
Theoretical Investigation of Anhydrofusarubin: Structural and Optical Properties

 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Methodological Part
3. Results and Discussion
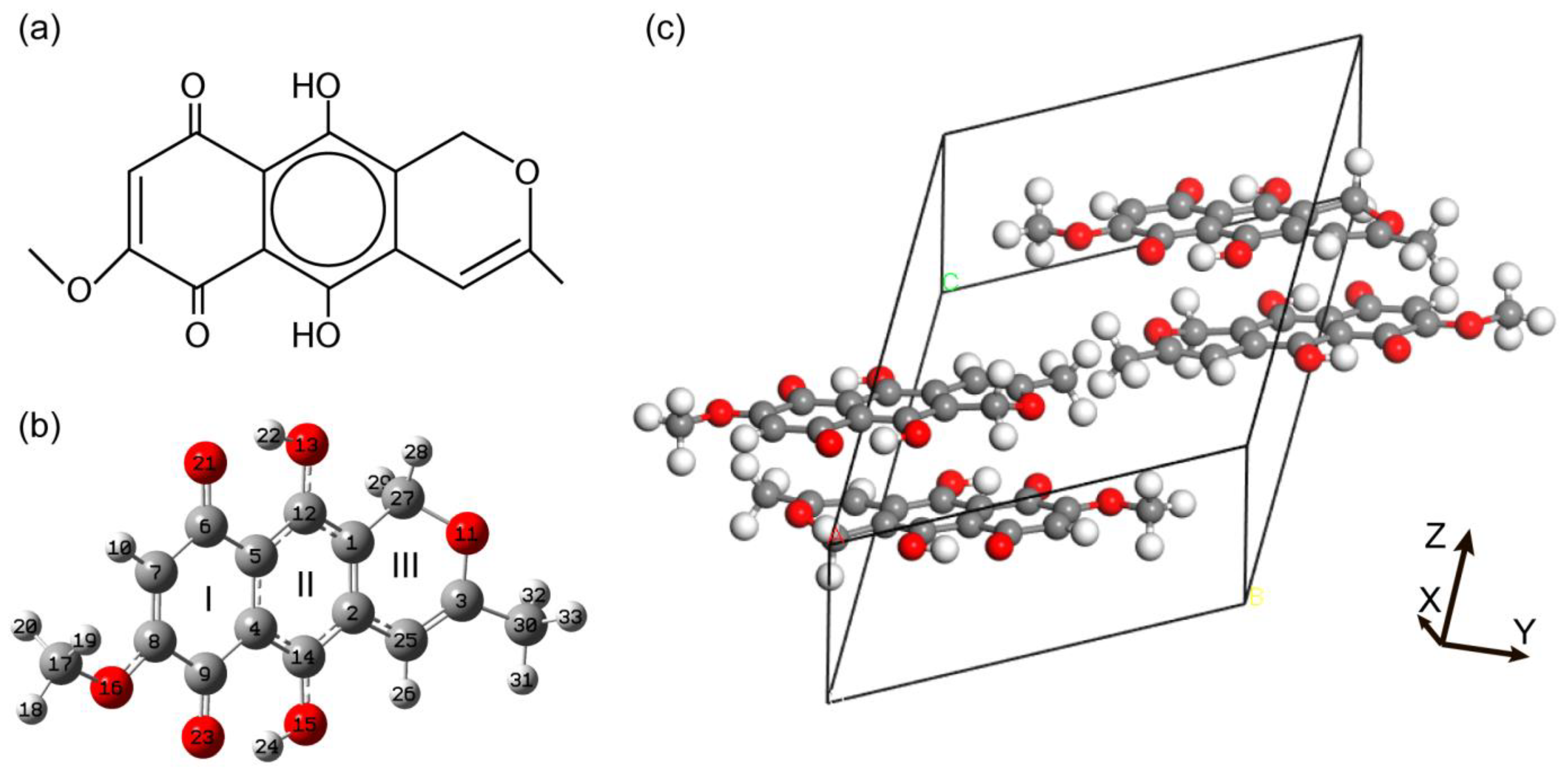
3.1. Structural Peculiarities
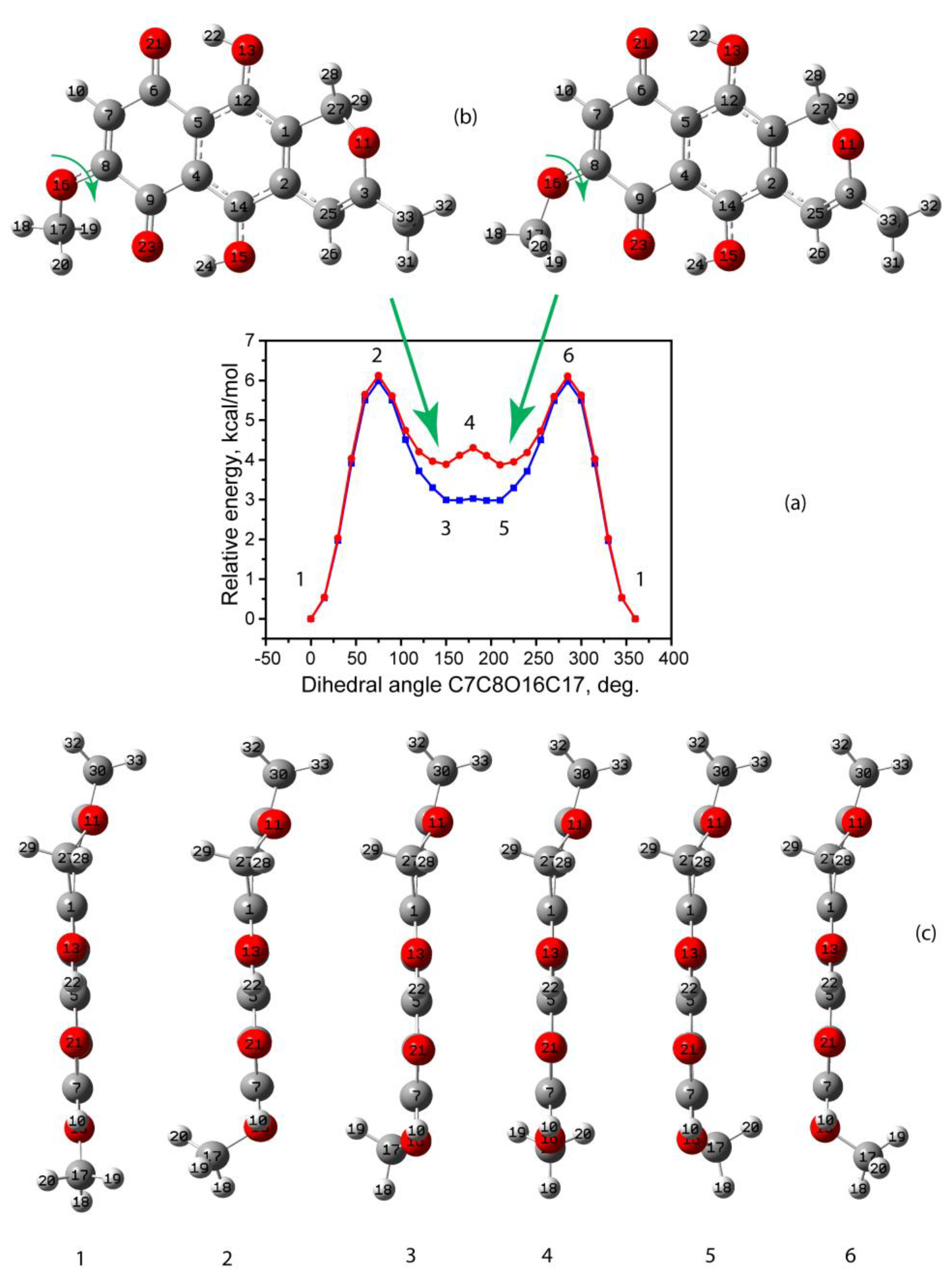
3.1.1. Conformational Enantiomers of Anhydrofusarubin
3.1.2. Tautomerism in Anhydrofusarubin
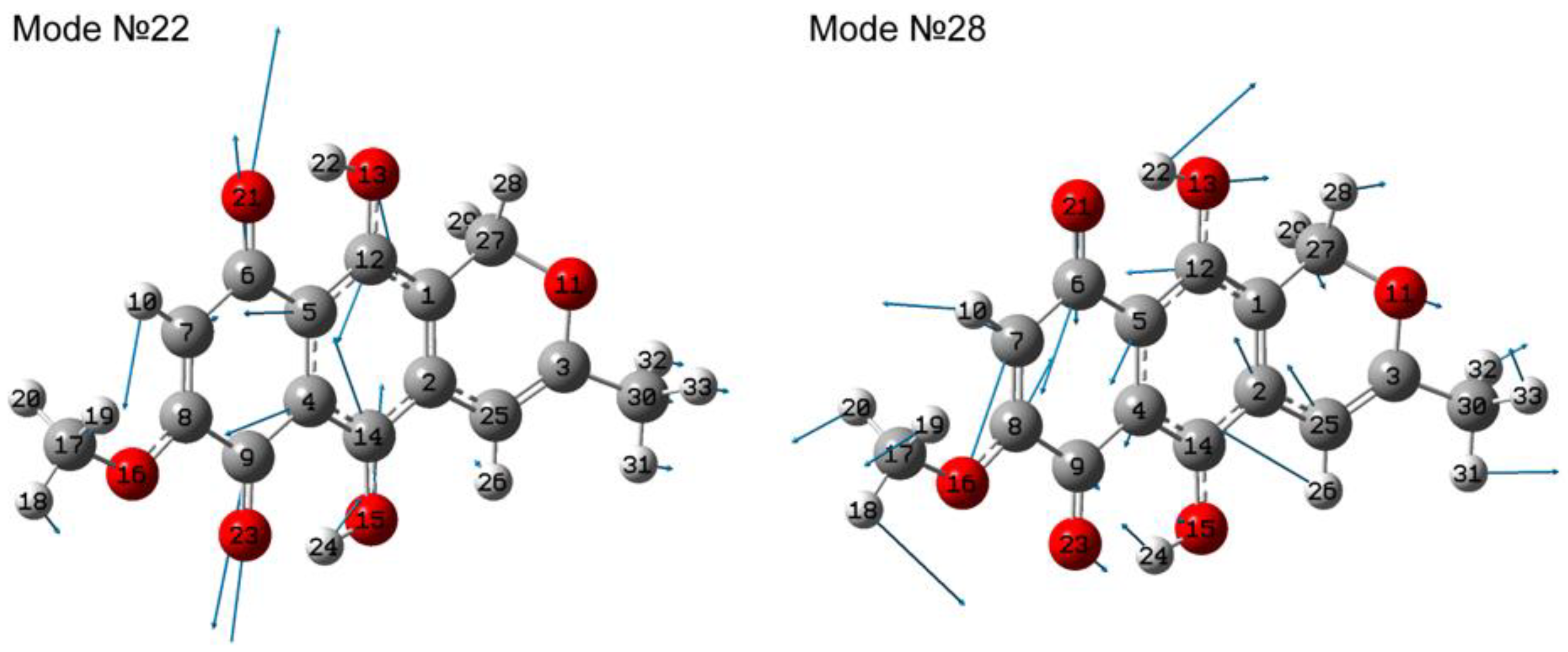
3.2. IR absorbance Spectroscopy
3.3. UV-Vis Absorbance Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Toghueo, R.M.K. Bioprospecting Endophytic Fungi from Fusarium Genus as Sources of Bioactive Metabolites. Mycology 2020, 11, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.M.; Mahmoud, B.K.; Millán-Aguiñaga, N.; Abdelmohsen, U.R.; Fouad, M.A. The Endophytic Fusarium Strains: A Treasure Trove of Natural Products. RSC Adv. 2023, 13, 1339–1369. [Google Scholar] [CrossRef] [PubMed]
- Kundu, A.; Mandal, A.; Saha, S.; Prabhakaran, P.; Walia, S. Fungicidal Activity and Molecular Modeling of Fusarubin Analogues from Fusarium oxysporum. Toxicol. Environ. Chem. 2020, 102, 78–91. [Google Scholar] [CrossRef]
- Daniel, J.J.; Zabot, G.L.; Tres, M.V.; Harakava, R.; Kuhn, R.C.; Mazutti, M.A. Fusarium Fujikuroi: A Novel Source of Metabolites with Herbicidal Activity. Biocatal. Agric. Biotechnol. 2018, 14, 314–320. [Google Scholar] [CrossRef]
- Spanic, V.; Katanic, Z.; Sulyok, M.; Krska, R.; Puskas, K.; Vida, G.; Drezner, G.; Šarkanj, B. Multiple Fungal Metabolites Including Mycotoxins in Naturally Infected and Fusarium-Inoculated Wheat Samples. Microorganisms 2020, 8, 578. [Google Scholar] [CrossRef]
- Cuperlovic-Culf, M.; Wang, L.; Forseille, L.; Boyle, K.; Merkley, N.; Burton, I.; Fobert, P.R. Metabolic Biomarker Panels of Response to Fusarium Head Blight Infection in Different Wheat Varieties. PLoS ONE 2016, 11, e0153642. [Google Scholar] [CrossRef]
