Defect Process, Dopant Behaviour and Li Ion Mobility in the Li2MnO3 Cathode Material
by
 and
and
Navaratnarajah Kuganathan
1,2,*, 
Efstratia N. Sgourou
3,4,
Yerassimos Panayiotatos
4 and
Alexander Chroneos
1,2
1
Department of Materials, Imperial College London, London SW72AZ, UK
2
Faculty of Engineering, Environment and Computing, Coventry University, Priory Street, Coventry CV15FB, UK
3
Solid State Physics Section, University of Athens, Panepistimiopolis Zografos, 15784 Athens, Greece
4
Department of Mechanical Engineering, University of West Attica, 12210 Athens, Greece
*
Author to whom correspondence should be addressed.
Energies 2019, 12(7), 1329; https://doi.org/10.3390/en12071329
Submission received: 6 March 2019
/
Revised: 29 March 2019
/
Accepted: 2 April 2019
/
Published: 7 April 2019
(This article belongs to the Special Issue Investigate Energy Related Materials Using Advanced Modelling Techniques)
Abstract
:Lithium manganite, Li2MnO3, is an attractive cathode material for rechargeable lithium ion batteries due to its large capacity, low cost and low toxicity. We employed well-established atomistic simulation techniques to examine defect processes, favourable dopants on the Mn site and lithium ion diffusion pathways in Li2MnO3. The Li Frenkel, which is necessary for the formation of Li vacancies in vacancy-assisted Li ion diffusion, is calculated to be the most favourable intrinsic defect (1.21 eV/defect). The cation intermixing is calculated to be the second most favourable defect process. High lithium ionic conductivity with a low activation energy of 0.44 eV indicates that a Li ion can be extracted easily in this material. To increase the capacity, trivalent dopants (Al3+, Co3+, Ga3+, Sc3+, In3+, Y3+, Gd3+ and La3+) were considered to create extra Li in Li2MnO3. The present calculations show that Al3+ is an ideal dopant for this strategy and that this is in agreement with the experiential study of Al-doped Li2MnO3. The favourable isovalent dopants are found to be the Si4+ and the Ge4+ on the Mn site.
1. Introduction
The next generation of high capacity energy storage systems require lithium ion cathode material exhibiting high energy density, low cost and non-toxicity. A variety of cathode materials have been examined in the past decade to improve the performance of the rechargeable Li ion batteries [1,2,3,4,5,6,7,8,9,10,11]. As very few of them exhibit promising results, there is a necessity to make breakthroughs in finding new materials.
“Layered” Li2MnO3 was recently investigated as a potential cathode material for Li ion batteries due to its high theoretical capacity of 285 mAhg−1 and first charge plateau of ~4.5 eV [12,13]. Furthermore, manganese is relatively safe, abundant and low-cost, making Li2MnO3 a very promising cathode material. However, the material suffered from poor structural stability during cycling and electrical conductivity [14,15]. Li2MnO3 was initially identified as an inactive material because the electrochemical activity of the material via the oxidation of Mn4+ to Mn5+ did not occur [16,17]. The electrochemical activity was reinvestigated later and it was determined that the extraction and reinsertion of Li is possible. Chen et al. [18] showed that theoretically Li extraction can be charge compensated by the formation of O2 from O2− ions in the lattice. Cho et al. [19] demonstrated that oxygen loss is energetically favourable during delithiation. Electrochemical performance was recently examined by doping Al on the Mn site, and it was shown that Al-doped Li2MnO3 exhibits an enhancement on the rate capability and cycling stability [20].
To optimize the performance of Li ion batteries, a more detailed fundamental understanding of existing materials is necessary. Computational modelling techniques have significantly contributed to the characterization of experimental structures, prediction of pathways of migrating ions and identification of promising dopants in a variety of oxide materials [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. In the present study, we examine the intrinsic defects process, Li ion diffusion paths and the effect of dopants on the Mn site in Li2MnO3.
