
Electrochemical Behavior and Voltammetric Determination of Two Synthetic Aroyl Amides Opioids
 ,
,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Synthesis of Aroyl Amides U-47700 and AH-7921
2.3. Glassy Carbon (GC) Electrode Pre-Treatment/Characterization
2.4. Electrochemical Analyses
2.4.1. Number of Electrons Involved in the Electrochemical Processes
2.4.2. Cyclic Voltammetry Measurements
2.5. Electrochemical Determination of Aroyl Amides
2.6. Evaluation of LOD, LOQ, and Statistical Parameters
2.7. Analysis of Aroyl Amides Capsules
2.8. Analysis of Aroyl Amides in Water Samples, Synthetic and Natural Urines
3. Results and Discussion
3.1. Electrode Characterization
3.2. Electrochemistry of the Examined Aroyl Amides
3.2.1. Cyclic Voltammetry and Oxidation Mechanism
3.2.2. Cyclic Voltammetry Plots
3.2.3. Determination of
3.2.4. Determination of the Charge Transfer Coefficient (α)
3.2.5. Determination of the Diffusion Coefficient (D)
3.2.6. Determination of the Kinetic Constant K°
3.2.7. Determination of the Reaction Order
3.3. Electrochemical Determination of Aroyl Amides
3.3.1. Analysis of the Drug Formulations in Presence of Possible Interfering Substances
3.3.2. Analysis of the Capsules
3.3.3. Analysis of Water and Natural and Synthetic Urines Spiked with the Title Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
List of Abbreviations and Compounds Name
| U-47700 | 3,4-dichloro-N-[2-(dimethylammino)ciclohexyl]-N-methyl-benzammide |
| AH-7921 | 3,4-Dicloro-N-[[1-(dimetilammino)cicloesil]metil]benzammide |
| CV | cyclic voltammetry |
| CPE | controlled potential electrolysis |
| DPV | differential pulsed voltammetry |
| ν | scan speed (mV·s−1) |
| Ep | peak potential (V) |
| ip | peak intensity (A) |
| Rc% | recovery factor % |
References
- Colvin, L.A.; Bull, F.; Hales, T.G. Perioperative opioid analgesia—When is enough too much? A review of opioid-induced tolerance and hyperalgesia. Lancet 2019, 393, 1558–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koepke, E.J.; Manning, E.L.; Miller, T.E.; Ganesh, A.; Williams, D.G.A.; Manning, M.W. The rising tide of opioid use and abuse: The role of the anesthesiologist. Perioper. Med. 2018, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercadante, S. Opioid Analgesics Adverse Effects: The Other Side of the Coin. Curr. Pharm. Des. 2019, 25, 3197–3202. [Google Scholar] [CrossRef]
- Hasegawa, K.; Minakata, K.; Suzuki, M.; Suzuki, O. U-47700 and Its Analogs: Non-Fentanyl Synthetic Opioids Impacting the Recreational Drug Market. Brain Sci. 2020, 10, 895. [Google Scholar]
- Lamy, F.R.; Daniulaityte, R.; Barratt, M.J.; Lokala, U.; Sheth, A.; Pardo, R.G.C. Etazene, safer than heroin and fentanyl: Non-fentanyl novel synthetic opioid listings on one darknet market. Drug Alcohol Depend. 2021, 225, 108790. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.; Szpot, P.; Zawadzki, M. Fatal intoxication with U-47700 in combination with other NPS (N-ethylhexedrone, adinazolam, 4-CIC, 4-CMC) confirmed by identification and quantification in autopsy specimens and evidences. Forensic Toxicol. 2021, 39, 493–505. [Google Scholar] [CrossRef]
- Baselt, R. Disposition of Toxic Drugs and Chemicals in Man, 11th ed.; Biomedical Publications: Foster City, CA, USA, 2007; p. 2208. [Google Scholar]
- Lukić, V.; Micić, R.; Arsić, B.; Nedović, B.; Radosavljević, Ž. Overview of the major classes of new psychoactive substances, psychoactive effects, analytical determination and conformational analysis of selected illegal drugs. Open Chem. 2021, 19, 60–106. [Google Scholar] [CrossRef]