- Shinha, K.K.; Bhatnagar, D. Mycotoxins in Agriculture and Food Safety; CRC Press: Boca Raton, FL, USA, 1998. [Google Scholar]
- Pankin, D.; Povolotckaia, A.; Kalinichev, A.; Povolotskiy, A.; Borisov, E.; Moskovskiy, M.; Gulyaev, A.; Lavrov, A.; Izmailov, A. Complex Spectroscopic Study for Fusarium Genus Fungi Infection Diagnostics of “Zalp” Cultivar Oat. Agronomy 2021, 11, 2402. [Google Scholar] [CrossRef]
- Gámiz-Gracia, L.; García-Campaña, A.M.; Arroyo-Manzanares, N. Application of LC–MS/MS in the Mycotoxins Studies. Toxins 2020, 12, 272. [Google Scholar] [CrossRef]
- Pankin, D.; Smirnov, M.; Povolotckaia, A.; Povolotskiy, A.; Borisov, E.; Moskovskiy, M.; Gulyaev, A.; Gerasimenko, S.; Aksenov, A.; Litvinov, M.; et al. DFT Modelling of Molecular Structure, Vibrational and UV-Vis Absorption Spectra of T-2 Toxin and 3-Deacetylcalonectrin. Materials 2022, 15, 649. [Google Scholar] [CrossRef]
- Makino, T.; Kato, K.; Lyozumi, H.; Honzawa, H.; Tachiiri, Y.; Hiramatsu, M. Ultraweak Luminescence Generated by Sweet Potato and Fusarium oxysporum Interactions Associated with a Defense Response. Photochem. Photobiol. 1996, 64, 953–956. [Google Scholar] [CrossRef]
- Moskovskiy, M.N.; Belyakov, M.V.; Dorokhov, A.S.; Boyko, A.A.; Belousov, S.V.; Noy, O.V.; Gulyaev, A.A.; Akulov, S.I.; Povolotskaya, A.; Efremenkov, I.Y. Design of Device for Optical Luminescent Diagnostic of the Seeds Infected by Fusarium. Agriculture 2023, 13, 619. [Google Scholar] [CrossRef]
- Pankin, D.; Povolotckaia, A.; Borisov, E.; Povolotskiy, A.; Borzenko, S.; Gulyaev, A.; Gerasimenko, S.; Dorochov, A.; Khamuev, V.; Moskovskiy, M. Investigation of Spectroscopic Peculiarities of Ergot-Infected Winter Wheat Grains. Foods 2023, 12, 3426. [Google Scholar] [CrossRef] [PubMed]