2. Computational Methods
Static atomistic calculations were performed on the crystal structure of Li2MnO3 and its defect structures using the General Utility Lattice Program (GULP) code [37]. This method is based on the classical Born model of ionic crystals. Interactions between ions include the long-range (i.e., Coulombic) ionic interactions and the short-range (i.e., electron–electron repulsion and van der Waals interactions) ionic interactions, with both being considered. Short-range repulsive forces were modelled using the Buckingham potentials (refer to Supplementary Information). The Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm [38] was applied to relax atom positions and lattice constants. In all optimized structures, forces on the atoms were smaller than 0.001 eV/Å. The point defects and migrating atoms were modelled using the Mott–Littleton method [39]. In this method, two spherical regions are defined, with the inner spherical region containing a number of ions greater than 700. In this region, ions are relaxed explicitly. Defect enthalpies in this simulation are expected to be overestimated as the ions are treated as spherical shapes with full charge at dilute limit. However, relative energies and trends remain consistent.
From a thermodynamic viewpoint, the defect parameters (for example, migration and formation energies) can be defined via the comparison of the real (defective) crystal to an isobaric or isochoric ideal (non-defective) crystal. These sets of defect formation parameters can be interconnected through thermodynamic relations as discussed in previous studies [40,41]. Here, the atomic scale calculations correspond to the isobaric parameters for the migration and formation processes [42,43].
3. Results
3.1. Bulk Li2MnO3 Structure
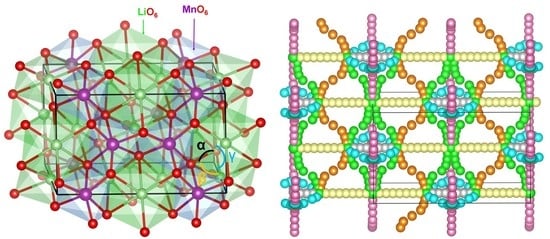
Figure 1 shows the experimentally observed crystal structure of Li2MnO3. This structure belongs to the monoclinic structure of the C2/m space group (lattice parameters a = 4.937 Å, b = 8.532 Å, c = 5.030 Å, α = 90.0°, β = 109.46° and γ = 90.0°) as reported by Strobel et al. [44] in their single crystal X-ray diffraction. The crystal structure of Li2MnO3 was subsequently reinvestigated by Boulineau et al. [45] and their reported structure was closer to the previous structure, with a small amount of cation mixing. Here, we used the crystal structure reported by Strobel et al. [44], as this model consisted of full occupancy atom positions. Both Li and Mn form edge-sharing distorted octahedral units with adjacent O atoms, as shown in the Figure 1.
Both atom positions and lattice constants were allowed to relax under constant pressure to obtain the equilibrium lattice structure. The calculated lattice constants were in excellent agreement with the experimental values, as reported in Table 1.
3.2. Intrinsic Defect Processes
Next, defect formation energies for the isolated vacancy, interstitial and anti-site defect were calculated, and were combined to calculate the Frenkel, Schottky and anti-site intrinsic defect reaction energies (Equations (1)–(8)). Intrinsic defect processes are useful to study the electrochemical behaviour of Li2MnO3. The following reactions, which were written by using Kröger–Vink notation [46], represent the Frenkel, Schottky and anti-site intrinsic defect processes:
We report the reaction energies for these intrinsic defect processes in Figure 2. The Li Frenkel was calculated to be the most thermodynamically favourable intrinsic defect process. This process increases the concentration of Li vacancies that can enhance vacancy-assisted Li ion diffusion in Li2MnO3. Other Frenkel defect processes exhibit highly endoergic energies, suggesting that they are unlikely to occur at operating temperatures. The second most favourable defect process is the Li–Mn anti-site. In this defect, a small percentage of Li on Mn sites () and Mn on Li sites ( would be observed at high temperatures. Anti-site defect has been observed in experimental and theoretical studies during the synthesis of as-prepared material and cycling [47,48,49,50,51,52]. The Li2O Schottky reaction (Equation (5)), leading to the formation of further and , is 5.16 eV per defect, indicating that this reaction can only take place at high temperatures.
