman, H.L. Thermodynamic Excess Functions for Electrolyte Solutions. J. Chem. Phys. 1960, 32, 1351–1362. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, X.; Wang, Y.; Wu, L.; Hu, Y. Experimental and Modeling of Conductivity for Electrolyte Solution Systems. ACS Omega 2020, 5, 22465–22474. [Google Scholar] [CrossRef]
- Jakobsen, H.A. Chemical Reactor Modeling; Springer: New York, NY, USA, 2014. [Google Scholar]
- Wilhelmsen, Ø.; Anantharaman, R.; Berstad, D.; Jordal, K. Multi-Scale modelling of a membrane reforming power cycle with CO2 capture. In 21st European Symposium on Computer Aided Process Engineering; Computer Aided Chemical Engineering; Pistikopoulos, E., Georgiadis, M., Kokossis, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 29, pp. 6–10. [Google Scholar] [CrossRef]
- Micale, G.; Cipollina, A.; Rizzuti, L. Seawater Desalination for Freshwater Production. In Seawater Desalination: Conventional and Renewable Energy Processes; Springer: Berlin/Heidelberg, Germany, 2009; Chapter 1; pp. 1–15. [Google Scholar] [CrossRef]
- Rumble, J.R. Aqueous solubility of inorganic compounds at various tempratures. In CRC Handbook of Chemistry and Physics, 101st ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2020. [Google Scholar]
- Veerman, J.; de Jong, R.M.; Saakes, M.; Metz, S.J.; Harmsen, G.J. Reverse electrodialysis: Comparison of six commercial membrane pairs on the thermodynamic efficiency and power density. J. Membr. Sci. 2009, 343, 7–15. [Google Scholar] [CrossRef]
- Burheim, O.S. Engineering Energy Storage; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Chen, X.; Jiang, C.; Zhang, Y.; Wang, Y.; Xu, T. Storable hydrogen production by Reverse Electro-Electrodialysis (REED). J. Membr. Sci. 2017, 544, 397–405. [Google Scholar] [CrossRef]
- Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrogen Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- Pitzer, K.S.; Mayorga, G. Thermodynammics of Electrolytes. II. Activity and Osmotic Coefficients for Strong Electrolytes with One or Both Ions Univalent. J. Phys. Chem. 1973, 77, 134–140. [Google Scholar] [CrossRef]
- Saluja, P.P.S.; Pitzer, K.S.; Phutela, R.C. High-temperature thermodynamic properties of several 1:1 electrolytes. Can. J. Chem. 1986, 64, 1278–1285. [Google Scholar] [CrossRef]
- Stokes, R.H.; Robinson, R.A. Ionic hydration and activity in electrolyte solutions. J. Am. Chem. Soc. 1948, 70, 1870–1878. [Google Scholar] [CrossRef]
- Lobo, V.M.M.; Quaresma, J.L. Handbook of Electrolyte Solutions, Part A; Elsevier: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Lobo, V.M.M.; Quaresma, J.L. Handbook of Electrolyte Solutions, Part B; Elsevier: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Dobos, D. Electrochemical Data; Elsevier: Amsterdam, The Netherlands, 1975. [Google Scholar]
- Clarke, E.C.W.; Glew, D.N. Evaluation of Debye–Hückel limiting slopes for water between 0 and 150 °C. J. Chem. Soc. Faraday Trans. 1980, 76, 1911–1916. [Google Scholar] [CrossRef]
- Silvester, L.F.; Pitzer, K.S. Thermodynamics of electrolytes. X. Enthalpy and the effect of temperature on the activity coefficients. J. Solut. Chem. 1978, 7, 327–337. [Google Scholar] [CrossRef]
- Chase, M. NIST-JANAF Thermochemical Tables, 4th ed.; American Institute of Physics: College Park, MD, USA, 1998. [Google Scholar]
- Ge, X.; Wang, X. Estimation of Freezing Point Depression, Boiling Point Elevation, and Vaporization Enthalpies of Electrolyte Solutions. Ind. Eng. Chem. Res. 2009, 48, 2229–2235. [Google Scholar] [CrossRef]
- Criss, C.M.; Millero, F.J. Modeling the Heat Capacities of Aqueous 1-1 Electrolyte Solutions with Pitzer’s Equations. J. Phys. Chem 1996, 100, 1288–1294. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Symbol | Value |
|---|---|---|
| Salt transference no. [43] | 0.45 | |
| Water transference no. [43] | 3 | |
| Salt diffusion coeff. [54] | m s | |
| Water diffusion coeff. [54] | m s | |
| IEM heat transfer coeff. | 1000 W mK | |
| CEM area resistance [32] | 2.96 cm | |
| AEM area resistance [32] | 1.55 cm | |
| IEM thickness [39] | 125 m | |
| IEM active area [32] | 100 cm | |
| Channel length [32] | 10 °Cm | |
| Channel height [32] | 10 °Cm | |
| Channel thickness [32] | 270 m | |
| Water mass flow | 0.27 g s | |
| Spacer shadow factor [32] | 1.56 | |
| Lumped overpotential [55] | 0.2 V |
| Precipitation | Evaporation | ||
|---|---|---|---|
| 0.14 | 0.38 | g mh | |
| 6750 | 1.7 | kWh g | |
| 3.8 | 10 | A m | |
| 286 | 30 | - | |
| 5.4 | 15 | W m |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solberg, S.B.B.; Zimmermann, P.; Wilhelmsen, Ø.; Lamb, J.J.; Bock, R.; Burheim, O.S. Heat to Hydrogen by Reverse Electrodialysis—Using a Non-Equilibrium Thermodynamics Model to Evaluate Hydrogen Production Concepts Utilising Waste Heat. Energies 2022, 15, 6011. https://doi.org/10.3390/en15166011
Solberg SBB, Zimmermann P, Wilhelmsen Ø, Lamb JJ, Bock R, Burheim OS. Heat to Hydrogen by Reverse Electrodialysis—Using a Non-Equilibrium Thermodynamics Model to Evaluate Hydrogen Production Concepts Utilising Waste Heat. Energies. 2022; 15(16):6011. https://doi.org/10.3390/en15166011
Chicago/Turabian StyleSolberg, Simon B. B., Pauline Zimmermann, Øivind Wilhelmsen, Jacob J. Lamb, Robert Bock, and Odne S. Burheim. 2022. "Heat to Hydrogen by Reverse Electrodialysis—Using a Non-Equilibrium Thermodynamics Model to Evaluate Hydrogen Production Concepts Utilising Waste Heat" Energies 15, no. 16: 6011. https://doi.org/10.3390/en15166011