- Alexandridoua, A.; Mouskeftar, T.; Raikos, N.; Gika, H.G. GC-MS analysis of underivatised new psychoactive substances in whole blood and urine. J. Chromatogr. B 2020, 1156, 122308. [Google Scholar] [CrossRef]
- Richeval, C.; Gaulier, J.-M.; Romeuf, L.; Allorge, D.; Gaillard, Y. Case report: Relevance of metabolite identification to detect new synthetic opioid intoxications illustrated by U-47700. Int. J. Leg. Med. 2019, 133, 133–142. [Google Scholar] [CrossRef]
- Rojek, S.; Romańczuk, A.; Kula, K.; Synowiec, K.; Kłys, M. Quantifcation of U-47700 and its metabolites: N-desmethyl-U-47700 and N,N-didesmethyl-U-47700 in 12 autopsy blood samples employing SPE/LC–ESI-MS-MS. Forensic Toxicol. 2019, 37, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Krotulski, A.J.; Mohr, A.L.A.; Papsun, D.M.; Logan, B.K. Metabolism of novel opioid agonists U-47700 and U-49900using human liver microsomes with confirmation in authenticurine specimens from drug users. Drug Test. Anal. 2018, 10, 127–136. [Google Scholar] [CrossRef]
- López-García, E.; Mastroianni, N.; Postigo, C.; Valcárcel, Y.; González-Alonso, S.; Barceló, D.; López de Alda, M. Simultaneous LC–MS/MS determination of 40 legal and illegal psychoactive drugs in breast and bovine milk. Food Chem. 2018, 245, 159–167. [Google Scholar] [CrossRef]
- Lowry, J.; Truver, M.T.; Swortwood, M.J. Quantification of seven novel synthetic opioids in blood using LC–MS/MS. Forensic Toxicol. 2019, 37, 215–223. [Google Scholar] [CrossRef]
- Truver, M.T.; Swortwood, M.J. Quantitative Analysis of Novel Synthetic Opioids, Morphine and Buprenorphine in Oral Fluid by LC–MS-MS. J. Anal. Toxicol. 2018, 42, 554–561. [Google Scholar] [CrossRef]
- Fleming, S.W.; Cooley, J.C.; Johnson, L.; Frazee, C.C.; Domanski, K.; Kleinschmidt, K.; Garg, U. Analysis of U-47700, a Novel Synthetic Opioid, in Human Urine by LC–MS–MS and LC–QToF. J. Anal. Toxicol. 2017, 41, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamowicz, P.; Bakhmut, Z.; Mikolajczyk, A. Screening procedure for 38 fentanyl analogues and five other new opioids in whole blood by liquid chromatography-tandem mass spectrometry. J. Appl. Toxicol. 2020, 40, 1033–1046. [Google Scholar] [CrossRef]
- Mohr, A.L.; Friscia, M.; Papsun, D.; Kacinko, S.L.; Buzby, D.; Logan, B.K. Analysis of Novel Synthetic Opioids U-47700, U-50488 and Furanyl Fentanyl by LC-MS/MS in Postmortem Casework. J. Anal. Toxicol. 2016, 40, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strayer, K.E.; Antonides, H.M.; Juhascik, M.P.; Daniulaityte, R.; Sizemore, I.E. LC-MS/MS-Based Method for the Multiplex Detection of 24 Fentanyl Analogues and Metabolites in Whole Blood at Sub ng mL–1 Concentrations. ACS Omega 2018, 3, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popławska, M.; Bednarek, E.; Naumczuk, B.; Kozerski, L.; Błażewicz, A. Identification and structure characterization of five synthetic opioids: 3,4-methylenedioxy-U-47700, o-methyl-acetylfentanyl, 2-thiophenefentanyl, benzoylfentanyl and benzoylbenzylfentanyl. Forensic Toxicol. 2021, 39, 45–58. [Google Scholar] [CrossRef]
- Bade, R.; Abdelaziz, A.; Nguyen, L.; Pandopulos, A.J.; White, J.M.; Gerber, C. Determination of 21 synthetic cathinones, phenethylamines, amphetamines and opioids in influent wastewater using liquid chromatography coupled to tandem mass spectrometry. Talanta 2020, 208, 120479. [Google Scholar] [CrossRef] [PubMed]
- Gerace, E.; Salomone, A.; Luciano, C.; Di Corcia, D.; Vincenti, M. First Case in Italy of Fatal Intoxication Involving the New Opioid U-47700. Front. Pharmacol. 2018, 9, 747. [Google Scholar] [CrossRef]