3.3. Lithium Ion Diffusion
There is a necessity to understand the lithium ion diffusion paths together with activation energies to assess Li2MnO3 as a potential high-capacity cathode material for Li ion batteries. Determining Li ion diffusion paths is extremely challenging experimentally. Classical pair potential-based simulation can provide valuable information about various possible Li ion diffusion paths and corresponding activation energies. Low activation energy is a key requirement for a promising high-rate battery material. Possible Li vacancy-assisted diffusion paths were constructed. Six different local Li hops (Figure 3) were identified for the Li vacancy migration. Table 2 reports the Li–Li separation, together with corresponding activation energies. Figure 4 shows the energy profile diagrams for activation energies of local Li hops.
Five possible long-range paths consisting of local Li hops with lower overall activation energies were identified (Table 3). The first long-range path (intra layer along the b axis) exhibited a slightly distorted linear pattern (A→E→E→A), with an overall activation energy of 0.59 eV. The second path connected local hops B and F, forming a curved trajectory (F→B→B→B) along the ab plane (intra layer), with an overall activation energy of 0.47 eV. The third long-range path (C→C→C→C) lay between layers (inter layer), with the Li local hops of C. The activation energy for this migration was calculated to be 0.44 eV. This was the lowest activation energy of the four intra layer Li migration paths. In the fourth long-range path (D→D→D→D), the Li ion migrated in the bc plane (intra layer), with an overall migration energy of 0.45 eV. Finally, the fifth long-range path (D→D→D→D) was located in the ab plane (intra layer) and the activation energy for this path was 0.47 eV. The current results show that the Li ion would diffuse fast in Li2MnO3 via intra layers or inter layers.
3.4. Trivalent Doping
The capacity of Li2MnO3 can be increased by incorporating additional Li in the form of interstitials into the as-prepared crystal structure. This would increase its applicability in rechargeable lithium batteries. Doping trivalent cations on the Mn site is an efficient engineering strategy to create Li interstitials in the lattice. In previous work, this strategy has been applied to Li and Na ion battery materials. In this work, we considered some trivalent dopants from different parts of the periodic table (early transition elements, post-transition elements and lanthanide elements). The selection of the prominent dopant Al3+ is due to its small size and low cost. Furthermore, there is an experimental report on the doping of Al3+ on the Mn site [20]. The solution of R2O3 (R = Al, Co, Ga, Sc, In, Y, Gd and La) was considered via the following process (in Kröger–Vink notation):
Figure 5 reports the solution enthalpies of R2O3 calculated using the classical pair-potential method. The present calculations show that the most favourable dopant on the Mn site is Al3+. This indicates that the extra lithium can be incorporated in the form of interstitials into Li2MnO3. The exact concentration of the composition can be provided by an experimental study. The possible composition of Al-doped Li2MnO3 is Li2+xMn1−xAlxO3 (x = 0.0, …, 1.0).
Xiang et al. [20] synthesised both Li2MnO3 and Al-doped Li2MnO3 and examined their rate capacities. Their study shows that there is a greater improvement in the Al-doped Li2MnO3 compared with that of pristine Li2MnO3. The second most favourable dopant is the Co3+ and its solution enthalpy is calculated to be 0.52 eV, only 0.07 eV higher than that of Al3+. Solution enthalpy increases gradually with the ionic radius of M3+ ions reflecting in the bond lengths and bond angles. The optimised bond lengths and bond angles of trivalent dopants occupying the Mn site and the octahedral MnO6 unit in the relaxed structure of undoped Li2MnO3 are shown in Figure 6. The highest solution enthalpy is calculated for La3+. This is due to the larger ionic radius difference between the La3+ and the Mn4+. The current solution enthalpy values for Sc3+, In3+, Y3+, Gd3+ and La3+ are highly endoergic, suggesting that they are unfavourable at operating temperatures.
