Flavivirus Nonstructural Protein NS5 Dysregulates HSP90 to Broadly Inhibit JAK/STAT Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
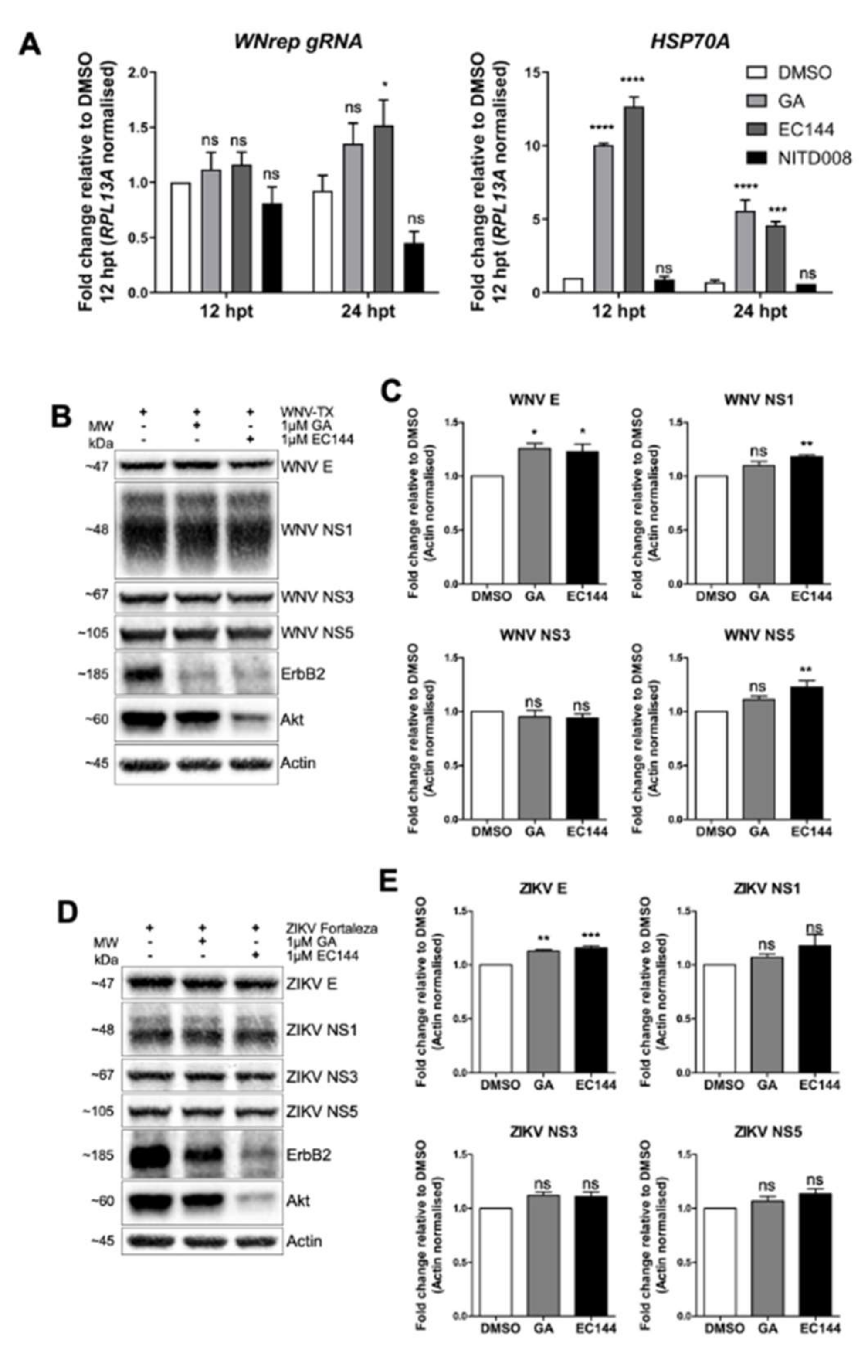
:1. Introduction
2. Materials and Methods
2.1. Contact for Reagent and Resource Sharing
2.2. Cell Lines
2.3. Virus Strains
2.4. Transcript Analysis by qRT-PCR
2.5. Protein Analysis by Western Blot
2.6. Immunofluorescence Assays
2.7. Conditioned Media Experiments
2.8. Generation of Recombinant DNA Constructs
2.9. Transfection of Nucleic Acids
2.10. Co-IP of Recombinant FLAG- and HA-tagged Proteins
2.11. ReCLIP Analysis of Endogenous Proteins
2.12. Inhibition of HSP90 in Replicon Cells and During WNV and ZIKV Infection
2.13. Proteasome Inhibition with MG-132
2.14. Generation of CRISPR-Targeted IFNAR1-/- PH5CH8 Cells
2.15. Generation of Huh7 WNrep Cells and Replicon-Cured Cells
2.16. Quantification and Statistical Analysis
3. Results
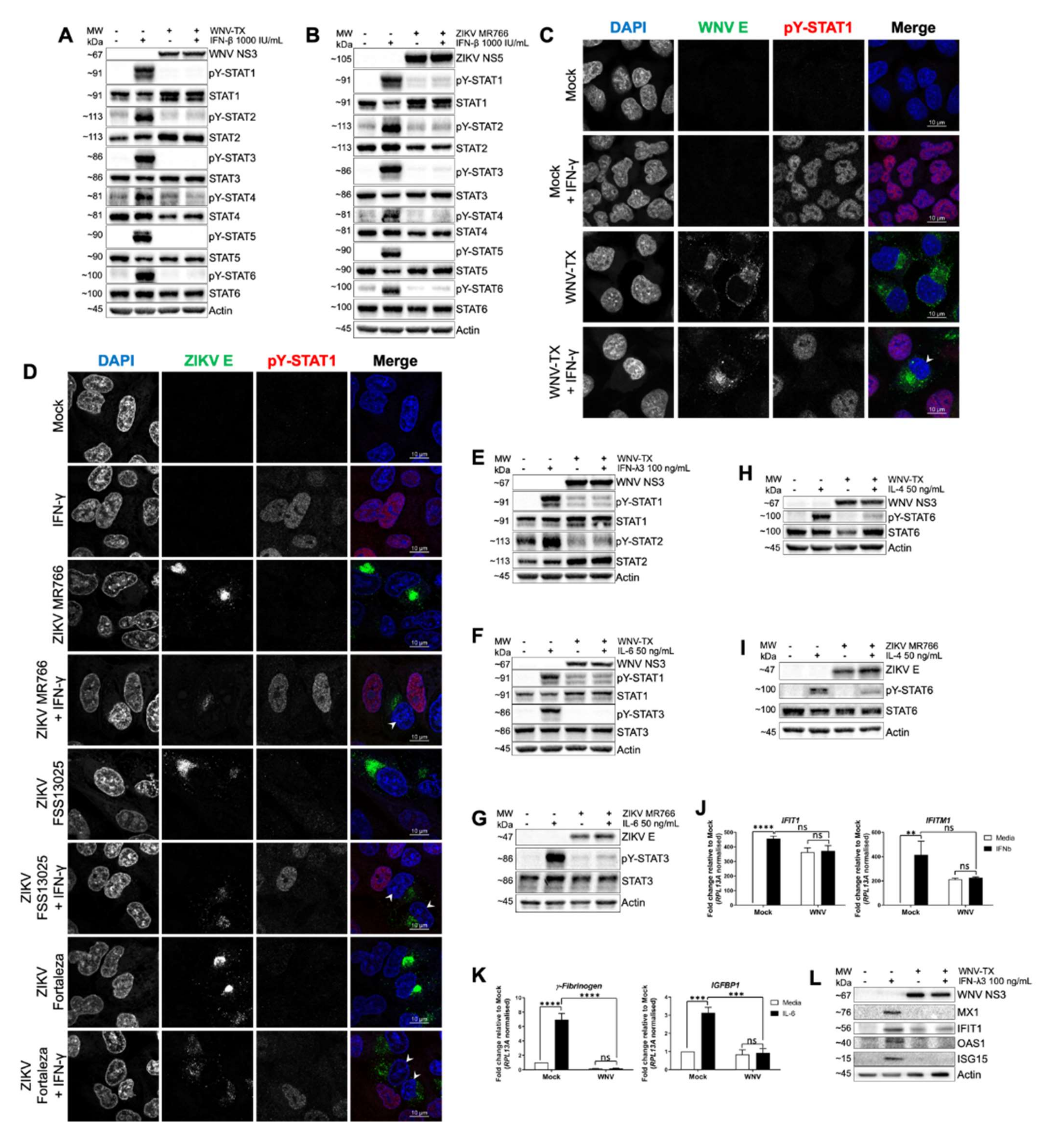
3.1. WNV and ZIKV Broadly Inhibited JAK/STAT Signaling Following Cytokine Stimulation of Target Cells
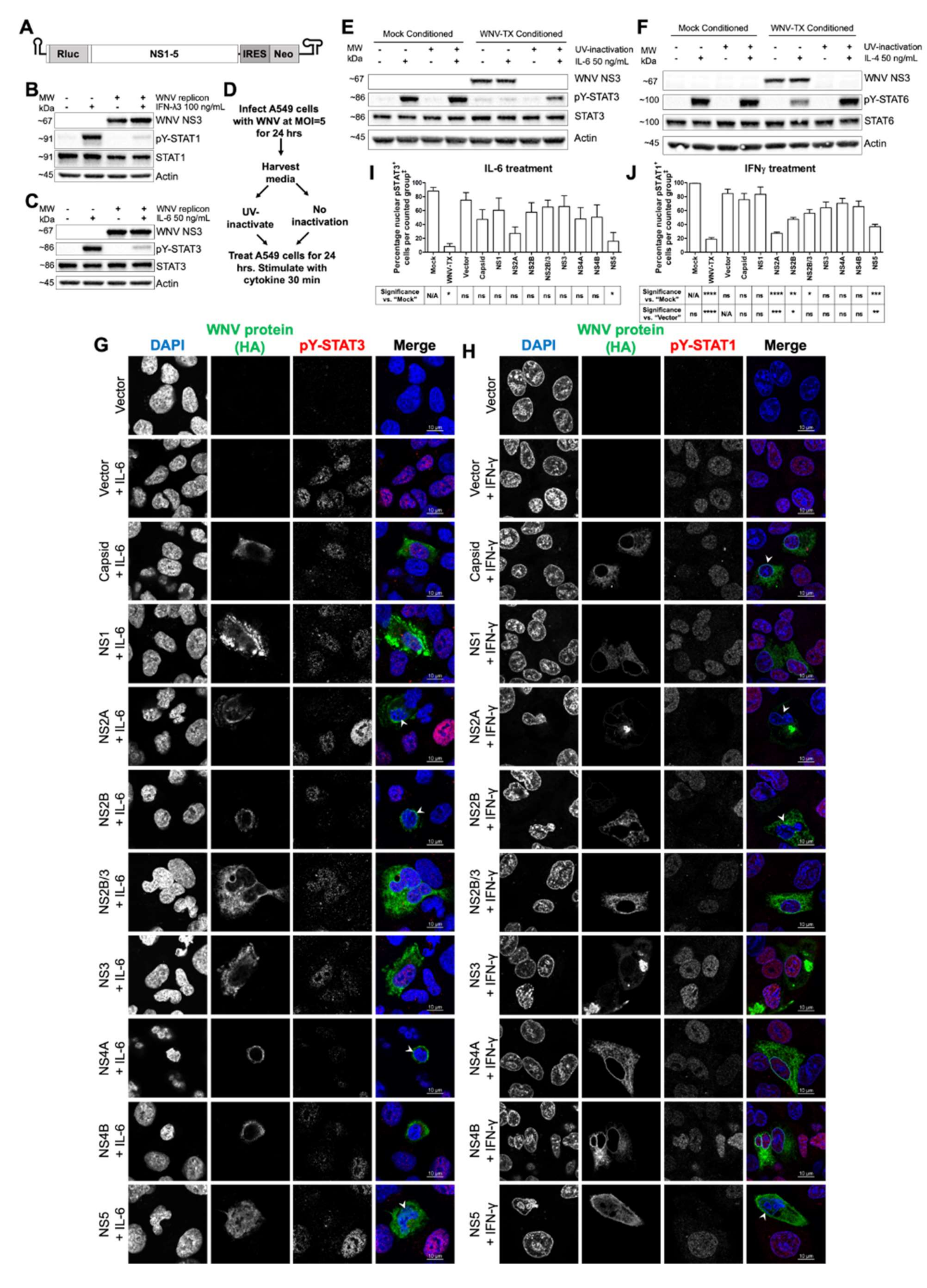
3.2. Cell-Intrinsic WNV NS Proteins mediated Broad JAK/STAT Inhibition
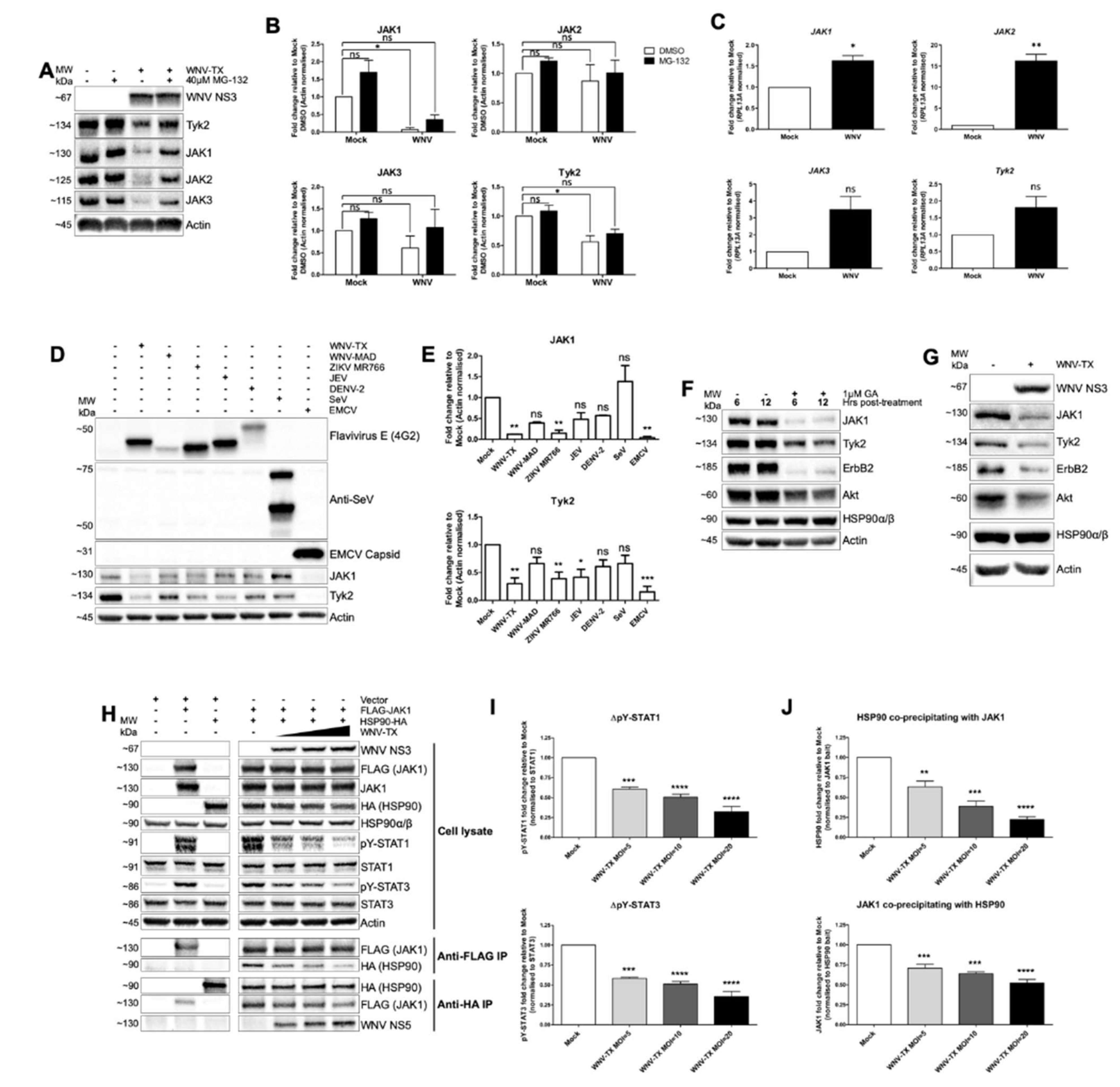
3.3. Flavivirus Infection Lead to JAK and Cytokine Receptor Degradation by the Proteasome
3.4. NS5 Disrupted HSP90-Client Kinase Interaction to Block JAK/STAT Signaling
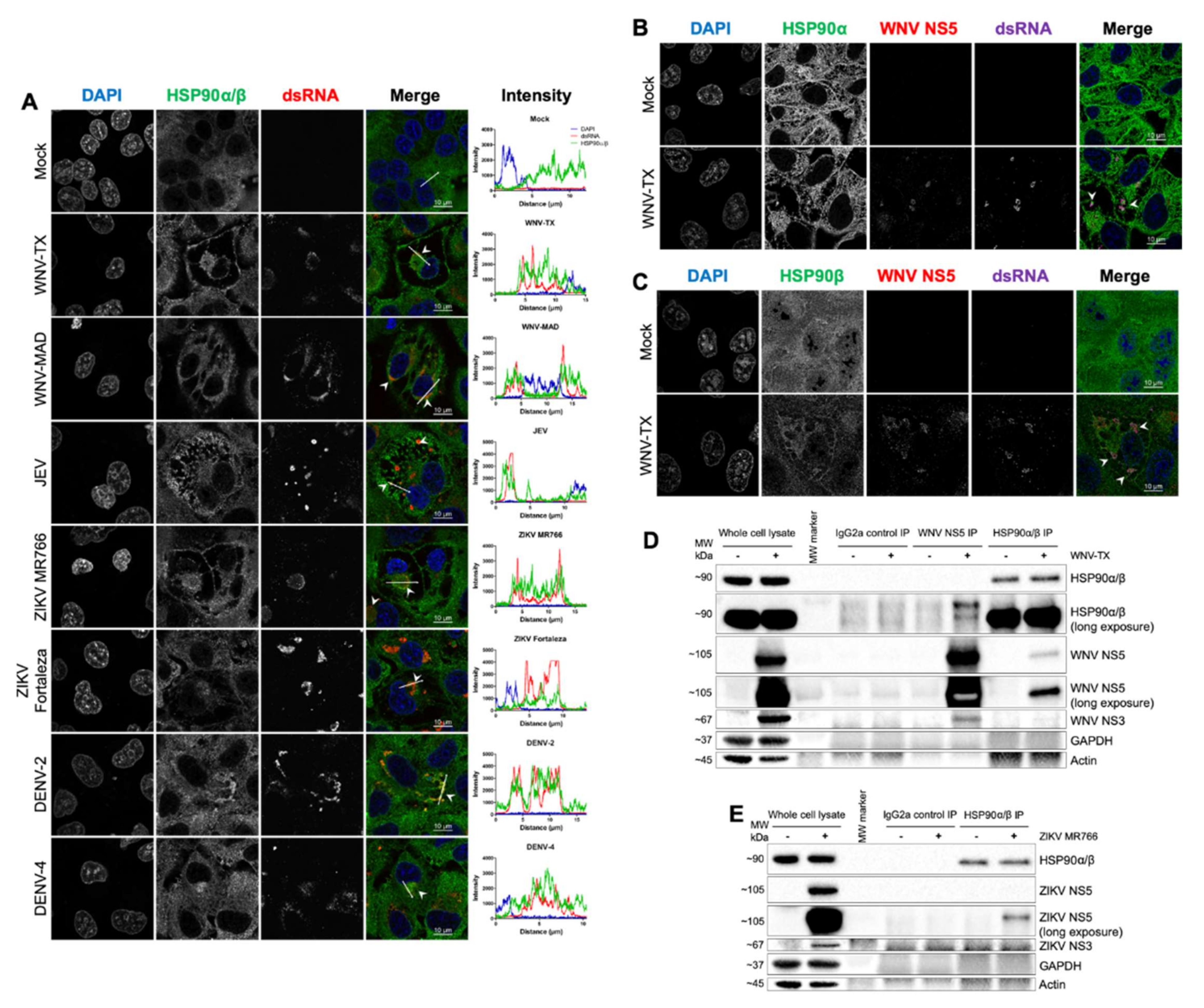
3.5. HSP90 Antagonism Was Found to be Linked to Interaction with Viral NS5 at Sites of RNA Replication
3.6. HSP90 Activity Was not Required to Support Flavivirus RNA Replication nor to Stabilize Viral Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guzman, M.G.; Gubler, D.J.; Izquierdo, A.; Martinez, E.; Halstead, S. Dengue infection. Nat. Rev. Dis. Prim. 2016, 2, 16055. [Google Scholar] [CrossRef] [PubMed]