- Nunes, C.N.; Pauluk, L.E.; Dos Anjos, V.E.; Lopes, M.C.; Quináia, S.P. New approach to the determination of contaminants of emerging concern in natural water: Study of alprazolam employing adsorptive cathodic stripping voltammetry. Anal. Bioanal. Chem. 2015, 407, 6171–6179. [Google Scholar] [CrossRef] [PubMed]
- Glasscott, M.W.; Vannoy, K.J.; Fernando, P.U.A.I.; Kosgei, G.K.; Moores, L.C.; Dick, J.E. Electrochemical sensors for the detection of fentanyl and its analogs: Foundations and recent advances. Trends Anal. Chem. 2020, 132, 116037. [Google Scholar] [CrossRef]
- Choìnska, M.K.; Šestáková, I.; Hrdlĭcka, V.; Skopalová, J.; Langmaier, J.; Maier, V.; Navrátil, T. Electroanalysis of Fentanyl and Its New Analogs: A Review. Biosensors 2022, 12, 26. [Google Scholar] [CrossRef]
- Merli, D.; Profumo, A.; Tinivella, S.; Protti, S. From smart drugs to smartphone: A colorimetric spot test for the analysis of the synthetic cannabinoid AB-001. Forensic Chem. 2019, 14, 100167. [Google Scholar] [CrossRef]
- Capucciati, A.; Cacciatore, L.; Protti, S.; Profumo, A.; Merli, D. Electrochemical analysis and characterization of psychoactive substances glaucine and tetrahydropalmatine. J. Electroanal. Chem. 2022, 907, 116032. [Google Scholar] [CrossRef]
- Zamboni, D.; Merli, D.; Protti, S.; Profumo, A. Electrochemistry and analytical determination of Lysergic Acid Diethylamide (LSD) via adsorbitive stripping voltammetry. Talanta 2014, 130, 456–461. [Google Scholar]
- Rustler, K.; Pockes, S.; König, B. Light-Switchable Antagonists for the Histamine H 1 Receptor at the Isolated Guinea Pig Ileum. ChemMedChem 2019, 14, 636–644. [Google Scholar] [CrossRef]
- Norman, I.; Harper, J.; Veitch, B.A. 1-(3,4-Dichlorobenzamidomethyl)-cyclohexyldimethylamine. US 518720, 17 August 1976. Available online: https://patentimages.storage.googleapis.com/38/15/18/e7e698218687c3/US3975443.pdf (accessed on 13 March 2023).
- Ott, J.; Spilhaug, M.M.; Maschauer, S.; Rafique, W.; Jakobsson, J.E.; Hartvig, K.; Hübner, H.; Gmeiner, P.; Prante, O.; Riss, P.J. Pharmacological Characterization of Low-to-Moderate Affinity Opioid Receptor Agonists and Brain Imaging with 18F-Labeled Derivatives in Rats. J. Med. Chem. 2020, 63, 9484–9499. [Google Scholar] [CrossRef]
- Yang, B.; ShenLanglois, K. Serotoninergic properties of new conformationally restricted benzamides. Eur. J. Med. Chem. 1996, 31, 231–239. [Google Scholar] [CrossRef]
- Merli, D.; Pretali, L.; Fasani, E.; Albini, A.; Profumo, A. Analytical Determination and Electrochemical Characterization of the Oxazolidinone Antibiotic Linezolid. Electroanalysis 2011, 23, 2364–2372. [Google Scholar] [CrossRef]
- Milanesi, C.L.; Protti, S.; Chiodi, D.; Profumo, A.; Merli, D. Electrochemical characterization and voltammetric determination of aryl piperazine emerging as designer drugs. J. Electroanal. Chem. 2021, 2021, 115480. [Google Scholar] [CrossRef]
- Hammerich, O.; Speiser, B. Organic Electrochemistry, 5th ed.; Hammerich, O., Speiser, B., Eds.; CRC Press (Taylor & Francis group): Boca Raton, FL, USA, 2016. [Google Scholar]
- Merli, D.; Profumo, A.; Dossi, C. An analytical method for Fe(II) and Fe(III) determination in pharmaceutical grade iron sucrose complex and sodium ferric gluconate complex. J. Pharm. Anal. 2012, 2, 450–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: http://www.mpl.loesungsfabrik.de/en/english-blog/method-validation/calibration-line-procedure (accessed on 6 February 2023).