Density functional theory calculations performed by Hoang et al. [53] show that Al and Fe are energetically favourable dopants on the Mn site, which agrees with our calculations. Kong et al. [54] used ab initio simulations to examine the thermodynamical stability of a variety of aliovalent dopants, including trivalent dopants Al3+ and Fe3+ on the Mn site. In their study, charge introduced by cation doping was compensated by anion doping (F and N) on the O site. In our study, negative charge introduced by trivalent dopants on the Mn site was compensated by positively charged Li interstitials, creating a Li2MnO3 material with a high capacity.
3.5. Tetravalent Doping
Here, we consider some isovalent dopants (Si4+, Ge4+, Ti4+, Sn4+, Zr4+ and Ce4+) on the Mn site. The following reaction equation was used to calculate the solution enthalpy:
Solution enthalpy increases with ionic radius. Exoergic solution enthalpy was calculated for Si4+ and Ge4+ (Figure 7). This was due to the smaller ionic radius of Si4+ (0.40 Å) compared with Mn4+ (0.53 Å). The higher charge density of Si4+ forms stronger Si–O bonds, as reported in Figure 8. The ionic radii of Ge4+ and Mn4+ are the same. This is reflected in the exoergic solution enthalpy. The optimised structures of MO6 units together with bond lengths and bond angles are shown in Figure 8. Endoergic solution enthalpies are observed for the other dopants. Solution enthalpy for CeO2 is highly endoergic, meaning that doping Ce on the Mn site is highly unlikely to occur.
4. Conclusions
In conclusion, we have used atomistic simulation techniques to examine intrinsic defects, Li ion diffusion pathways with activation energies and favourable trivalent and tetravalent dopants on the Mn site in Li2MnO3. The lowest defect energy process was calculated to be the Li Frenkel, which will ensure the number of Li vacancies that are necessary for vacancy-assisted Li diffusion. The second most favourable defect process was found to be the cation mixing. Diffusion of lithium with the low activation energy of 0.44 eV suggests that high ionic conductivity would be observed in Li2MnO3. Doping of Al on the Mn site is an efficient strategy to increase the Li content, as reported in the experiment. The favourable isovalent dopants were calculated to be the Si4+ and Ge4+. These theoretical predictions require experimental verification.
Supplementary Materials
The following are available online at https://www.mdpi.com/1996-1073/12/7/1329/s1, Table S1: Interatomic potential parameters used in the atomistic simulations of Li2MnO3.
Author Contributions
Computation, N.K.; writing, N.K.; analysis and editing, E.N.S., Y.P. and A.C.
Funding
The research leading to these results received funding from the European Union’s H2020 Programme under Grant Agreement no 824072–HARVESTORE.
Acknowledgments
We acknowledge the computational facilities and support provided by the High Performance Computing Center at Imperial College.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Armand, M.; Tarascon, J.M. Building better batteries. Nature 2008, 451, 652. [Google Scholar] [CrossRef] [PubMed]
- Whittingham, M.S. Lithium batteries and cathode materials. Chem. Rev. 2004, 104, 4271–4302. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Nytén, A.; Abouimrane, A.; Armand, M.; Gustafsson, T.; Thomas, J.O. Electrochemical performance of Li2FeSiO4 as a new Li-battery cathode material. Electrochem. Commun. 2005, 7, 156–160. [Google Scholar] [CrossRef]