- Roehrig, J. West Nile Virus in the United States—A Historical Perspective. Viruses 2013, 5, 3088–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, N.R.; Azevedo, R.D.S.D.S.; Kraemer, M.U.G.; Souza, R.; Cunha, M.S.; Hill, S.C.; Theze, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I.; et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016, 352, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaZear, H.M.; Diamond, M.S. Zika Virus: New Clinical Syndromes and Its Emergence in the Western Hemisphere. J. Virol. 2016, 90, 4864–4875. [Google Scholar] [CrossRef] [Green Version]
- França, G.V.A.; Schuler-Faccini, L.; Oliveira, W.; Henriques, C.M.P.; Carmo, E.H.; Pedi, V.D.; Nunes, M.L.; De Castro, M.C.; Serruya, S.; Silveira, M.F.; et al. Congenital Zika virus syndrome in Brazil: A case series of the first 1501 livebirths with complete investigation. Lancet 2016, 388, 891–897. [Google Scholar] [CrossRef] [Green Version]
- Melo, A.S.D.O.; Aguiar, R.; Ribeiro, S.T.C.; Batista, A.G.M.; Ferreira, T.; Sampaio, V.V.; Moura, S.R.M.; Rabello, L.P.; Gonzaga, C.E.; Ximenes, R.; et al. Congenital Zika Virus Infection: Beyond Neonatal Microcephaly. JAMA Neurol. 2016, 73, 1407–1416. [Google Scholar] [CrossRef]
- Walker, C.L.; Little, M.-T.E.; Roby, J.A.; Armistead, B.; Gale, M.; Rajagopal, L.; Nelson, B.R.; Ehinger, N.; Mason, B.; Nayeri, U.; et al. Zika virus and the nonmicrocephalic fetus: Why we should still worry. Am. J. Obstet. Gynecol. 2019, 220, 45–56. [Google Scholar] [CrossRef]