- Dorokhov, A.; Moskovskiy, M.; Belyakov, M.; Lavrov, A.; Khamuev, V. Detection of Fusarium Infected Seeds of Cereal Plants by the Fluorescence Method. PLoS ONE 2022, 17, e0267912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yu, X.; Wen, K.; Li, C.; Mujtaba Mari, G.; Jiang, H.; Shi, W.; Shen, J.; Wang, Z. Multiplex Lateral Flow Immunoassays Based on Amorphous Carbon Nanoparticles for Detecting Three Fusarium Mycotoxins in Maize. J. Agric. Food Chem. 2017, 65, 8063–8071. [Google Scholar] [CrossRef]
- Pillay, A.; Rousseau, A.L.; Fernandes, M.A.; de Koning, C.B. The Synthesis of the Pyranonaphthoquinones Dehydroherbarin and Anhydrofusarubin Using Wacker Oxidation Methodology as a Key Step and Other Unexpected Oxidation Reactions with Ceric Ammonium Nitrate and Salcomine. Org. Biomol. Chem. 2012, 10, 7809. [Google Scholar] [CrossRef]
- Wu, Q.; Patocka, J.; Nepovimova, E.; Kuca, K. A Review on the Synthesis and Bioactivity Aspects of Beauvericin, a Fusarium Mycotoxin. Front. Pharmacol. 2018, 9, 1338. [Google Scholar] [CrossRef]
- Adeleke, B.; Babalola, O. Pharmacological Potential of Fungal Endophytes Associated with Medicinal Plants: A Review. J. Fungi 2021, 7, 147. [Google Scholar] [CrossRef]
- Tatum, J.H.; Baker, R.A. Naphthoquinones Produced by Fusarium Solani Isolated from Citrus. Phytochemistry 1983, 22, 543–547. [Google Scholar] [CrossRef]
- Shao, C.-L.; Wang, C.-Y.; Deng, D.-S.; She, Z.-G.; Gu, Y.-C.; Lin, Y.-C. Crystal Structure of a Marine Natural Compound, Anhydrofusarubin. Chin. J. Struct. Chem. 2008, 27, 824–828. [Google Scholar]
- Tatum, J.H.; Baker, R.A.; Berry, R.E. Metabolites of Fusarium Solani. Phytochemistry 1989, 28, 283–284. [Google Scholar] [CrossRef]
