Probing Carbon Utilization of Cordyceps militaris by Sugar Transportome and Protein Structural Analysis


 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
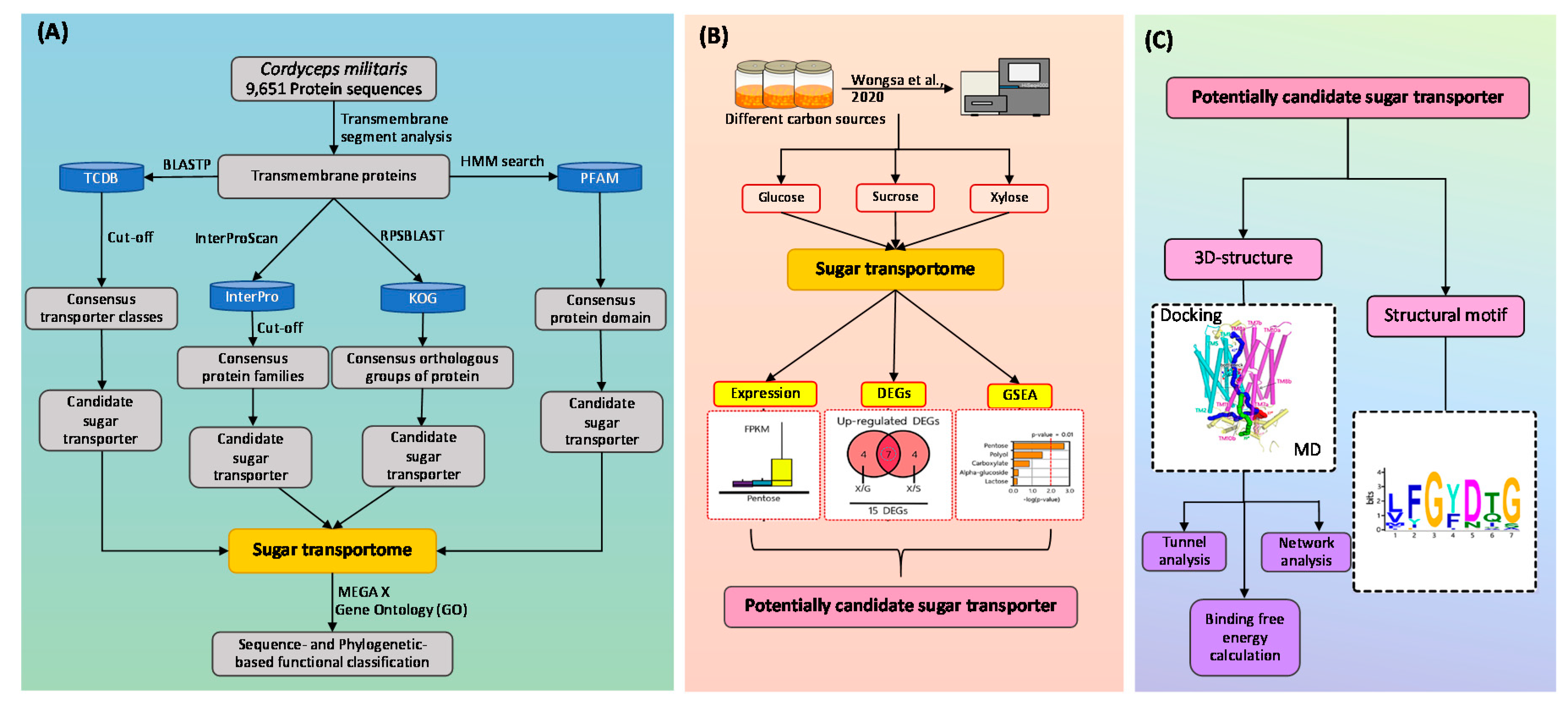
2.1. Construction of Sugar Transportome
2.2. Multiple Sequence Alignment and Structural Motif Analysis
2.3. Gene Set Enrichment Analysis
2.4. Structural Modeling and Molecular Docking
2.5. Bilayer Construction and Molecular Dynamics Simulation
2.6. Transport Pathway Identification and Residue Interaction Network Analysis
2.7. Binding Free Energy Calculation
3. Results
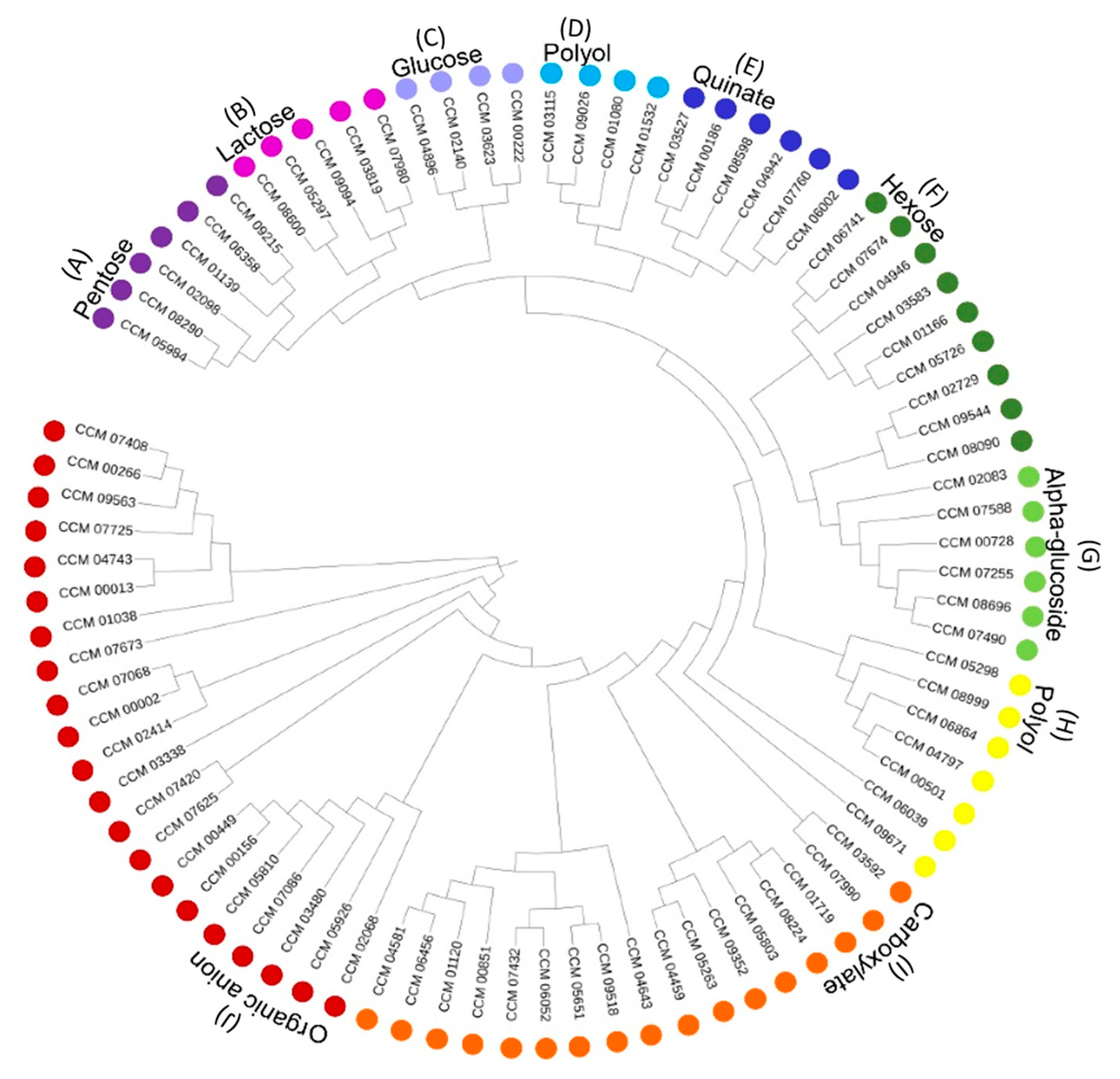