- Nishimura, S.-I.; Hayase, S.; Kanno, R.; Yashima, M.; Nakayama, N.; Yamada, A. Structure of Li2FeSiO4. J. Am. Chem. Soc. 2008, 130, 13212–13213. [Google Scholar] [CrossRef]
- Armstrong, A.R.; Kuganathan, N.; Islam, M.S.; Bruce, P.G. Structure and lithium transport pathways in li2fesio4 cathodes for lithium batteries. J. Am. Chem. Soc. 2011, 133, 13031–13035. [Google Scholar] [CrossRef]
- Masquelier, C.; Croguennec, L. Polyanionic (phosphates, silicates, sulfates) frameworks as electrode materials for rechargeable Li (or Na) batteries. Chem. Rev. 2013, 113, 6552–6591. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, G.; Liu, Z.; Li, H.; Liu, Y.; Wang, Z.; Li, X.; Shih, K.; Mai, L. Li3V(MoO4)3 as a novel electrode material with good lithium storage properties and improved initial coulombic efficiency. Nano Energy 2018, 44, 272–278. [Google Scholar] [CrossRef]
- Recham, N.; Chotard, J.N.; Dupont, L.; Delacourt, C.; Walker, W.; Armand, M.; Tarascon, J.M. A 3.6 V lithium-based fluorosulphate insertion positive electrode for lithium-ion batteries. Nat. Mater. 2009, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Afyon, S.; Wörle, M.; Nesper, R. A lithium-rich compound Li7Mn(BO3)3 containing Mn2+ in tetrahedral coordination: A cathode candidate for lithium-ion batteries. Angew. Chemie Inter. Ed. 2013, 52, 12541–12544. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.-I.; Nakamura, M.; Natsui, R.; Yamada, A. New lithium iron pyrophosphate as 3.5 V class cathode material for lithium ion battery. J. Am. Chem. Soc. 2010, 132, 13596–13597. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, M.M.; Johnson, C.S.; Vaughey, J.T.; Li, N.; Hackney, S.A. Advances in manganese-oxide ‘composite’ electrodes for lithium-ion batteries. J. Mater. Chem. 2005, 15, 2257–2267. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Kim, Y. Challenges for rechargeable Li batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Rana, J.; Stan, M.; Kloepsch, R.; Li, J.; Schumacher, G.; Welter, E.; Zizak, I.; Banhart, J.; Winter, M. Structural changes in Li2MnO3 cathode material for Li-ion batteries. Adv. Energy Mater. 2014, 4, 1300998. [Google Scholar] [CrossRef]
- Francis Amalraj, S.; Markovsky, B.; Sharon, D.; Talianker, M.; Zinigrad, E.; Persky, R.; Haik, O.; Grinblat, J.; Lampert, J.; Schulz-Dobrick, M.; et al. Study of the electrochemical behavior of the “inactive” Li2MnO3. Electrochim. Acta 2012, 78, 32–39. [Google Scholar] [CrossRef]
- Ammundsen, B.; Paulsen, J. Novel lithium-ion cathode materials based on layered manganese oxides. Adv. Mater. 2001, 13, 943–956. [Google Scholar] [CrossRef]
- Robertson, A.D.; Bruce, P.G. The origin of electrochemical activity in Li2MnO3. Chem. Commun. 2002, 2790–2791. [Google Scholar] [CrossRef]
- Chen, H.; Islam, M.S. Lithium extraction mechanism in Li-Rich Li2MnO3 involving oxygen hole formation and dimerization. Chem. Mater. 2016, 28, 6656–6663. [Google Scholar] [CrossRef]
- Cho, E.; Kim, K.; Jung, C.; Seo, S.-W.; Min, K.; Lee, H.S.; Park, G.-S.; Shin, J. Overview of the oxygen behavior in the degradation of Li2MnO3 cathode material. J. Phys. Chem. C 2017, 121, 21118–21127. [Google Scholar] [CrossRef]
- Xiang, Y.; Wu, X. Enhanced electrochemical performances of Li2MnO3 cathode materials by Al doping. Ionics 2018, 24, 83–89. [Google Scholar] [CrossRef]
- Kuganathan, N.; Islam, M.S. Li2MnSiO4 lithium battery material: Atomic-scale study of defects, lithium mobility, and trivalent dopants. Chem. Mater. 2009, 21, 5196–5202. [Google Scholar] [CrossRef]