- Waldorf, K.A.; Stencel-Baerenwald, J.E.; Kapur, R.P.; Studholme, C.; Boldenow, E.; Vornhagen, J.; Baldessari, A.; Dighe, M.K.; Thiel, J.; Merillat, S.; et al. Fetal brain lesions after subcutaneous inoculation of Zika virus in a pregnant nonhuman primate. Nat. Med. 2016, 22, 1256–1259. [Google Scholar] [CrossRef]
- Waldorf, K.A.; Nelson, B.R.; Stencel-Baerenwald, J.E.; Studholme, C.; Kapur, R.P.; Armistead, B.; Walker, C.L.; Merillat, S.; Vornhagen, J.; Tisoncik-Go, J.; et al. Congenital Zika virus infection as a silent pathology with loss of neurogenic output in the fetal brain. Nat. Med. 2018, 24, 368–374. [Google Scholar] [CrossRef]
- Loo, Y.-M.; Gale, M. Immune Signaling by RIG-I-like Receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Suthar, M.S.; Ma, D.Y.; Thomas, S.; Lund, J.M.; Zhang, N.; Daffis, S.; Rudensky, A.Y.; Bevan, M.J.; Clark, E.A.; Kaja, M.-K.; et al. IPS-1 Is Essential for the Control of West Nile Virus Infection and Immunity. PLOS Pathog. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2013, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suthar, M.S.; Diamond, M.S.; Jr, M.G. West Nile virus infection and immunity. Nat. Rev. Genet. 2013, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Best, S.M. The Many Faces of the Flavivirus NS5 Protein in Antagonism of Type I Interferon Signaling. J. Virol. 2017, 91, e01970–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, M.S. Mechanisms of Evasion of the Type I Interferon Antiviral Response by Flaviviruses. J. Interf. Cytokine Res. 2009, 29, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.D.; Seeger, C. Differential Effects of Mutations in NS4B on West Nile Virus Replication and Inhibition of Interferon Signaling. J. Virol. 2007, 81, 11809–11816. [Google Scholar] [CrossRef] [Green Version]