3.1. Characteristics and Consensus Features of Sugar Transportome
3.2. Functional Classification of C. militaris Sugar Transporters
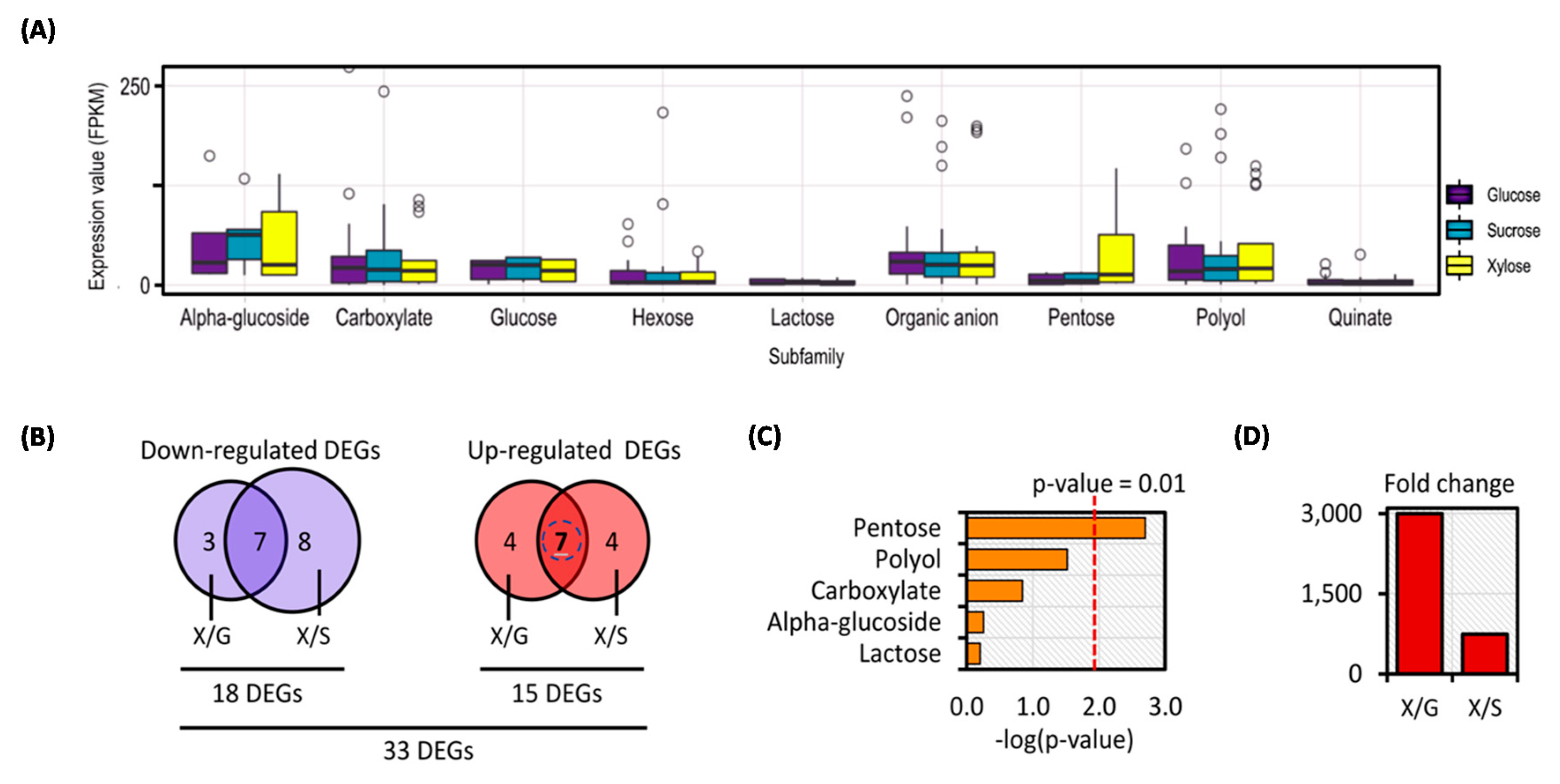
3.3. Transcriptional Response of C. militaris Sugar Transportome in Different Carbon Sources Uncovered Potentially Candidate Sugar Transporter for Xylose Utilization
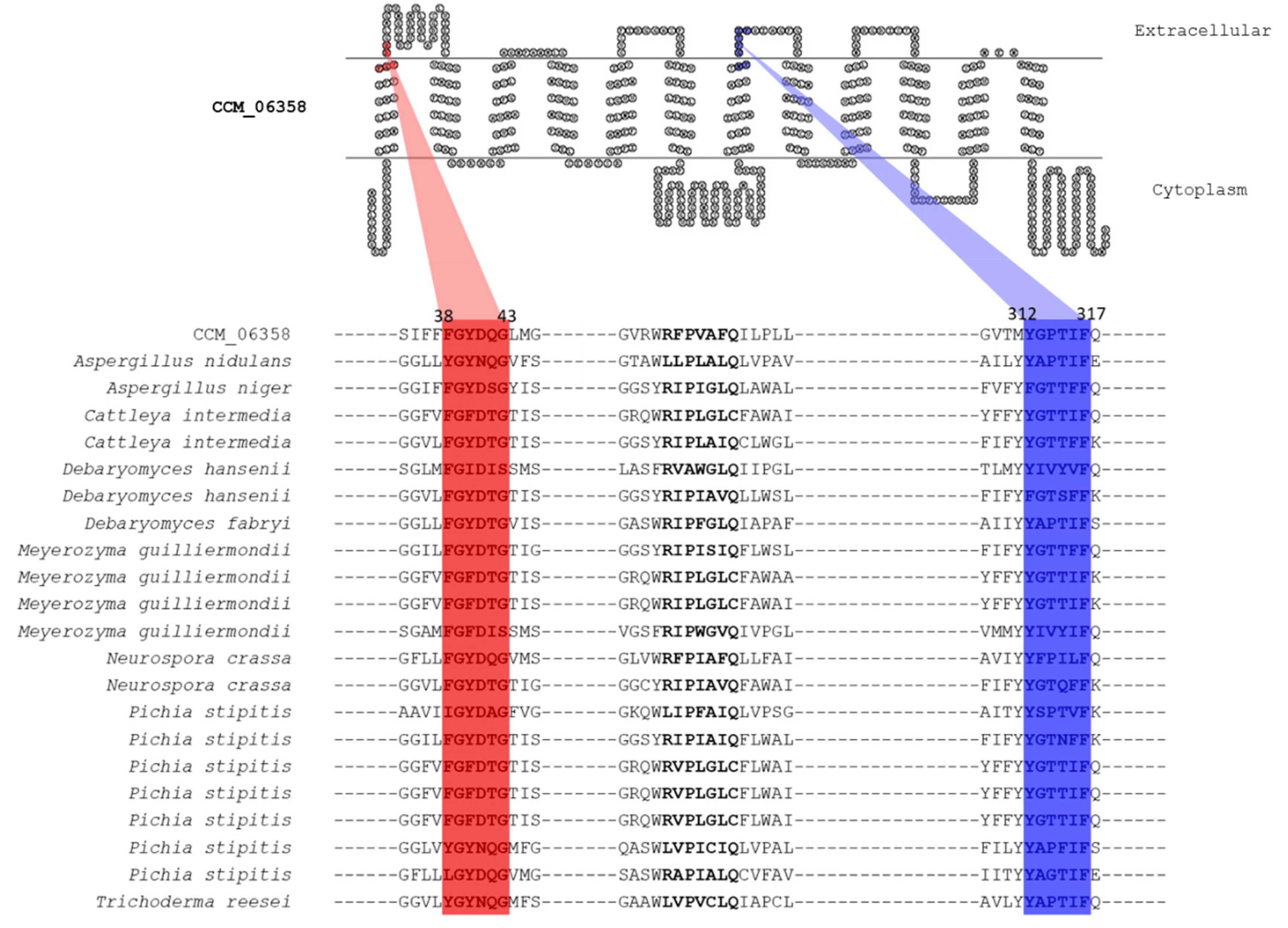
3.4. Putative Xylose Transport Function of CCM_06358 Revealed by the Presence of Highly Conserved Structural Motifs
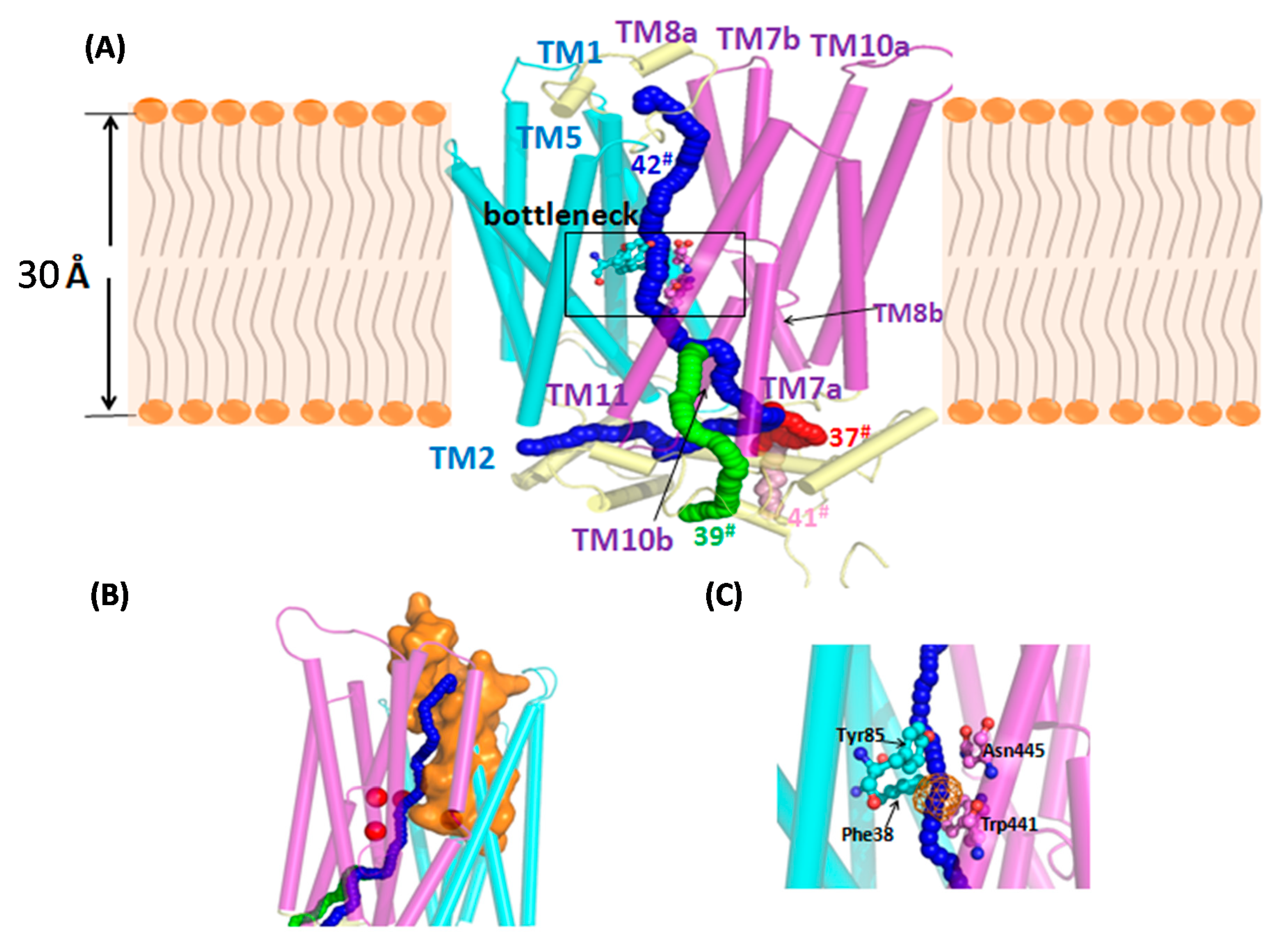
3.5. Annotated Molecular Structure and Transport Pathway of a Selected Pentose Transporter, CCM_06358