- Sarigul, N.; Korkmaz, F.; Kurultak, İ. A New Artificial Urine Protocol to Better Imitate Human Urine. Sci. Rep. 2019, 9, 20159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Cao, X. Anodic oxidation of hydrazine on glassy carbon modified by macrociclic transition metal complexes. J. Electroanal. Chem. 1991, 309, 147–158. [Google Scholar] [CrossRef]
- Zanello, P. Inorganic Electrochemistry—Theory, Practice and Application; Royal Society of Chemistry: Cambridge, UK, 2003. [Google Scholar]
- Fotouhi, L.; Hajilari, F.; Heravi, M.M. Electrochemical Behavior of Some Thiotriazoles in Aqueous-Alcoholic Media at GCE. Electroanalysis 2002, 14, 1728–1732. [Google Scholar] [CrossRef]
- Gowda, J.I.; Nandibewoor, S.T. Electrochemical Behavior of Paclitaxel and Its Determination at Glassy Carbon Electrode. Asian J. Pharm. Sci. 2014, 9, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Masui, M.; Sayo, H.; Tsuda, Y. Anodic Oxidation of Amines. Part I. Cyclic Voltammetry of Aliphatic Amines at a Stationary Glassy-Carbon Electrode. J. Chem. Soc. B Phys. Org. 1968, 973–976. [Google Scholar] [CrossRef]
- Adenier, A.; Chehimi, M.M.; Gallardo, I.; Pinson, J.; Vilà, N. Electrochemical Oxidation of Aliphatic Amines and Their Attachment to Carbon and Metal Surfaces. Langmuir 2004, 20, 8243–8253. [Google Scholar] [CrossRef]
- Masui, M.; Hiroteru Sayo, H. Anodic Oxidation of Amines. Part 2. Electrochemical Dealkylation of Aliphatic Tertiary Amines. J. Chem. Soc. B 1971, 1593–1596. [Google Scholar] [CrossRef]
- Hull, L.A.; Davis, G.T.; Rosenblatt, D.H.; Mann, C.K. Oxidations of amines. VII. Chemical and electrochemical correlations. Phys. Chem. 1969, 73, 2142–2146. [Google Scholar] [CrossRef]
- Adhoum, N.; Monser, L. Determination of trimebutine in pharmaceuticals by differential pulse voltammetry at a glassy carbon electrode. J. Pharm. Biomed. Anal. 2005, 38, 619–623. [Google Scholar] [CrossRef]
- Amatore, C.; Farsang, G.; Maisonhaute, E.; Simon, P. Voltammetric investigation of the anodic dimerization of p-halogenoanilines in DMF Reactivity of their electrogenerated cation radicals. J. Electroanal. Chem. 1999, 462, 55–62. [Google Scholar] [CrossRef]
- Alfonso-Súarez, P.; Kolliopoulos, V.A.; Smith, J.P.; Banks, C.E.; Jones, A.M. An Experimentalists Guide to Electrosynthesis: The Shono Oxidation. Tetrahedron Lett. 2015, 56, 6863–6867. [Google Scholar] [CrossRef]
- Yunhua, W.; Xiaobo, J.; Shengshui, H. Studies on electrochemical oxidation of azithromycin and its interaction with bovine serum albumin. Bioelectrochemistry 2004, 64, 91–97. [Google Scholar]
- Bontempelli, G.; Dossi, N.; Toniolo, R. Linear Sweep and Cyclic, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering. Available online: https://doi.org/10.1016/B978-0-12-409547-2.12200-0 (accessed on 30 December 2022).
