
Identifiability Analysis of Degradation Model Parameters from Transient CO2 Release in Low-Temperature PEM Fuel Cell under Various AST Protocols


Abstract
:1. Introduction
2. Materials and Methods
2.1. Modeling Framework
Modeling Transport Phenomena in Connecting Pipes
2.2. Experimental
2.2.1. Fuel Cell Setup
2.2.2. ASTs Measurements
2.3. Optimization Procedure
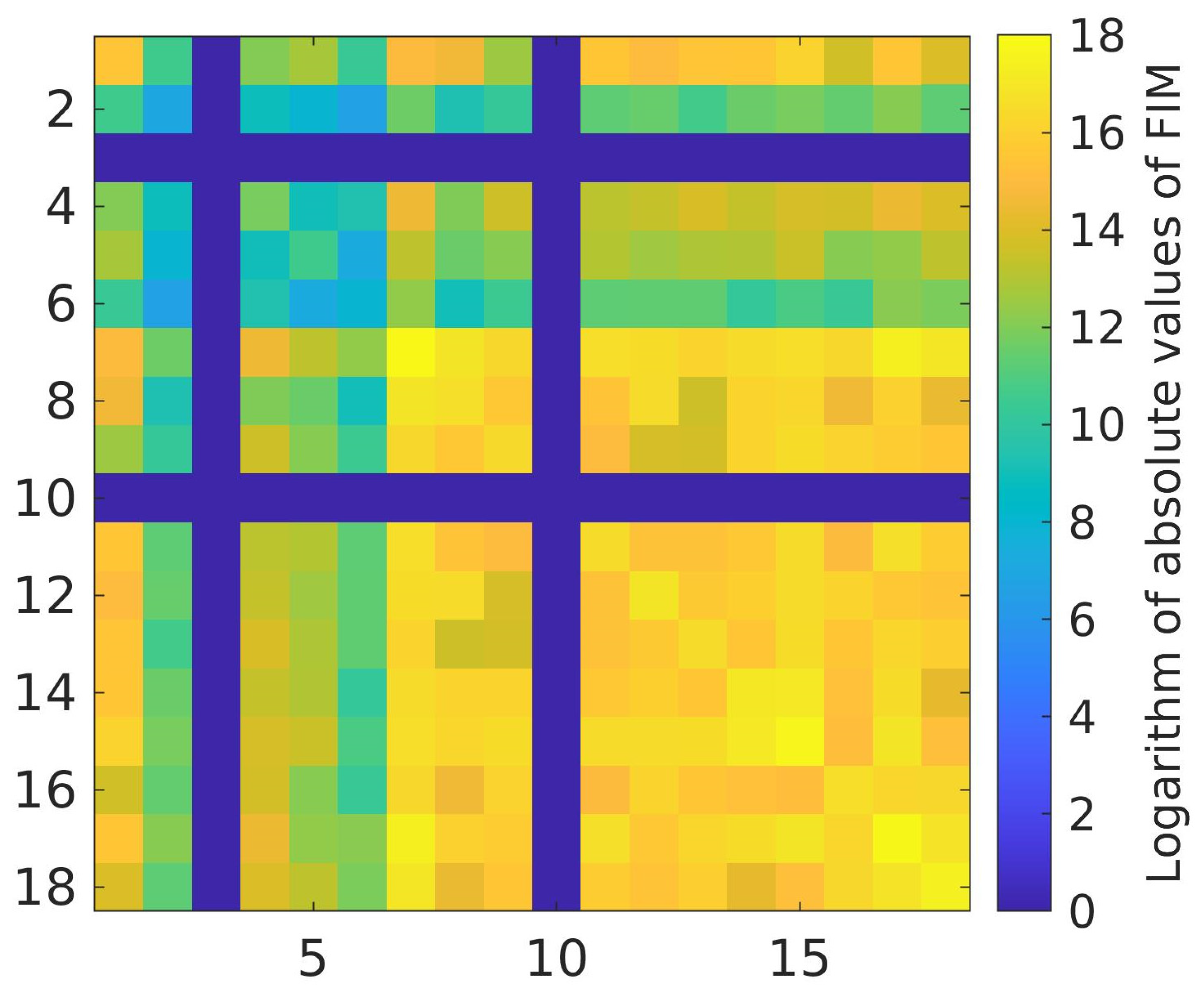
2.4. Calibration Parameter Sensitivity and Intercorrelation
3. Results and Discussion
3.1. Degradation Model Calibration and Validation
3.2. Calibration Parameter Sensitivity and Intercorrelation
Proposed Reduction of the Set of Calibration Parameters of the Model
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Appendix A. Model Parameters

{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Units | Description | Source |
|---|---|---|---|---|
| 21,090 | Pt density | Ref. [46] | ||
| 14,170 | Pt–OH density | Calc. from Ref. [46] | ||
| 14,100 | Pt=O density | Ref. [46] | ||
| 2000 | Carbon support density | - | ||
| 2.73 | Pt surface tension | Ref. [46] | ||
| 1.34 | Pt–OH surface tension | Calc. from Ref. [46] | ||
| 1 | Pt=O surface tension | Ref. [46] | ||
| 0.195 | Pt molar mass | Ref. [46] | ||
| 0.212 | Pt–OH molar mass | Calc. from Ref. [46] | ||
| 0.211 | Pt=O molar mass | Ref. [46] | ||
| 0.012 | Carbon molar mass | - | ||
| 0.018 | Water molar mass | - | ||
| 30,000 | Pt oxide interaction | Ref. [46] | ||
| 0.3 | - | Pt oxidation reversibility | Ref. [20] | |
| 2.15 | Pt surface site density | Ref. [46] | ||
| 4.6 | Carbon surface site density | Refs. [4,32] | ||
| 1000 | Reference concentration | Ref. [46] |
| Parameter | Value | Units | Description |
|---|---|---|---|
| 2.4744 | Pt→Pt–OH reaction rate | ||
| 28,351 | Pt–OH→Pt=O reaction rate | ||
| 3.9306 | Pt→Pt2+ reaction rate | ||
| 156.71 | C→C–OH reaction rate | ||
| 392.56 | C–OH→CO reaction rate | ||
| 12,422 | CO2 production reaction rate | ||
| 0.1786 | C–OH→CO2 reaction rate | ||
| 0.3910 | - | Pt→Pt–OH transfer coefficient | |
| 0.5080 | - | Pt–OH→Pt=O transfer coefficient | |
| 0.4792 | - | Pt→Pt2+ transfer coefficient | |
| 0.5290 | - | C→C–OH transfer coefficient | |
| 0.5031 | - | C–OH→CO transfer coefficient | |
| 0.5201 | - | CO2 production transfer coefficient | |
| 0.5498 | - | C–OH→CO2 transfer coefficient | |
| 0.1985 | - | Initial free Pt surface | |
| 0.4181 | - | Initial surface covered with OH | |
| 0.2068 | - | Initial free C surface | |
| 0.1929 | - | itial surface covered with OH |
References
- Hart, D.; Lehner, F.; Jones, S.; Lewis, J.; Klippenstein, M. E4tech Fuel Cell Industry Review 2018; E4tech: London, UK, 2018. [Google Scholar]