- Keller, B.C.; Fredericksen, B.L.; Samuel, M.A.; Mock, R.E.; Mason, P.W.; Diamond, M.S.; Gale, M., Jr. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J. Virol. 2006, 80, 9424–9434. [Google Scholar] [CrossRef] [Green Version]
- Laurent-Rolle, M.; Boer, E.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 Protein of the Virulent West Nile Virus NY99 Strain Is a Potent Antagonist of Type I Interferon-Mediated JAK-STAT Signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef] [Green Version]
- Daffis, S.; LaZear, H.M.; Liu, W.J.; Audsley, M.; Engle, M.; Khromykh, A.A.; Diamond, M.S. The Naturally Attenuated Kunjin Strain of West Nile Virus Shows Enhanced Sensitivity to the Host Type I Interferon Response. J. Virol. 2011, 85, 5664–5668. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, R.L.; MacKenzie, J.M. West Nile Virus Differentially Modulates the Unfolded Protein Response To Facilitate Replication and Immune Evasion. J. Virol. 2010, 85, 2723–2732. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Wang, X.J.; Mokhonov, V.; Shi, P.-Y.; Randall, R.; Khromykh, A.A. Inhibition of Interferon Signaling by the New York 99 Strain and Kunjin Subtype of West Nile Virus Involves Blockage of STAT1 and STAT2 Activation by Nonstructural Proteins. J. Virol. 2005, 79, 1934–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Jordán, J.L.; Laurent-Rolle, M.; Ashour, J.; Martínez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; García-Sastre, A. Inhibition of Alpha/Beta Interferon Signaling by the NS4B Protein of Flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuessler, A.; Funk, A.; LaZear, H.M.; Cooper, D.; Torres, S.; Daffis, S.; Jha, B.K.; Kumagai, Y.; Takeuchi, O.; Hertzog, P.; et al. West Nile Virus Noncoding Subgenomic RNA Contributes to Viral Evasion of the Type I Interferon-Mediated Antiviral Response. J. Virol. 2012, 86, 5708–5718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuai, K.; Liu, B. Regulation of JAK–STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef]