- Sandford, C.; Edwards, M.A.; Klunder, K.J.; Hickey, D.P.; Li, M.; Barman, K.; Sigman, M.S.; White, H.S.; Minteer, S.D. A synthetic chemist’s guide to electroanalytical tools for studying reaction mechanisms. Chem. Sci. 2019, 10, 6404–6422. [Google Scholar] [CrossRef] [Green Version]
- Keerti, M.N.; Sharanappa, T.N. Electrochemical behaviour of chalcone at a glassy carbon electrode and its analytical applications. Am. J. Anal. Chem. 2012, 3, 656–663. [Google Scholar]
- Patil, S.M.; Bagoji, A.M.; Konnur, S.B.; Gokavi, N.M.; Nandibewoor, S.T. Electro-analysis of Orphenadrine Hydrochloride by Graphene Modified Glassy Carbon Electrode and Its Oxidation Mechanism. Anal. Bioanal. Electrochem. 2021, 13, 190–201. [Google Scholar]
- Galus, Z.; Adams, R.N. Anodic Oxidation Studies of N,N-Dimethylaniline. II. Stationary and Rotated Disk Studies at Inert Electrodes. J. Am. Chem. Soc. 1962, 84, 2061–2065. [Google Scholar] [CrossRef]
- Mizoguchi, T.; Adams, R.N. Anodic Oxidation Studies of N,N-Dimethylaniline. I. Voltammetric and Spectroscopic Investigations at Platinum Electrodes. J. Am. Chem. Soc. 1961, 84, 2058–2061. [Google Scholar] [CrossRef]
- Rambaran, K.A.; Fleming, S.W.; An, J.; Burkhart, S.; Furmaga, J.; Kleinschmidt, K.J.; Spiekerman, A.M.; Alzghari, S.K. U-47700: A Clinical Review of the Literature. J. Emerg. Med. 2017, 53, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Concheiro, M.; Chesser, R.; Pardi, J.; Cooper, G. Postmortem Toxicology of New Synthetic Opioids. Front. Pharmacol. 2018, 9, 1210. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AH-7920 | U-47700 | |||||
|---|---|---|---|---|---|---|
| Quantification Methods | LOD ng∙mL–1 | Linear Range ng∙mL–1 | R2 | LOD ng∙mL–1 | Linear Range ng∙mL–1 | R2 |
| LC-MS/MS * [15] | 5 | 1–2500 | 0.985 | 5 | 1–2500 | 0.985 |