- Tadpetch, K.; Vijitphan, P. Synthesis of 8-0-Methylfusarubin, 8-0-Methylanhydrofusarubin, Fusarubin and Anhydrofusarubin. Doctoral Dissertation, Prince of Songkla University, Hat Yai, Thailand, 2019. [Google Scholar]
- Suzuki, M.; Nishida, N.; Ishihara, A.; Nakajima, H. New 3-O-Alkyl-4a,10a-dihydrofusarubins Produced by Fusarium sp. Mj-2. Biosci. Biotechnol. Biochem. 2013, 77, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.H.; Sohrab, M.H.; Rony, S.R.; Tareq, F.S.; Hasan, C.M.; Mazid, M.A. Cytotoxic and Antibacterial Naphthoquinones from an Endophytic Fungus, Cladosporium sp. Toxicol. Rep. 2016, 3, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, J. Dictionary of Natural Products, Supplement 1; Chapman & Hall: London, UK, 1994. [Google Scholar]
- Khan, N.; Afroz, F.; Begum, M.N.; Roy Rony, S.; Sharmin, S.; Moni, F.; Mahmood Hasan, C.; Shaha, K.; Sohrab, M.H. Endophytic Fusarium Solani: A Rich Source of Cytotoxic and Antimicrobial Napthaquinone and Aza-Anthraquinone Derivatives. Toxicol. Rep. 2018, 5, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Adorisio, S.; Fierabracci, A.; Muscari, I.; Liberati, A.; Cannarile, L.; Thuy, T.; Sung, T.; Sohrab, H.; Hasan, C.; Ayroldi, E.; et al. Fusarubin and Anhydrofusarubin Isolated from A Cladosporium Species Inhibit Cell Growth in Human Cancer Cell Lines. Toxins 2019, 11, 503. [Google Scholar] [CrossRef]
- Ammar, M.S.; Gerber, N.N.; Mcdaniel, L.E. New Antibiotic Pigments Related to Fusarubin from Fusarium Solani (MART.) SACC. I. Fermentation, Islation, and Antimicrobial Activities. J. Antibiot. 1979, 32, 679–684. [Google Scholar] [CrossRef]