- Pandy, A.; Yang, Z.; Gummalla, M.; Atrazhev, V.V.; Kuzminyh, N.Y.; Sultanov, V.I.; Burlatsky, S. A Carbon Corrosion Model to Evaluate the Effect of Steady State and Transient Operation of a Polymer Electrolyte Membrane Fuel Cell. J. Electrochem. Soc. 2013, 160, F972–F979. [Google Scholar] [CrossRef] [Green Version]
- Darling, R.; Meyers, J. Kinetic Model of Platinum Dissolution in PEMFCs. J. Electrochem. Soc. 2003, 150. [Google Scholar] [CrossRef]
- Bi, W.; Fuller, T.F. Modeling of PEM Fuel Cell Pt/C Catalyst Degradation. J. Power Sources 2008, 178, 188–196. [Google Scholar] [CrossRef]
- Reimer, U.; Schumacher, B.; Lehnert, W. Accelerated Degradation of High-Temperature Polymer Electrolyte Fuel Cells: Discussion and Empirical Modeling. J. Electrochem. Soc. 2014, 162, F153–F164. [Google Scholar] [CrossRef]
- Kim, J.; Kim, M.; Kang, T.; Sohn, Y.-J.; Song, T.; Choi, K.H. Degradation Modeling and Operational Optimization for Improving the Lifetime of High-Temperature PEM (Proton Exchange Membrane) Fuel Cells. Energy 2014, 66, 41–49. [Google Scholar] [CrossRef]
- Pohl, E.; Maximini, M.; Bauschulte, A.; vom Schloß, J.; Hermanns, R.T.E. Degradation Modeling of High Temperature Proton Exchange Membrane Fuel Cells Using Dual Time Scale Simulation. J. Power Sources 2015, 275, 777–784. [Google Scholar] [CrossRef]
- Tavčar, G.; Katrašnik, T. A Real Time Capable Quasi 3D System Level Model of PEM Fuel Cells. Fuel Cells 2020, 20, 17–32. [Google Scholar] [CrossRef]
- Kregar, A.; Tavčar, G.; Kravos, A.; Katrašnik, T. Predictive System-Level Modeling Framework for Transient Operation and Cathode Platinum Degradation of High Temperature Proton Exchange Membrane Fuel Cells. Appl. Energy 2020, 263, 114547. [Google Scholar] [CrossRef]
- Promislow, K.; Chang, P.; Haas, H.; Wetton, B. Two-Phase Unit Cell Model for Slow Transients in Polymer Electrolyte Membrane Fuel Cells. J. Electrochem. Soc. 2008, 155, A494. [Google Scholar] [CrossRef]
- Meyers, J.P.; Darling, R.M. Model of Carbon Corrosion in PEM Fuel Cells. J. Electrochem. Soc. 2006, 153, A1432. [Google Scholar] [CrossRef]
- Takeuchi, N.; Fuller, T.F. Modeling and Investigation of Design Factors and Their Impact on Carbon Corrosion of PEMFC Electrodes. J. Electrochem. Soc. 2008, 155, B770. [Google Scholar] [CrossRef]
- Macauley, N.; Papadias, D.D.; Fairweather, J.; Spernjak, D.; Langlois, D.; Ahluwalia, R.; More, K.L.; Mukundan, R.; Borup, R.L. Carbon Corrosion in PEM Fuel Cells and the Development of Accelerated Stress Tests. J. Electrochem. Soc. 2018, 165, F3148–F3160. [Google Scholar] [CrossRef]
- Malek, K.; Franco, A.A. Microstructure-Based Modeling of Aging Mechanisms in Catalyst Layers of Polymer Electrolyte Fuel Cells. J. Phys. Chem. B 2011, 115, 8088–8101. [Google Scholar] [CrossRef] [PubMed]