- Bauer, S.; Kerr, B.J.; Patterson, P.H. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat. Rev. Neurosci. 2007, 8, 221–232. [Google Scholar] [CrossRef]
- Taguwa, S.; Maringer, K.; Li, X.; Bernal-Rubio, D.; Rauch, J.N.; Gestwicki, J.E.; Andino, R.; Fernández-Sesma, A.; Frydman, J. Defining Hsp70 Subnetworks in Dengue Virus Replication Reveals Key Vulnerability in Flavivirus Infection. Cell 2015, 163, 1108–1123. [Google Scholar] [CrossRef] [Green Version]
- Taguwa, S.; Yeh, M.-T.; Rainbolt, T.K.; Nayak, A.; Shao, H.; Gestwicki, J.E.; Andino, R.; Frydman, J. Zika Virus Dependence on Host Hsp70 Provides a Protective Strategy against Infection and Disease. Cell Rep. 2019, 26, 906–920. [Google Scholar] [CrossRef] [Green Version]
- Pujhari, S.; Brustolin, M.; Macias, V.; Nissly, R.; Nomura, M.; Kuchipudi, S.; Rasgon, J.L. Heat shock protein 70 (Hsp70) mediates Zika virus entry, replication, and egress from host cells. Emerg. Microbes Infect. 2019, 8, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.L.; Friedman, D.B.; Yu, H.; Carnahan, R.H.; Reynolds, A.B. ReCLIP (Reversible Cross-Link Immuno-Precipitation): An Efficient Method for Interrogation of Labile Protein Complexes. PLoS ONE 2011, 6, e16206. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Gale, M.; Keller, B.; Huang, H.; Brown, M.S.; Goldstein, J.L.; Ye, J. Identification of FBL2 As a Geranylgeranylated Cellular Protein Required for Hepatitis C Virus RNA Replication. Mol. Cell 2005, 18, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.A.; Young, P. The