- Vijitphan, P.; Rukachaisirikul, V.; Muanprasat, C.; Iawsipo, P.; Panprasert, J.; Tadpetch, K. Unified Synthesis and Cytotoxic Activity of 8-O-Methylfusarubin and Its Analogues. Org. Biomol. Chem. 2019, 17, 7078–7087. [Google Scholar] [CrossRef]
- Hasan, S.; Ansari, M.; Ahmad, A.; Mishra, M. Major Bioactive Metabolites from Marine Fungi: A Review. Bioinformation 2015, 11, 176–181. [Google Scholar] [CrossRef]
- Gerber, N.N.; Ammar, M.S. New Antibiotic Pigments Related to Fusarubin from Fusarium Solani (MART.) SACC. II. Structure Elucidations. J. Antibiot. 1979, 32, 685–688. [Google Scholar] [CrossRef]
- De Gussem, K. Mycotechnology: Present Status & Future Prospects; I K International Publishing: Delhi, India, 2007; pp. 288–301. [Google Scholar]
- Volkov, V.V.; Perry, C.C. Fungal Pigments on Paper: Raman and Quantum Chemistry Studies of Alternaria Sp. Dyes Pigments 2021, 195, 109719. [Google Scholar] [CrossRef]
- Pankin, D.; Povolotckaia, A.; Borisov, E.; Belyakov, M.; Borzenko, S.; Gulyaev, A.; Moskovskiy, M. Theoretical Modelling of Structure, Vibrational and UV–Vis Absorbance Spectra of Rubrofusarin Molecule. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2023, 293, 122469. [Google Scholar] [CrossRef]
- Zięba, S.; Piotrowska, A.; Mizera, A.; Ławniczak, P.; Markiewicz, K.H.; Gzella, A.; Dubis, A.T.; Łapiński, A. Spectroscopic and Structural Study of a New Conducting Pyrazolium Salt. Molecules 2021, 26, 4657. [Google Scholar] [CrossRef]
- Sutradhar, D.; Chandra, A.K.; Zeegers-Huyskens, T. Theoretical Study of the Interaction of Fluorinated Dimethyl Ethers and the ClF and HF Molecules. Comparison between Halogen and Hydrogen Bonds. Int. J. Quantum Chem. 2016, 116, 670–680. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01. 2010. Available online: https://www.scienceopen.com/document?vid=45b5a7ba-f6ee-40ce-b346-7407f99a540d (accessed on 25 October 2023).