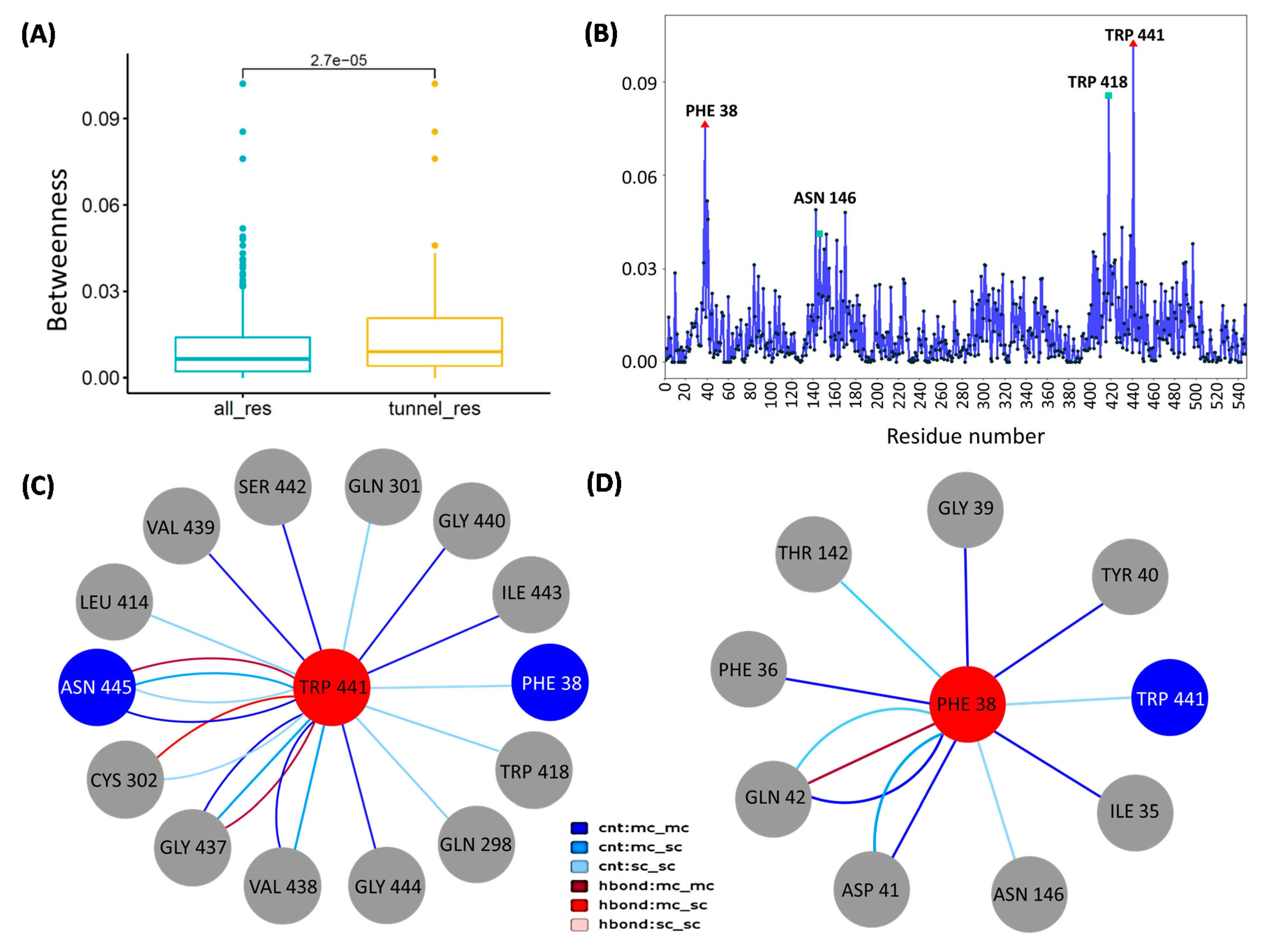
3.6. Network Analysis Identified Peculiar Residues of CCM_06358 with a Key Role in Transport Pathways
3.7. Verification of Functional Roles of Key Residues of CCM_06358 by Binding Free Energy Calculation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cui, J.D. Biotechnological production and applications of Cordyceps militaris, a valued traditional Chinese medicine. Crit. Rev. Biotechnol. 2015, 35, 475–484. [Google Scholar] [CrossRef]
- Molnár, I.; Gibson, D.M.; Krasnoff, S.B. Secondary metabolites from entomopathogenic Hypocrealean fungi. Nat. Prod. Rep. 2010, 27, 1241–1275. [Google Scholar] [CrossRef]