flavivirus NS1 protein: Molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antivir. Res. 2013, 98, 192–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jen, G.; Detjen, B.M.; Thach, R.E. Shutoff of HeLa cell protein synthesis by encephalomyocarditis virus and poliovirus: A comparative study. J. Virol. 1980, 35, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, L.; Tomasi, T.B. The Heat Shock Protein 90-CDC37 Chaperone Complex Is Required for Signaling by Types I and II Interferons. J. Boil. Chem. 2005, 281, 1876–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoof, N.; Von Bonin, F.; Truemper, L.; Kube, D. HSP90 is essential for Jak-STAT signaling in classical Hodgkin lymphoma cells. Cell Commun. Signal. 2009, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.-S.; Lee, L.-C.; Sun, F.-C.; Chao, C.-C.; Fu, H.-W.; Lai, Y.-K. Involvement of calcium in the differential induction of heat shock protein 70 by heat shock protein 90 inhibitors, geldanamycin and radicicol, in human non-small cell lung cancer H460 cells. J. Cell. Biochem. 2005, 97, 156–165. [Google Scholar] [CrossRef]
- Srisutthisamphan, K.; Jirakanwisal, K.; Ramphan, S.; Tongluan, N.; Kuadkitkan, A.; Smith, D.R. Hsp90 interacts with multiple dengue virus 2 proteins. Sci. Rep. 2018, 8, 4308. [Google Scholar] [CrossRef] [Green Version]
- Bocchini, C.E.; Kasembeli, M.M.; Roh, S.-H.; Tweardy, D.J. Contribution of chaperones to STAT pathway signaling. JAK-STAT 2014, 3, e970459. [Google Scholar] [CrossRef]
- Lu, C.-Y.; Chang, Y.-C.; Hua, C.-H.; Chuang, C.; Huang, S.-H.; Kung, S.-H.; Hour, M.-J.; Lin, C.-W. Tubacin, an HDAC6 Selective Inhibitor, Reduces the Replication of the Japanese Encephalitis Virus via the Decrease of Viral RNA Synthesis. Int. J. Mol. Sci. 2017, 18, 954. [Google Scholar] [CrossRef] [Green Version]