- Fisher, C.A.J.; Kuganathan, N.; Islam, M.S. Defect chemistry and lithium-ion migration in polymorphs of the cathode material Li2MnSiO4. J. Mater. Chem. A 2013, 1, 4207–4214. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Anurakavan, S.; Iyngaran, P.; Chroneos, A. Li3SbO4 lithium-ion battery material: Defects, lithium ion diffusion and tetravalent dopants. Mater. Chem. Phys. 2019, 225, 34–41. [Google Scholar] [CrossRef]
- Kordatos, A.; Kuganathan, N.; Kelaidis, N.; Iyngaran, P.; Chroneos, A. Defects and lithium migration in Li2CuO2. Sci. Rep. 2018, 8, 6754. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Chroneos, A. Defects, dopants and sodium mobility in Na2MnSiO4. Sci. Rep. 2018, 8, 14669. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Defects and dopant properties of Li3V2(PO4)3. Sci. Rep. 2019, 9, 333. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Ganeshalingam, S.; Chroneos, A. Defects, dopants and lithium mobility in Li9V3(P2O7)3 (PO4)2. Sci. Rep. 2018, 8, 8140. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Iyngaran, P.; Chroneos, A. Lithium diffusion in Li5FeO4. Sci. Rep. 2018, 8, 5832. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Chroneos, A. Li2SnO3 as a cathode material for lithium-ion batteries: Defects, lithium ion diffusion and dopants. Sci. Rep. 2018, 8, 12621. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Chroneos, A. Defect chemistry and Li-ion diffusion in Li2RuO3. Sci. Rep. 2019, 9, 550. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Kordatos, A.; Kelaidis, N.; Chroneos, A. Defects, lithium mobility and tetravalent dopants in the Li3NbO4 cathode material. Sci. Rep. 2019, 9, 2192. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Kordatos, A.; Fitzpatrick, M.E.; Vovk, R.V.; Chroneos, A. Defect process and lithium diffusion in Li2TiO3. Solid State Ionics 2018, 327, 93–98. [Google Scholar] [CrossRef]
- Kuganathan, N.; Tsoukalas, L.H.; Chroneos, A. Defects, dopants and Li-ion diffusion in Li2SiO3. Solid State Ionics 2019, 335, 61–66. [Google Scholar] [CrossRef]
- Araújo, C.M.; Blomqvist, A.; Scheicher, R.H.; Chen, P.; Ahuja, R. Superionicity in the hydrogen storage material Li2NH: Molecular dynamics simulations. Phys. Rev. B 2009, 79, 172101. [Google Scholar] [CrossRef]
- Seymour, I.D.; Chroneos, A.; Kilner, J.A.; Grimes, R.W. Defect processes in orthorhombic LnBaCo2O5.5 double perovskites. Phys. Chem. Chem. Phys. 2011, 13, 15305–15310. [Google Scholar] [CrossRef] [PubMed]
- Jay, E.E.; Rushton, M.J.D.; Chroneos, A.; Grimes, R.W.; Kilner, J.A. Genetics of superionic conductivity in lithium lanthanum titanates. Phys. Chem. Chem. Phys. 2015, 17, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Gale, J.D.; Rohl, A.L. The general utility lattice program (GULP). Molec. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Gale, J.D. GULP: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Mott, N.F.; Littleton, M.J. Conduction in polar crystals. I. Electrolytic conduction in solid salts. Trans. Faraday Soc. 1938, 34, 485–499. [Google Scholar] [CrossRef]
- Varotsos, P. Defect volumes and the equation of state in α-PbF2. Phys. Rev. B 2007, 76, 092106. [Google Scholar] [CrossRef]
- Varotsos, P. Comparison of models that interconnect point defect parameters in solids with bulk properties. J. Appl. Phys. 2007, 101, 123503. [Google Scholar] [CrossRef]
- Chroneos, A.; Vovk, R.V. Modeling self-diffusion in UO2 and ThO2 by connecting point defect parameters with bulk properties. Solid State Ionics 2015, 274, 1–3. [Google Scholar] [CrossRef]