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Kristallogr. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type Density Functional Constructed with a Long-range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of Crystals with the Quasi-Newton Method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Cambridge Crystallographic Data Centre. Available online: https://www.ccdc.cam.ac.uk/Structures/Search?Compound=anhydrofusarubin&DatabaseToSearch=Published (accessed on 25 October 2023).
- Caballero, B.; Trugo, L.; Finglas, P. (Eds.) Encyclopedia of Food Sciences and Nutrition; Elsevier Science B.V.: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Mayo, D.W.; Miller, F.A.; Hannah, R.W. (Eds.) Course Notes on the Interpretation of Infrared and Raman Spectra; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; ISBN 9780471690085. [Google Scholar]
- Paul, S.O.; Schutte, C.J.H.; Hendra, P.J. The Fourier Transform Raman and Infrared Spectra of Naphthazarin. Spectrochim. Acta Part A Mol. Spectrosc. 1990, 46, 323–329. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Thrane, U. Standardized High-Performance Liquid Chromatography of 182 Mycotoxins and Other Fungal Metabolites Based on Alkylphenone Retention Indices and UV—VIS Spectra (Diodearray Detection). J. Chromatogr. A 1987, 404, 195–214. [Google Scholar] [CrossRef]
- Onda, K.; Yamochi, H.; Koshihara, S. Diverse Photoinduced Dynamics in an Organic Charge-Transfer Complex Having Strong Electron–Phonon Interactions. Acc. Chem. Res. 2014, 47, 3494–3503. [Google Scholar] [CrossRef]
- Parsaee, Z.; Mohammadi, K.; Ghahramaninezhad, M.; Hosseinzadeh, B. A Novel Nano-Sized Binuclear Nickel(II) Schiff Base Complex as a Precursor for NiO Nanoparticles: Synthesis, Characterization, DFT Study and Antibacterial Activity. New J. Chem. 2016, 40, 10569–10583. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Theoretical Geometry for Single Molecule | XRD Data from [20,43] | Theoretical Geometry for Molecules in Crystal Asymmetrical Unit | |||||
|---|---|---|---|---|---|---|---|
| Bond, Å | 1st Approach | 2nd Approach | Molecule 1 | Molecule 2 | GGA-PBE-G6 Molecule 2 | GGA-PBE-G6 Molecule 1 | |
| Ring I | C7-C6 | 1.458 | 1.461 | 1.448 | 1.443 | 1.451 | 1.445 |
| C6-C5 | 1.455 | 1.460 | 1.445 | 1.451 | 1.450 | 1.449 | |
| C5-C4 | 1.420 | 1.423 | 1.421 | 1.421 | 1.419 | 1.421 | |
| C4-C9 | 1.454 | 1.458 | 1.443 | 1.446 | 1.445 | 1.447 | |
| C9-C8 | 1.498 | 1.502 | 1.492 | 1.492 | 1.487 | 1.491 | |
| C8-C7 | 1.352 | 1.348 | 1.334 | 1.334 | 1.369 | 1.371 | |
| C6-O21 | 1.248 | 1.237 | 1.256 | 1.256 | 1.275 | 1.278 | |
| C9-O23 | 1.236 | 1.226 | 1.239 | 1.239 | 1.260 | 1.259 | |
