
Severe Juvenile-Onset Systemic Lupus Erythematosus: A Case Series-Based Review and Update
 , and
, and 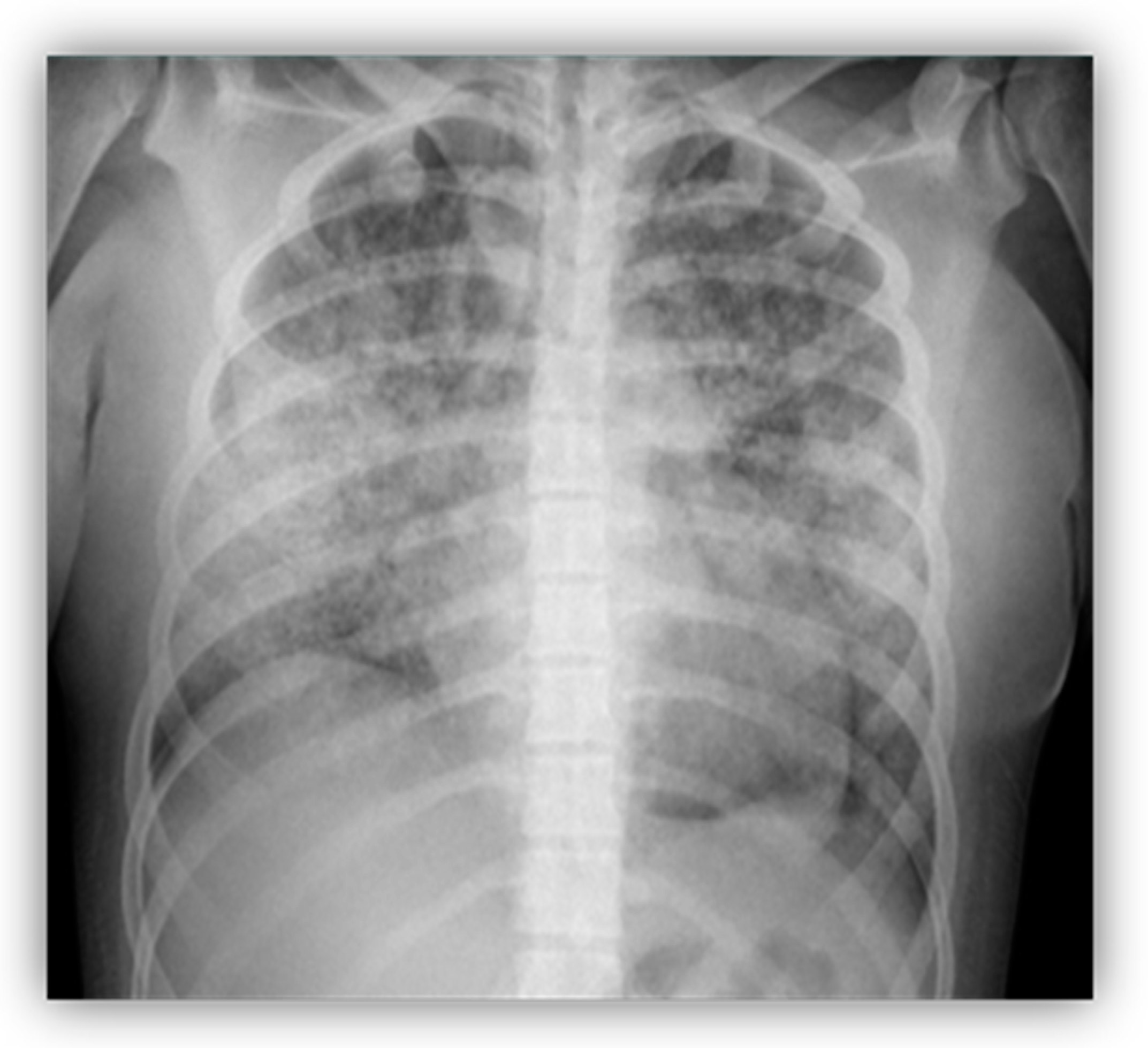{kind=link}
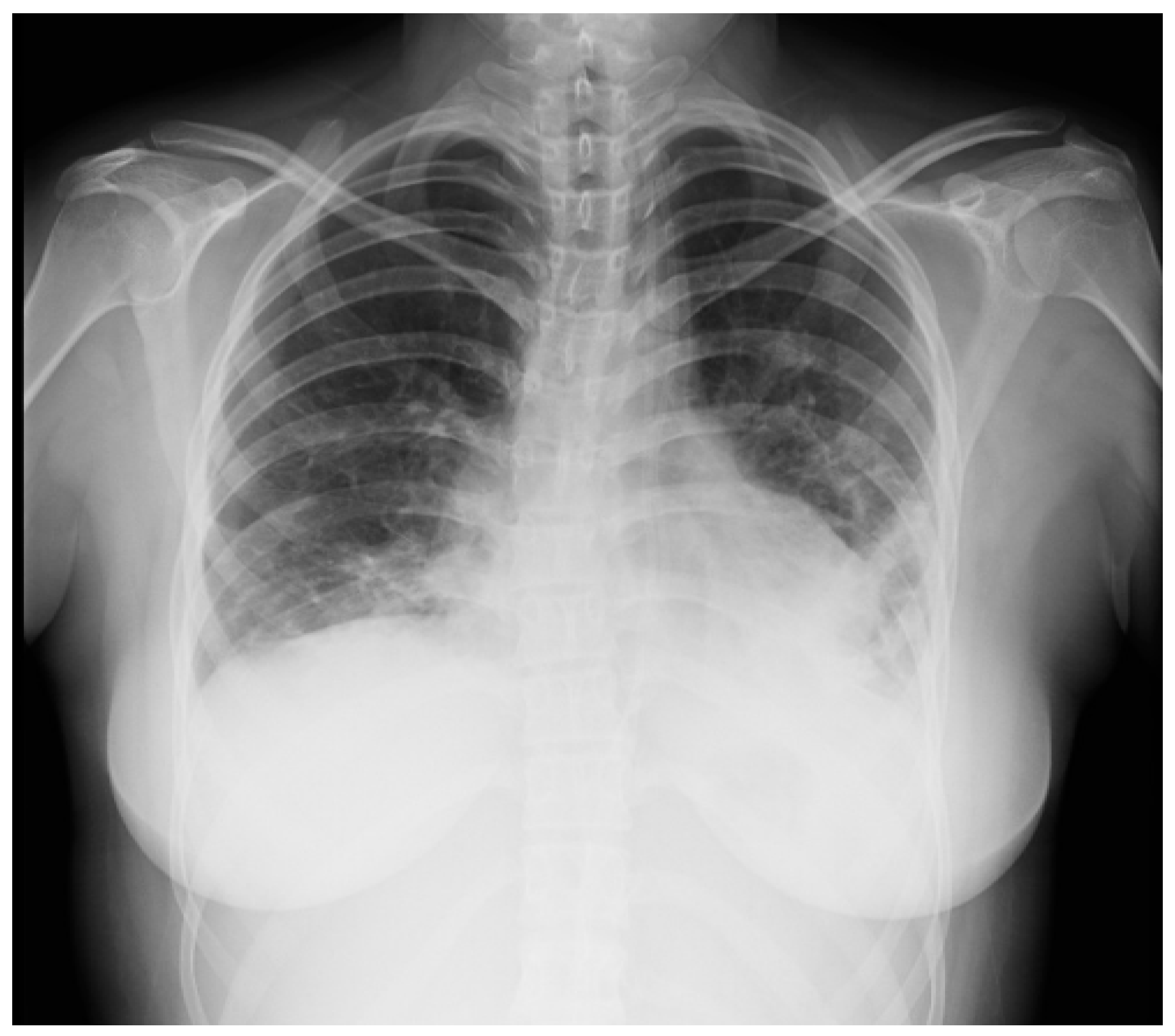{kind=link}
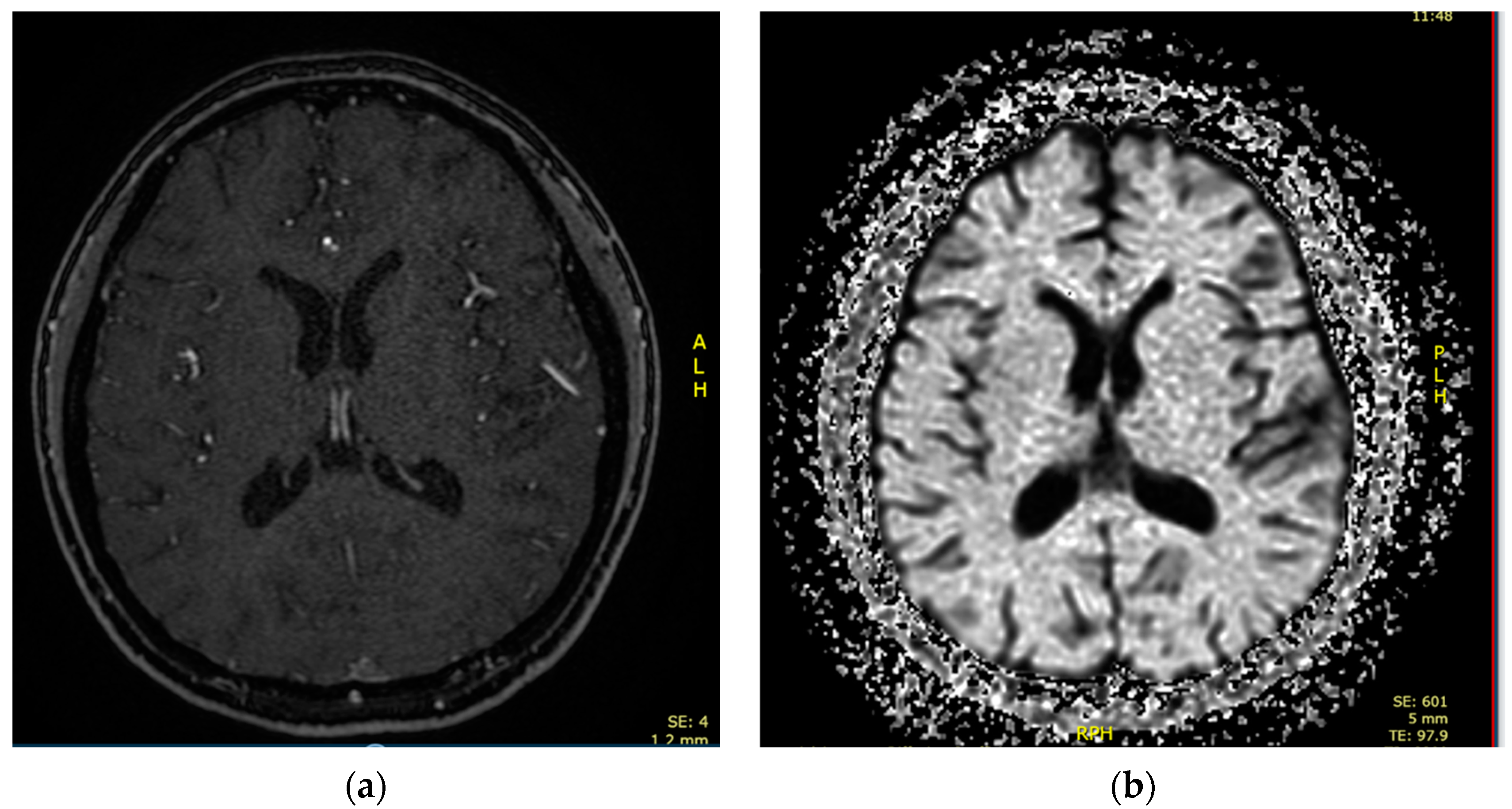{kind=link}
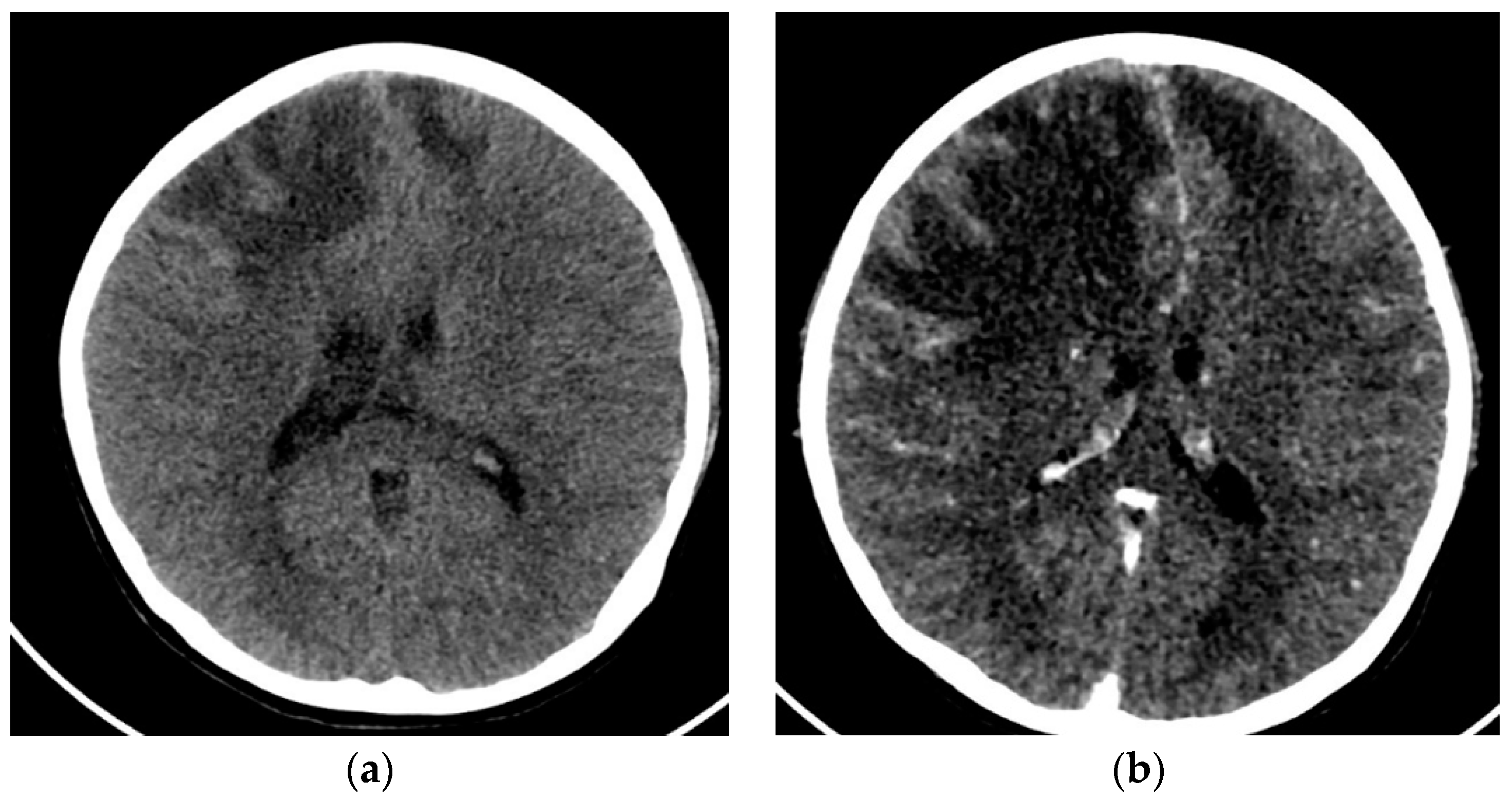{kind=link}
{kind=link}
Abstract
:1. Introduction
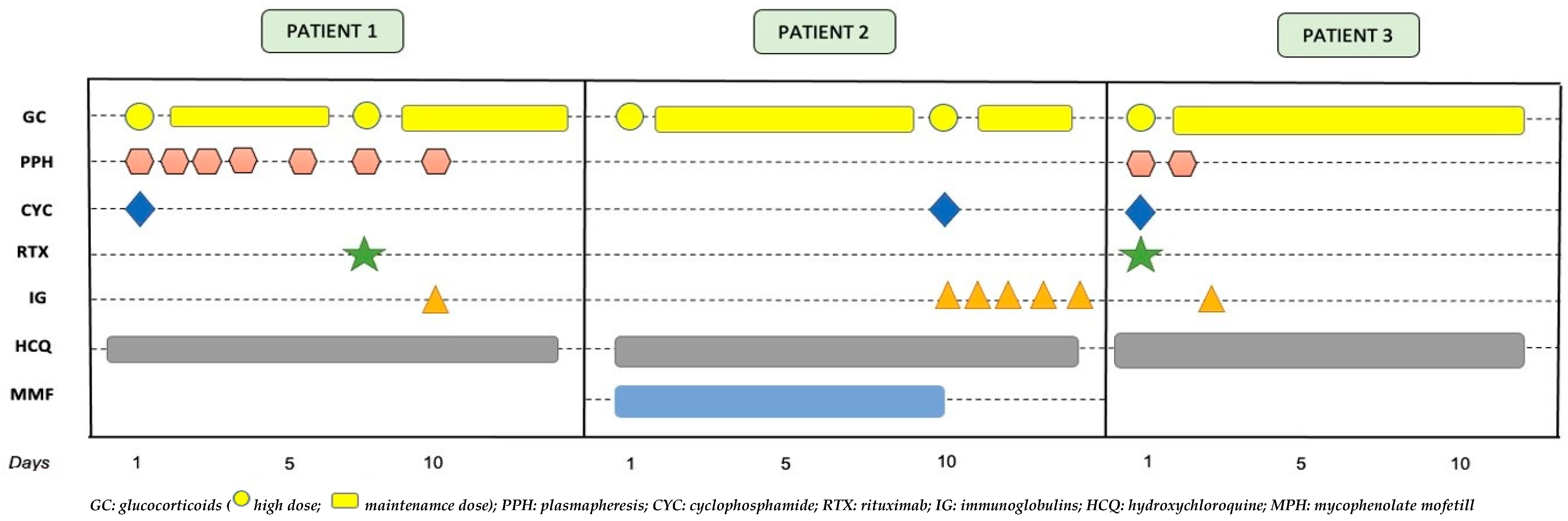
2. Case Presentations
2.1. Case 1
2.2. Case 2
2.3. Case 3
3. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Groot, N.; de Graeff, N.; Avcin, T.; Bader-Meunier, B.; Brogan, P.; Dolezalova, P.; Feldman, B.; Kone-Paut, I.; Lahdenne, P.; Marks, S.D.; et al. European Evidence-Based Recommendations for Diagnosis and Treatment of Childhood-Onset Systemic Lupus Erythematosus: The SHARE Initiative. Ann. Rheum. Dis. 2017, 76, 1788–1796. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.I.; Gladman, D.D.; Ibañez, D.; Urowitz, M.D.; Silverman, E.D. Difference in Disease Features between Childhood-Onset and Adult-Onset Systemic Lupus Erythematosus. Arthritis Rheum. 2008, 58, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Charras, A.; Smith, E.; Hedrich, C.M. Systemic Lupus Erythematosus in Children and Young People. Curr. Rheumatol. Rep. 2021, 23, 20. [Google Scholar] [CrossRef]
- Sandling, J.K.; Rosenberg, L.H.; Farias, F.H.G.; Alexsson, A.; Leonard, D.; Kozyrev, S.; Murén, E.; Karlsson, Å.; Mathioudaki, A.; Pucholt, P.; et al. Targeted Next-Generation Sequencing Suggests Novel Risk Loci in Juvenile Onset Systemic Lupus Erythematosus. In Proceedings of the 11th European Lupus Meeting, Düsseldorf, Germany, 21–24 March 2018. [Google Scholar]
- Aringer, M.; Costenbader, K.; Daikh, D.; Brinks, R.; Mosca, M.; Ramsey-Goldman, R.; Smolen, J.S.; Wofsy, D.; Boumpas, D.T.; Kamen, D.L.; et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.D.; Lythgoe, H.; Midgley, A.; Beresford, M.W.; Hedrich, C.M. Juvenile-Onset Systemic Lupus Erythematosus: Update on Clinical Presentation, Pathophysiology and Treatment Options. Clin. Immunol. 2019, 209, 108274. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, N.; Morgan, T.A.; Galloway, J.; Ionnoau, Y.; Beresford, M.W.; Isenberg, D.A. Differences in Disease Phenotype and Severity in SLE across Age Groups. Lupus 2016, 25, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Fanouriakis, A.; Kostopoulou, M.; Alunno, A.; Aringer, M.; Bajema, I.; Boletis, J.N.; Cervera, R.; Doria, A.; Gordon, C.; Govoni, M.; et al. 2019 Update of the EULAR Recommendations for the Management of Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2019, 78, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Serrano, J.; Pedroza, J.; Regalado, J.; Robledo, J.; Reyes, E.; Sifuentes-Osornio, J.; Flores-Suárez, L. High Prevalence of Infections in Patients with Systemic Lupus Erythematosus and Pulmonary Haemorrhage. Lupus 2008, 17, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Kasap-Demir, B.; Bayram, G.; Önder-Siviş, Z.; Güneş-Tatlı, B.; Anıl, B.A. Diffuse Alveolar Hemorrhage as a Life-Threatening Feature of Juvenile Onset Systemic Lupus Erythematosus: A Case-Based Review. Mediterr. J. Rheumatol. 2022, 33, 81. [Google Scholar] [CrossRef] [PubMed]
- Sarecka-Hujar, B.; Kopyta, I. Antiphospholipid Syndrome and Its Role in Pediatric Cerebrovascular Diseases: A Literature Review. World J. Clin. Cases 2020, 8, 1806–1817. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.S.; Diab, K.J.; Carlos, W.G.; Mathur, P. Intrapulmonary Recombinant Factor VII as an Effective Treatment for Diffuse Alveolar Hemorrhage: A Case Series. J. Bronchol. Interv. Pulmonol. 2016, 23, 255–258. [Google Scholar] [CrossRef]
- Senken, B.; Whitehead, A. Catastrophic Antiphospholipid Syndrome Presenting as a Stroke in an 11-Year-Old with Lupus. Case Rep. Pediatr. 2022, 2022, 7890566. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huerta-Calpe, S.; Del Castillo-Velilla, I.; Felipe-Villalobos, A.; Jordan, I.; Hernández-Platero, L. Severe Juvenile-Onset Systemic Lupus Erythematosus: A Case Series-Based Review and Update. Children 2023, 10, 852. https://doi.org/10.3390/children10050852
Huerta-Calpe S, Del Castillo-Velilla I, Felipe-Villalobos A, Jordan I, Hernández-Platero L. Severe Juvenile-Onset Systemic Lupus Erythematosus: A Case Series-Based Review and Update. Children. 2023; 10(5):852. https://doi.org/10.3390/children10050852
Chicago/Turabian StyleHuerta-Calpe, Sergi, Ignacio Del Castillo-Velilla, Aida Felipe-Villalobos, Iolanda Jordan, and Lluïsa Hernández-Platero. 2023. "Severe Juvenile-Onset Systemic Lupus Erythematosus: A Case Series-Based Review and Update" Children 10, no. 5: 852. https://doi.org/10.3390/children10050852