- Chroneos, A. Connecting point defect parameters with bulk properties to describe diffusion in solids. Appl. Phys. Rev. 2016, 3, 041304. [Google Scholar] [CrossRef]
- Strobel, P.; Lambert-Andron, B. Crystallographic and magnetic structure of Li2MnO3. J. Solid State Chem. 1988, 75, 90–98. [Google Scholar] [CrossRef]
- Boulineau, A.; Croguennec, L.; Delmas, C.; Weill, F. Reinvestigation of Li2MnO3 structure: Electron diffraction and high resolution TEM. Chem. Mater. 2009, 21, 4216–4222. [Google Scholar] [CrossRef]
- Kröger, F.A.; Vink, H.J. Relations between the concentrations of imperfections in crystalline solids. In Solid State Physics; Seitz, F., Turnbull, D., Eds.; Academic Press: New York, NY, USA, 1956; Volume 3, pp. 307–435. [Google Scholar]
- Politaev, V.V.; Petrenko, A.A.; Nalbandyan, V.B.; Medvedev, B.S.; Shvetsova, E.S. Crystal structure, phase relations and electrochemical properties of monoclinic Li2MnSiO4. J. Solid State Chem. 2007, 180, 1045–1050. [Google Scholar] [CrossRef]
- Ensling, D.; Stjerndahl, M.; Nytén, A.; Gustafsson, T.; Thomas, J.O. A comparative XPS surface study of Li2FeSiO4/C cycled with LiTFSI- and LiPF6-based electrolytes. J. Mater. Chem. 2009, 19, 82–88. [Google Scholar] [CrossRef]
- Liu, H.; Choe, M.-J.; Enrique, R.A.; Orvañanos, B.; Zhou, L.; Liu, T.; Thornton, K.; Grey, C.P. Effects of antisite defects on li diffusion in LiFePO4 revealed by Li isotope exchange. J. Phys. Chem. C 2017, 121, 12025–12036. [Google Scholar] [CrossRef]
- Kempaiah Devaraju, M.; Duc Truong, Q.; Hyodo, H.; Sasaki, Y.; Honma, I. Synthesis, characterization and observation of antisite defects in LiNiPO4 nanomaterials. Sci. Rep. 2015, 5, 11041. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Na3V(PO4)2 cathode material for Na ion batteries: Defects, dopants and Na diffusion. Solid State Ionics 2019, 336, 75–79. [Google Scholar] [CrossRef]
- Kuganathan, N.; Iyngaran, P.; Vovk, R.; Chroneos, A. Defects, dopants and Mg diffusion in MgTiO3. Sci. Rep. 2019, 9, 4394. [Google Scholar] [CrossRef] [PubMed]
- Hoang, K. Doping Li-rich cathode material Li2MnO3: Interplay between lattice site preference, electronic structure, and delithiation mechanism. Phys. Rev. Mater. 2017, 1, 075404. [Google Scholar] [CrossRef]
- Kong, F.; Longo, R.C.; Park, M.-S.; Yoon, J.; Yeon, D.-H.; Park, J.-H.; Wang, W.-H.; Kc, S.; Doo, S.-G.; Cho, K. Ab initio study of doping effects on LiMnO2 and Li2MnO3 cathode materials for Li-ion batteries. J. Mater. Chem. A 2015, 3, 8489–8500. [Google Scholar] [CrossRef]
Figure 1.
Crystal structure of Li2MnO3 (space group C 2/m).

Figure 2.
Energetics of intrinsic defect process calculated in monoclinic Li2MnO3.

Figure 3.
Different Li trajectory views (a) and (b) possible long-range lithium vacancy migration paths considered. Yellow, light blue, purple, green, grey and light brown color atoms correspond to different Li hopping trajectories.
Figure 3.
Different Li trajectory views (a) and (b) possible long-range lithium vacancy migration paths considered. Yellow, light blue, purple, green, grey and light brown color atoms correspond to different Li hopping trajectories.

Figure 4.
Six different energy profiles (as shown in Figure 3) of Li vacancy hopping between two adjacent Li sites in Li2MnO3.
Figure 4.
Six different energy profiles (as shown in Figure 3) of Li vacancy hopping between two adjacent Li sites in Li2MnO3.

Figure 5.
Enthalpy of solution of R2O3 (R = Al, Co, Ga, Sc, In, Y, Gd and La) with respect to the R3+ ionic radius in Li2MnO3.