| C8-O16 | 1.335 | 1.326 | 1.339 | 1.339 | 1.343 | 1.339 | |
| O16-C17 | 1.426 | 1.418 | 1.439 | 1.438 | 1.443 | 1.446 | |
| Ring II | C5-C12 | 1.401 | 1.393 | 1.393 | 1.389 | 1.414 | 1.415 |
| C12-C1 | 1.407 | 1.409 | 1.402 | 1.407 | 1.413 | 1.412 | |
| C1-C2 | 1.386 | 1.379 | 1.378 | 1.378 | 1.397 | 1.394 | |
| C2-C14 | 1.424 | 1.422 | 1.418 | 1.418 | 1.433 | 1.432 | |
| C14-C4 | 1.401 | 1.394 | 1.394 | 1.394 | 1.413 | 1.411 | |
| C12-O13 | 1.339 | 1.335 | 1.344 | 1.344 | 1.341 | 1.341 | |
| C14-O15 | 1.336 | 1.333 | 1.334 | 1.334 | 1.334 | 1.334 | |
| Ring III | C1-C27 | 1.501 | 1.501 | 1.498 | 1.492 | 1.504 | 1.506 |
| C27-O11 | 1.441 | 1.428 | 1.430 | 1.430 | 1.451 | 1.456 | |
| O11-C3 | 1.357 | 1.352 | 1.351 | 1.351 | 1.359 | 1.355 | |
| C3-C25 | 1.348 | 1.346 | 1.326 | 1.326 | 1.359 | 1.359 | |
| C25-C2 | 1.446 | 1.450 | 1.443 | 1.443 | 1.438 | 1.437 | |
| C3-C30 | 1.491 | 1.490 | 1.489 | 1.488 | 1.485 | 1.487 | |
| Angle, o | |||||||
| Ring I | C7-C6-C5 | 118.5 | 118.7 | 119.0 | 119.2 | 118.9 | 119.0 |
| C6-C5-C4 | 120.4 | 120.3 | 119.9 | 119.9 | 120.4 | 120.3 | |
| C5-C4-C9 | 120.7 | 120.5 | 120.5 | 120.2 | 120.6 | 120.9 | |
| C4-C9-C8 | 117.4 | 117.5 | 117.5 | 117.5 | 117.8 | 117.1 | |
| C9-C8-C7 | 121.2 | 121.3 | 121.6 | 121.6 | 121.2 | 121.5 | |
| C7-C6-O21 | 119.7 | 119.7 | 119.8 | 120.0 | 120.6 | 120.5 | |
| C8-C9-O23 | 119.6 | 119.6 | 119.6 | 119.6 | 120.5 | 120.6 | |
| C9-C8-O16 | 112.2 | 112.1 | 111.8 | 111.8 | 112.7 | 112.7 | |
| C8-C7-H10 | 122.7 | 122.8 | 119.1 | 119.3 | 121.6 | 121.8 | |
| C8-O16-C17 | 118.0 | 118.0 | 117.1 | 117.2 | 116.9 | 116.6 | |
| Ring II | C5-C12-C1 | 119.6 | 119.4 | 120.1 | 120.5 | 119.9 | 120.1 |
| C12-C1-C2 | 121.4 | 121.4 | 121.1 | 120.4 | 121.2 | 120.6 | |
| C1-C2-C14 | 119.5 | 119.7 | 120.0 | 120.0 | 119.1 | 119.8 | |
| C2-C14-C4 | 119.7 | 119.5 | 119.8 | 119.8 | 119.8 | 119.9 | |
| C14-C4-C5 | 120.2 | 120.3 | 120.1 | 120.0 | 120.3 | 119.8 | |
| C4-C14-C15 | 122.4 | 123.3 | 123.7 | 123.7 | 121.4 | 122.2 | |
| C14-C15-H24 | 106.2 | 107.5 | 104.8 | 103.1 | 104.8 | 105.9 | |
| C5-C12-O13 | 122.0 | 123.0 | 121.8 | 122.0 | 120.7 | 120.7 | |
| C12-O13-H22 | 105.5 | 106.8 | 101.8 | 100.9 | 103.6 | 103.3 | |
| Ring III | C1-C27-O11 | 112.3 | 111.9 | 114.2 | 114.1 | 113.6 | 114.0 |
| C27-O11-C3 | 116.4 | 116.1 | 118.4 | 118.0 | 117.8 | 118.9 | |
| O11-C3-C25 | 122.2 | 122.0 | 122.3 | 122.3 | 123.0 | 122.9 | |
| C3-C25-C2 | 119.4 | 118.9 | 120.7 | 120.7 | 119.6 | 120.1 | |
| C25-C2-C1 | 118.6 | 118.4 | 118.5 | 118.2 | 119.2 | 119.1 | |
| C2-C1-C27 | 117.1 | 116.7 | 118.3 | 118.5 | 118.3 | 119.1 | |
| C1-C27-H_eq | 111.6 | 111.8 | 108.7 | 108.7 | 112.2 | 112.3 | |