- Franco, A. A Physical Multiscale Model of the Electrochemical Dynamics in a Polymer Electrolyte Fuel Cell—An Infinite Dimensional Bond Graph Approach. Ph.D. Thesis, Université Claude Bernard Lyon, Villeurbanne, France, 2005. [Google Scholar]
- Redmond, E.L.; Setzler, B.P.; Alamgir, F.M.; Fuller, T.F. Elucidating the Oxide Growth Mechanism on Platinum at the Cathode in PEM Fuel Cells. Phys. Chem. Chem. Phys. 2014, 16, 5301–5311. [Google Scholar] [CrossRef]
- Borup, R.L.; Papadias, D.D.; Mukundan, R.; Spernjak, D.; Langlois, D.A.; Ahluwalia, R.; More, K.L.; Grot, S. Carbon Corrosion in PEM Fuel Cells during Drive Cycle Operation. Ecs Meet. Abstr. 2015. [Google Scholar] [CrossRef]
- Franco, A.A.; Coulon, R.; Ferreira de Morais, R.; Cheah, S.K.; Kachmar, A.; Gabriel, M.A. Multi-Scale Modeling-Based Prediction of PEM Fuel Cells MEA Durability under Automotive Operating Conditions. Ecs Trans. 2019, 25, 65–79. [Google Scholar] [CrossRef]
- Darling, R.M.; Meyers, J.P. Mathematical Model of Platinum Movement in PEM Fuel Cells. J. Electrochem. Soc. 2005, 152, A242. [Google Scholar] [CrossRef]
- Li, Y.; Moriyama, K.; Gu, W.; Arisetty, S.; Wang, C.Y. A One-Dimensional Pt Degradation Model for Polymer Electrolyte Fuel Cells. J. Electrochem. Soc. 2015, 162, F834–F842. [Google Scholar] [CrossRef] [Green Version]
- Ruckenstein, E.; Pulvermacher, B. Growth Kinetics and the Size Distributions of Supported Metal Crystallites. J. Catal. 1973, 29, 224–245. [Google Scholar] [CrossRef]
- Holby, E.F.; Sheng, W.; Shao-Horn, Y.; Morgan, D. Pt Nanoparticle Stability in PEM Fuel Cells: Influence of Particle Size Distribution and Crossover Hydrogen. Energy Environ. Sci. 2009, 2, 865–871. [Google Scholar] [CrossRef]
- Baroody, H.A.; Stolar, D.B.; Eikerling, M.H. Modelling-Based Data Treatment and Analytics of Catalyst Degradation in Polymer Electrolyte Fuel Cells. Electrochim. Acta 2018, 283, 1006–1016. [Google Scholar] [CrossRef]
- Urchaga, P.; Kadyk, T.; Rinaldo, S.G.; Pistono, A.O.; Hu, J.; Lee, W.; Richards, C.; Eikerling, M.H.; Rice, C.A. Catalyst Degradation in Fuel Cell Electrodes: Accelerated Stress Tests and Model-Based Analysis. Electrochim. Acta 2015, 176, 1500–1510. [Google Scholar] [CrossRef]
- Kregar, A.; Kravos, A.; Katrašnik, T. Methodology for Evaluation of Contributions of Ostwald Ripening and Particle Agglomeration to Growth of Catalyst Particles in PEM Fuel Cells. Fuel Cells 2020, 20, 487–498. [Google Scholar] [CrossRef]
- Kregar, A.; Tavčar, G.; Kravos, A.; Katrašnik, T. Predictive Virtual Modelling Framework for Performance and Platinum Degradation Modelling of High Temperature PEM Fuel Cells. Energy Procedia 2019, 158, 1817–1822. [Google Scholar] [CrossRef]
- Chandan, A.; Hattenberger, M.; El-kharouf, A.; Du, S.; Dhir, A.; Self, V.; Pollet, B.G.; Ingram, A.; Bujalski, W. High Temperature (HT) Polymer Electrolyte Membrane Fuel Cells (PEMFC)—A Review. J. Power Sources 2013, 231, 264–278. [Google Scholar] [CrossRef]