- Cui, Z.Y.; Park, S.J.; Jo, E.; Hwang, I.H.; Lee, K.B.; Kim, S.W.; Kim, D.J.; Joo, J.C.; Hong, S.H.; Lee, M.G.; et al. Cordycepin induces apoptosis of human ovarian cancer cells by inhibiting CCL5-mediated Akt/NF-κB signaling pathway. Cell Death Discov. 2018, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Fong, Y.C.; Lee, C.Y.; Chen, M.Y.; Tsai, H.C.; Hsu, H.C.; Tang, C.H. CCL5 increases lung cancer migration via PI3K, Akt and NF-kappaB pathways. Biochem. Pharmacol. 2009, 77, 794–803. [Google Scholar] [CrossRef]
- Takahashi, S.; Tamai, M.; Nakajima, S.; Kato, H.; Johno, H.; Nakamura, T.; Kitamura, M. Blockade of adipocyte differentiation by cordycepin. Br. J. Pharmacol. 2012, 167, 561–575. [Google Scholar] [CrossRef] [Green Version]
- Qi, G.; Zhou, Y.; Zhang, X.; Yu, J.; Li, X.; Cao, X.; Wu, C.; Guo, P. Cordycepin promotes browning of white adipose tissue through an AMP-activated protein kinase (AMPK)-dependent pathway. Acta Pharm. Sin. B 2019, 9, 135–143. [Google Scholar] [CrossRef]
- Das, S.K.; Masuda, M.; Sakurai, A.; Sakakibara, M. Medicinal uses of the mushroom Cordyceps militaris: Current state and prospects. Fitoterapia 2010, 81, 961–968. [Google Scholar] [CrossRef]