| LC-MS/MS [16] | - | - | - | 1 | 1–1250 | 0.9983 |
| LC-MS/MS [17] | 0.02 | 0.01–20 | 0.9992 | 0.01 | 0.01–20 | 0.9990 |
| UHPLC-MS/MS # [22] | - | - | - | 0.6 | 0–250 | 0.998 |
| LC-MS/MS [18] | - | - | - | 0.5 | 1–100 | 0.9995 |
| LC-MS/MS [11] | 0.042 | 0.100−10.0 | - | 0.019 | 0.100−10.0 | - |
| LC-MS/MS [19] | - | - | - | 0.5 | 1–1000 | 0.9997 |
| LC-MS/MS ° [20] | qualitative | qualitative | ||||
| Compound | D cm2∙s−1 | E0′ mV | Ep1 mV | Ep2 mV | Peak 1 Intensity μA | Peak 2 Intensity μA | ln K° | α | Reaction Order |
|---|---|---|---|---|---|---|---|---|---|
| U-47700 | 1.40 × 10−5 | +980 (i) | +980 | +1300 | 41 ± 5 | 56 ± 7 | −16.8 | 0.73 | 1 |
| AH-7921 | 2.40 × 10−5 | +1100 (i) | +1100 | +1300 | 30 ± 6 | 31 ± 5 | −15.4 | 0.70 | 1 |
| U-47700 | |
| E vs. log v | E/V = (0.091 ± 0.013)·log (v/V·s−1) + (1.144 ± 0.047) |
| log i vs. log v | log (i/A) = (0.504 ± 0.010)·log (v/V·s−1) − (4.103 ± 0.032) |
| i vs. v1/2 | i/A = (1.003 ± 0.021) × 10−4 v1/2/V1/2·s−1/2 + (3.004 ± 0.032)∙10−6 |
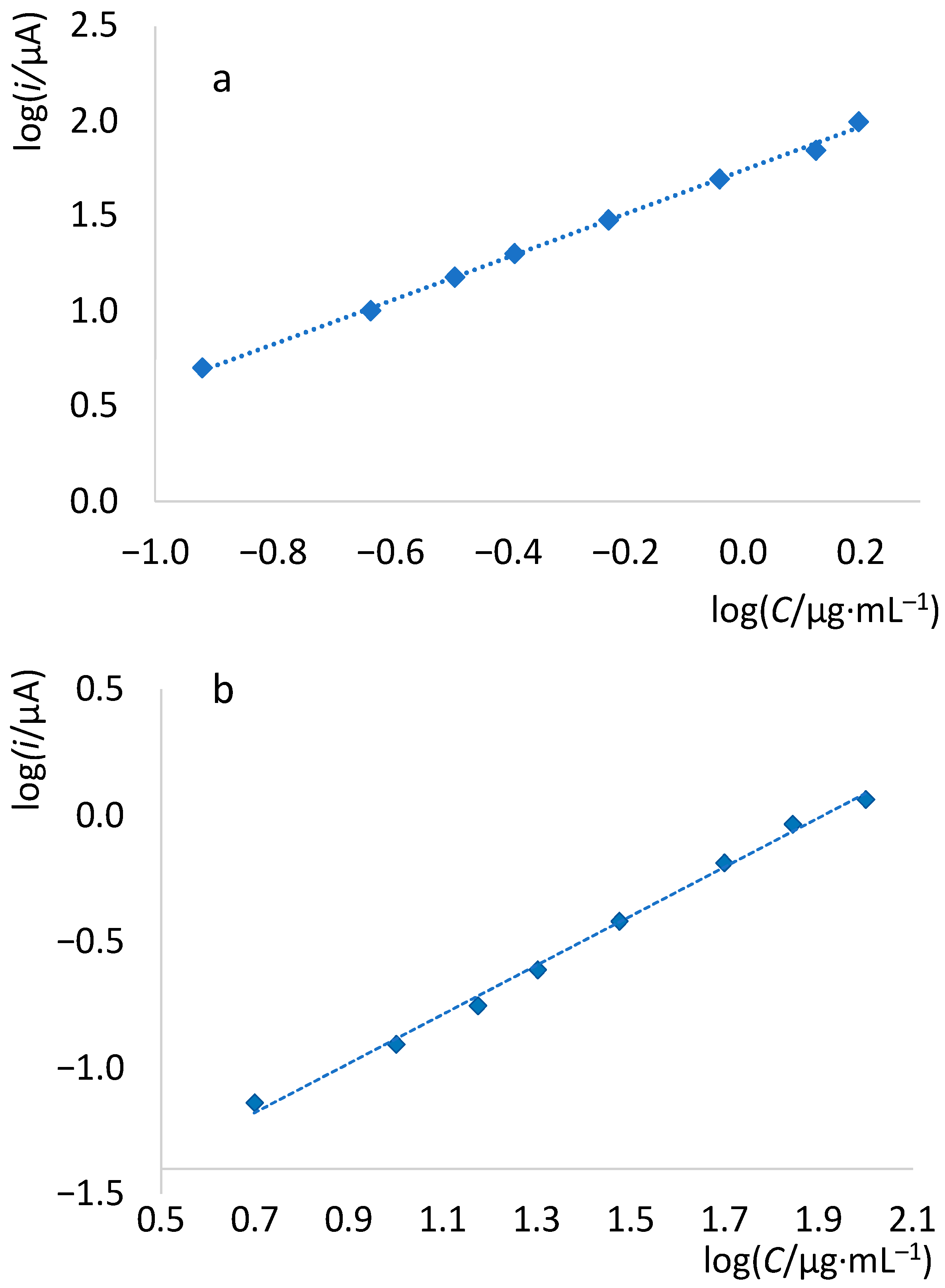
| log i vs. log C | log (i/A) = (3.153 ± 0.032)·log (C/mg∙L−1) + (1.753 ± 0.019) |
| i·v−1/2 vs. v1/2 | i·v−1/2/A·V−1/2·s1/2 = −(1.004 ± 0.042) × 10−5/V1/2·s−1/2 + (1.42 ± 0.28) × 10−4 |
| AH-7921 | |
| E vs. log v | E/V = (0.093 ± 0.014)·log (v/V·s−1) + (1.080 ± 0.032) |
| log i vs. log v | log (i/A) = (0.531 ± 0.023)·log (v/V·s−1) − (3.941 ± 0.043) |
| i vs. v1/2 | i/A = (7.003 ± 0.041) × 10−5 v1/2/V1/2 ·s−1/2 + (4.002 ± 0.053)∙10−4 |
| log i vs. log C | log (i/A) = (0.972 ± 0.031)·log (C/mg∙L−1) + (1.821 ± 0.040) |
| i·v−1/2 vs. v1/2 | i·v−1/2/A·V−1/2·s1/2 = −(1.002 ± 0.050) × 10−5/V1/2·s−1/2 +(9.001 ± 0.033) × 10−5 |
| Compound | Calibration Curve | Correlation Coefficient R2 | LOD μg∙mL−1 | LOQ μg∙mL−1 | Intraday Precision % | Interday Precision % | Ep1 mV | Ep2 mV |