- House, J.E. Principles of Chemical Kinetics; Bibliyografya Ve Indeks; Wm. C. Brown; Academic Press; Elsevier: London, UK, 2007; ISBN 978-0-697-32881-6. [Google Scholar]
- Tavčar, G.; Katrašnik, T. An Innovative Hybrid 3D Analytic-Numerical Approach for System Level Modelling of PEM Fuel Cells. Energies 2013, 6, 5426–5485. [Google Scholar] [CrossRef] [Green Version]
- Tavčar, G.; Katrašnik, T. An Innovative Hybrid 3D Analytic-Numerical Model for Air Breathing Parallel Channel Counter-Flow PEM Fuel Cells. Acta Chim. Slov. 2014, 61, 284–301. [Google Scholar]
- Tavčar, G.; Katrašnik, T. A Computationally Efficient Hybrid 3D Analytic-Numerical Approach for System Level Modelling of PEM Fuel Cells. In Proceedings of the 5th European PEFC and H2 Forum 2015, Lucerne, Switzerland, 30 June–3 July 2015. [Google Scholar]
- Sanders, I.J.; Peeten, T.L. Carbon Black: Production, Properties, and Uses; Chemical Engineering Methods and Technology; Nova Science Publishers, Nova Press: Hauppauge, NY, USA, 2011; ISBN 978-1-61209-535-6. [Google Scholar]
- Cherstiouk, O.V.; Simonov, A.N.; Moseva, N.S.; Cherepanova, S.V.; Simonov, P.A.; Zaikovskii, V.I.; Savinova, E.R. Microstructure Effects on the Electrochemical Corrosion of Carbon Materials and Carbon-Supported Pt Catalysts. Electrochim. Acta 2010, 55, 8453–8460. [Google Scholar] [CrossRef]
- Bezmalinović, D.; Radošević, J.; Barbir, F. Initial Conditioning of Polymer Eelectrolyte Membrane Fuel Cell by Temperature and Potential Cycling. Acta Chim. Slov. 2015, 62, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Tavčar, G.; Katrašnik, T. A Computationally Efficient Hybrid 3D Analytic-Numerical Approach for Modelling Species Transport in a Proton Exchange Membrane Fuel Cell. J. Power Sources 2013, 236, 321–340. [Google Scholar] [CrossRef]
- Tatschl, R.; Fink, C.; Tavčar, G.; Urthaler, P.; Katrašnik, T. A Scalable PEM Fuel Cell Modelling Approach to Support FCEV Component and System Development. In Proceedings of the 5th European Battery, Hybrid and Fuel Cell Electric Vehicle Congress, Geneva, Switzerland, 14–16 March 2017. [Google Scholar]
- Fink, C.; Fouquet, N. Three-Dimensional Simulation of Polymer Electrolyte Membrane Fuel Cells with Experimental Validation. Electrochim. Acta 2011, 56, 10820–10831. [Google Scholar] [CrossRef]
- Storn, R.; Price, K. Differential Evolution—A Simple and Efficient Heuristic for Global Optimization over Continuous Spaces. J. Glob. Optim. 1997, 11, 341–359. [Google Scholar] [CrossRef]
- The Mathworks, I., MATLAB Optimization Toolbox; The MathWorks: Matlab: Natick, MA, USA, 2018; Available online: https://www.mathworks.com/help/optim/ (accessed on 19 July 2021).