Figure 5.
Enthalpy of solution of R2O3 (R = Al, Co, Ga, Sc, In, Y, Gd and La) with respect to the R3+ ionic radius in Li2MnO3.

Figure 6.
Distorted octahedral MnO6 units in the relaxed structure of undoped Li2MnO3.

Figure 7.
Enthalpy of solution of RO2 (R = Si, Ge, Ti, Sn, Zr and Ce) with respect to the R4+ ionic radius in Li2MnO3.
Figure 7.
Enthalpy of solution of RO2 (R = Si, Ge, Ti, Sn, Zr and Ce) with respect to the R4+ ionic radius in Li2MnO3.

Figure 8.
Distorted octahedral MnO6 units in the relaxed structure of undoped Li2MnO3 and the coordination formed by the isovalent (tetravalent) dopants on the Mn site with neighbour oxygen.
Figure 8.
Distorted octahedral MnO6 units in the relaxed structure of undoped Li2MnO3 and the coordination formed by the isovalent (tetravalent) dopants on the Mn site with neighbour oxygen.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated structural parameters and corresponding experimental values [44] reported for monoclinic (C2/m) Li2MnO3.
Table 1.
Calculated structural parameters and corresponding experimental values [44] reported for monoclinic (C2/m) Li2MnO3.
| Parameter | Calculated | Experiment [44] | |∆|(%) |
|---|---|---|---|
| a (Å) | 4.8715 | 4.9370 | 1.33 |
| b (Å) | 8.3519 | 8.5320 | 2.11 |
| c (Å) | 5.0177 | 5.0300 | 0.24 |
| α (°) | 90.0 | 90.0 | 0.00 |
| β (°) | 110.07 | 109.46 | 0.56 |
| γ (°) | 90.0 | 90.0 | 0.00 |
Table 2.
Calculated Li–Li separations and activation energies using classical pair-potential method for the lithium ion migration between two adjacent Li sites (as shown in Figure 3).
Table 2.
Calculated Li–Li separations and activation energies using classical pair-potential method for the lithium ion migration between two adjacent Li sites (as shown in Figure 3).
| Migration Path | Li–Li Separation (Å) | Activation Energy (eV) |
|---|---|---|
| A | 2.65 | 0.27 |
| B | 2.77 | 0.37 |
| C | 2.83 | 0.44 |
| D | 2.84 | 0.45 |
| E | 2.85 | 0.59 |
| F | 2.87 | 0.47 |
Table 3.
Possible long-range Li ion diffusion paths and their corresponding overall activation energies.
Table 3.
Possible long-range Li ion diffusion paths and their corresponding overall activation energies.
| Long-Range Path | Direction | Overall Activation Energy (eV) |
|---|---|---|
| A→E→E→A | b axis | 0.59 |
| F→B→B→ B | ab plane | 0.47 |
| C→C→C→C | ac plane | 0.44 |
| D→D→D→D | bc plane | 0.45 |
| F→F→F→F | ab plane | 0.47 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kuganathan, N.; Sgourou, E.N.; Panayiotatos, Y.; Chroneos, A. Defect Process, Dopant Behaviour and Li Ion Mobility in the Li2MnO3 Cathode Material. Energies 2019, 12, 1329. https://doi.org/10.3390/en12071329
AMA Style
Kuganathan N, Sgourou EN, Panayiotatos Y, Chroneos A. Defect Process, Dopant Behaviour and Li Ion Mobility in the Li2MnO3 Cathode Material. Energies. 2019; 12(7):1329. https://doi.org/10.3390/en12071329
Chicago/Turabian StyleKuganathan, Navaratnarajah, Efstratia N. Sgourou, Yerassimos Panayiotatos, and Alexander Chroneos. 2019. "Defect Process, Dopant Behaviour and Li Ion Mobility in the Li2MnO3 Cathode Material" Energies 12, no. 7: 1329. https://doi.org/10.3390/en12071329
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.