|---|---|---|---|---|---|---|---|---|
| U-47700 | i/μA = (0.01212 ± 0.00043)·(C/μg·L−1) + (0.012 ± 0.020) | 0.993 | 0.2 | 0.6 | 7 | 6 | +880 | +1150 |
| AH-7921 | i/μA = (0.01533 ± 0.00083)·(C/μg·L−1) + (0.083 ± 0.02) | 0.990 | 0.2 | 0.5 | 9 | 8 | +970 | +1150 |
| Water | Synthetic Urine | Urine | Capsules | |||||
|---|---|---|---|---|---|---|---|---|
| Compound | Concentration μg∙mL−1 a | Rc % | Concentration μg∙mL−1 a | Rc % | Concentration μg∙mL−1 a | Rc % | Amount mg | Rc % |
| U-47700 | 0.5 (100) | 111 | 0.5 (100) | 102 | 0.5 (100) | 73 | 10 | 86–99 |
| 0.5 (200) | 111 | 0.5 (100) | 102 | 0.5 (200) | 97 | |||
| 1 (50) | 115 | 1 (50) | 101 | 2 (50) | 86 | |||
| 20 (10) | 87 | 2 (50) | 105 | 5 (50) | 95 | |||
| AH-7921 | 0.5 (100) | 98 | 0.5 (100) | 76 | 0.5 (100) | 104 | 10 | 83–99 |
| 0.5 (200) | 115 | 0.5 (200) | 89 | 0.5 (200) | 96 | |||
| 1 (50) | 100 | 1 (50) | 85 | 2 (50) | 103 | |||
| 20 (10) | 110 | 2 (50) | 81 | 5 (50) | 106 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capucciati, A.; Burato, A.; Bersani, C.; Protti, S.; Profumo, A.; Merli, D. Electrochemical Behavior and Voltammetric Determination of Two Synthetic Aroyl Amides Opioids. Chemosensors 2023, 11, 198. https://doi.org/10.3390/chemosensors11030198
Capucciati A, Burato A, Bersani C, Protti S, Profumo A, Merli D. Electrochemical Behavior and Voltammetric Determination of Two Synthetic Aroyl Amides Opioids. Chemosensors. 2023; 11(3):198. https://doi.org/10.3390/chemosensors11030198
Chicago/Turabian StyleCapucciati, Andrea, Anna Burato, Chiara Bersani, Stefano Protti, Antonella Profumo, and Daniele Merli. 2023. "Electrochemical Behavior and Voltammetric Determination of Two Synthetic Aroyl Amides Opioids" Chemosensors 11, no. 3: 198. https://doi.org/10.3390/chemosensors11030198