- Ziehn, T.; Tomlin, A.S. GUI–HDMR—A Software Tool for Global Sensitivity Analysis of Complex Models. Environ. Model. Softw. 2009, 24, 775–785. [Google Scholar] [CrossRef]
- Cherevko, S.; Keeley, G.P.; Geiger, S.; Zeradjanin, A.R.; Hodnik, N.; Kulyk, N.; Mayrhofer, K.J.J. Dissolution of Platinum in the Operational Range of Fuel Cells. ChemElectroChem 2015, 2, 1471–1478. [Google Scholar] [CrossRef] [Green Version]
- Kay, S.M. Fundamentals of Statistical Signal Processing: Estimation Theory; Prentice-Hall, Inc.: Upper Saddle River, NJ, USA, 1993; ISBN 0-13-345711-7. [Google Scholar]
- Kravos, A.; Ritzberger, D.; Hametner, C.; Jakubek, S.; Katrašnik, T. Methodology for Efficient Parametrisation of Electrochemical PEMFC Model for Virtual Observers: Model Based Optimal Design of Experiments Supported by Parameter Sensitivity Analysis. Int. J. Hydrogen Energy 2021, 46, 13832–13844. [Google Scholar] [CrossRef]
- Kravos, A.; Ritzberger, D.; Tavcar, G.; Hametner, C.; Jakubek, S.; Katrašnik, T. Thermodynamically Consistent Reduced Dimensionality Electrochemical Model for Proton Exchange Membrane Fuel Cell Performance Modelling and Control. J. Power Sources 2020, 454, 227930. [Google Scholar] [CrossRef]
- Multi-Year Research, Development, and Demonstration Plan, Section 3.4 Fuel Cells; Fuel Cell Technologies Office U.S. DOE: Washington, DC, USA, 2016.
- Strahl, S.; Husar, A.; Franco, A.A. Electrode Structure Effects on the Performance of Open-Cathode Proton Exchange Membrane Fuel Cells: A Multiscale Modeling Approach. Int. J. Hydrogen Energy 2014, 39, 9752–9767. [Google Scholar] [CrossRef] [Green Version]



| AST No. | Uupper (V) | Ulower (V) | tupper (min) | tlower (min) | Number of Cycles |
|---|---|---|---|---|---|
| 1 | 0.95 | 0.6 | 0.5 | 0.5 | 20 |
| 2 | 5 | 5 | 20 | ||
| 3 | 5 | 0.5 | 20 |
| AST No. | RMSD1 | RMSD3 |
|---|---|---|
| 1 | 0.3587 | 0.3959 |
| 2 | 0.4028 | 0.4541 |
| 3 | 0.7075 | 2.1347 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kravos, A.; Kregar, A.; Mayer, K.; Hacker, V.; Katrašnik, T. Identifiability Analysis of Degradation Model Parameters from Transient CO2 Release in Low-Temperature PEM Fuel Cell under Various AST Protocols. Energies 2021, 14, 4380. https://doi.org/10.3390/en14144380
Kravos A, Kregar A, Mayer K, Hacker V, Katrašnik T. Identifiability Analysis of Degradation Model Parameters from Transient CO2 Release in Low-Temperature PEM Fuel Cell under Various AST Protocols. Energies. 2021; 14(14):4380. https://doi.org/10.3390/en14144380
Chicago/Turabian StyleKravos, Andraž, Ambrož Kregar, Kurt Mayer, Viktor Hacker, and Tomaž Katrašnik. 2021. "Identifiability Analysis of Degradation Model Parameters from Transient CO2 Release in Low-Temperature PEM Fuel Cell under Various AST Protocols" Energies 14, no. 14: 4380. https://doi.org/10.3390/en14144380