- Carpp, L.N.; Rogers, R.S.; Moritz, R.L.; Aitchison, J.D. Quantitative Proteomic Analysis of Host-virus Interactions Reveals a Role for Golgi Brefeldin A Resistance Factor 1 (GBF1) in Dengue Infection. Mol. Cell. Proteom. 2014, 13, 2836–2854. [Google Scholar] [CrossRef] [Green Version]
- Kovanich, D.; Saisawang, C.; Sittipaisankul, P.; Ramphan, S.; Kalpongnukul, N.; Somparn, P.; Pisitkun, T.; Smith, D.R. Analysis of the Zika and Japanese Encephalitis Virus NS5 Interactomes. J. Proteome Res. 2019, 18, 3203–3218. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Zhong, Y.; Sanborn, M.A.; Teoh, T.C.; Ruan, J.; Yusof, R.; Hang, J.; Henderson, M.J.; Fang, S. Small molecule grp94 inhibitors with antiviral activity against Dengue and Zika virus. Antiviral Res. 2019, 104590. [Google Scholar] [CrossRef] [PubMed]
- Emeny, J.M.; Morgan, M.J. Regulation of the Interferon System: Evidence that Vero Cells have a Genetic Defect in Interferon Production. J. Gen. Virol. 1979, 43, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Desmyter, J.; Melnick, J.L.; Rawls, W.E. Defectiveness of Interferon Production and of Rubella Virus Interference in a Line of African Green Monkey Kidney Cells (Vero). J. Virol. 1968, 2, 955–961. [Google Scholar] [CrossRef] [Green Version]
- Michlmayr, D.; Andrade, P.; Gonzalez, K.; Balmaseda, A.; Harris, E. CD14+CD16+ monocytes are the main target of Zika virus infection in peripheral blood mononuclear cells in a paediatric study in Nicaragua. Nat. Microbiol. 2017, 2, 1462–1470. [Google Scholar] [CrossRef]