| C1-C27-H_ax | 110.3 | 109.8 | 108.7 | 108.7 | 109.9 | 110.3 | |
| C3-C25-H26 | 120.2 | 120.4 | 119.8 | 119.7 | 119.9 | 120.2 | |
| C25-C3-C30 | 125.7 | 125.7 | 126.2 | 126.1 | 125.5 | 125.2 | |
| H29-C27-H28 | 108.5 | 108.7 | 107.6 | 107.6 | 107.5 | 107.6 | |
| Dihedral angle, o | |||||||
| C12-C1-C27-O11 | 148.7 | 146.9 | 158.7 | −158.4 | −159.1 | 162.5 | |
| C1-C27-O11-C3 | 42.7 | 45.5 | 32.6 | −33.8 | −34.3 | 28.6 | |
| C27-O11-C3-C25 | −23.7 | −25.3 | −21.2 | 22.3 | 21.4 | −18.6 | |
| O11-C3-C25-C2 | −6.2 | −6.6 | −0,1 | 0.5 | 1.3 | −0.9 | |
| C3-C25-C2-C14 | −162.4 | −160.9 | −171.1 | 169.6 | 171.1 | −171.3 | |
| C3-C25-C2-C1 | 14.5 | 15.6 | 7.6 | −9.7 | −9.0 | 8.5 | |
| C27-O11-C3-C33 | 160.1 | 158.4 | 163.0 | −161.3 | −161.9 | 164.3 | |
| Theoretical Results for GP Single Molecule | Experimental Data | ||||
|---|---|---|---|---|---|
| [45] | [28] | ||||
| Peak number | Oscillator strength | Orbitals with >10% contribution (contribution percent) | Wavelength, nm (energy, eV) | Wavelength, nm | Wavelength, nm |
| Peak 1 | 0.2284 | 71–>78 (18%) 73–>78 (49%) | 203 (6.1006) | 215 | |
| Peak 2 | 0.1630 | 74–>78 (81%) | 241 (5.1536) | 235 | 237 |
| Peak 3 | 0.1579and0.1844 | 74–>77 (29%) 75–>78 (62%) and 75–>77 (87%) | 285 (4.3561) and 298 (4.1562) | 288 | 290 |
| Peak 4 | 0.0374 | 73–>76 (85%) | 351 (3.5301) | 345 | |
| Peak 5 | 0.1371 | 75–>76 (93%) | 560 (2.2151) | 540 * and 570 | 545 |
| Estimated Value | Method 1 |
|---|---|
| LUMO energy, eV | −3.15 |
| HOMO energy, eV | −5.75 |
| LUMO-HOMO, eV | 2.60 |
| Electron affinity, eV | 3.15 |
| Ionization potential, eV | 5.75 |
| Chemical hardness, eV | 1.30 |
| Electronegativity, eV | 4.45 |
| Electrophilicity index | 7.62 |
| Chemical softness, eV−1 | 0.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pankin, D.; Povolotckaia, A.; Smirnov, M.; Borisov, E.; Gulyaev, A.; Dorochov, A.; Novikov, V.; Kuznetsov, S.; Noy, O.; Belousov, S.; et al. Theoretical Investigation of Anhydrofusarubin: Structural and Optical Properties. Crystals 2023, 13, 1556. https://doi.org/10.3390/cryst13111556
Pankin D, Povolotckaia A, Smirnov M, Borisov E, Gulyaev A, Dorochov A, Novikov V, Kuznetsov S, Noy O, Belousov S, et al. Theoretical Investigation of Anhydrofusarubin: Structural and Optical Properties. Crystals. 2023; 13(11):1556. https://doi.org/10.3390/cryst13111556
Chicago/Turabian StylePankin, Dmitrii, Anastasia Povolotckaia, Mikhail Smirnov, Evgenii Borisov, Anatoly Gulyaev, Alexey Dorochov, Vasiliy Novikov, Sergey Kuznetsov, Oleg Noy, Sergey Belousov, and et al. 2023. "Theoretical Investigation of Anhydrofusarubin: Structural and Optical Properties" Crystals 13, no. 11: 1556. https://doi.org/10.3390/cryst13111556