- Balsitis, S.J.; Flores, D.; Coloma, J.; Beatty, P.R.; Alava, A.; McKerrow, J.H.; Castro, G.; Harris, E. Tropism of dengue virus in mice and humans defined by viral nonstructural protein 3-specific immunostaining. Am. J. Trop. Med. Hyg. 2009, 80, 416–424. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, J.S.; Gubler, D.J.; Petersen, L.R. Emerging flaviviruses: The spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat. Med. 2004, 10, S98–S109. [Google Scholar] [CrossRef]
- Robert, J. Evolution of heat shock protein and immunity. Dev. Comp. Immunol. 2003, 27, 449–464. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Boil. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Paradkar, P.; Duchemin, J.-B.; Rodriguez-Andres, J.; Trinidad, L.; Walker, P.J. Cullin4 Is Pro-Viral during West Nile Virus Infection of Culex Mosquitoes. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roby, J.A.; Esser-Nobis, K.; Dewey-Verstelle, E.C.; Fairgrieve, M.R.; Schwerk, J.; Lu, A.Y.; Soveg, F.W.; Hemann, E.A.; Hatfield, L.D.; Keller, B.C.; et al. Flavivirus Nonstructural Protein NS5 Dysregulates HSP90 to Broadly Inhibit JAK/STAT Signaling. Cells 2020, 9, 899. https://doi.org/10.3390/cells9040899
Roby JA, Esser-Nobis K, Dewey-Verstelle EC, Fairgrieve MR, Schwerk J, Lu AY, Soveg FW, Hemann EA, Hatfield LD, Keller BC, et al. Flavivirus Nonstructural Protein NS5 Dysregulates HSP90 to Broadly Inhibit JAK/STAT Signaling. Cells. 2020; 9(4):899. https://doi.org/10.3390/cells9040899
Chicago/Turabian StyleRoby, Justin A., Katharina Esser-Nobis, Elyse C. Dewey-Verstelle, Marian R. Fairgrieve, Johannes Schwerk, Amy Y. Lu, Frank W. Soveg, Emily A. Hemann, Lauren D. Hatfield, Brian C. Keller, and et al. 2020. "Flavivirus Nonstructural Protein NS5 Dysregulates HSP90 to Broadly Inhibit JAK/STAT Signaling" Cells 9, no. 4: 899. https://doi.org/10.3390/cells9040899