- Wongsa, B.; Raethong, N.; Chumnanpuen, P.; Wong-ekkabut, J.; Laoteng, K.; Vongsangnak, W. Alternative metabolic routes in channeling xylose to cordycepin production of Cordyceps militaris identified by comparative transcriptome analysis. Genomics 2020, 112, 629–636. [Google Scholar] [CrossRef]
- Raethong, N.; Laoteng, K.; Vongsangnak, W. Uncovering global metabolic response to cordycepin production in Cordyceps militaris through transcriptome and genome-scale network-driven analysis. Sci. Rep. 2018, 8, 9250. [Google Scholar] [CrossRef]
- Farinas, C.S.; Florencio, C.; Badino, A.C. On-site production of cellulolytic enzymes by the sequential cultivation method. Methods Mol. Biol. 2018, 1796, 273–282. [Google Scholar]
- Teeravivattanakit, T.; Baramee, S.; Phitsuwan, P.; Sornyotha, S.; Waeonukul, R.; Pason, P.; Tachaapaikoon, C.; Poomputsa, K.; Kosugi, A.; Sakka, K.; et al. Chemical pretreatment-independent saccharifications of xylan and cellulose of rice straw by bacterial weak lignin-binding xylanolytic and cellulolytic enzymes. Appl. Environ. Microbiol. 2017, 83, e01522-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.O.; Yun, J.W. A comparative study on the production of exopolysaccharides between two entomopathogenic fungi Cordyceps militaris and Cordyceps sinensis in submerged mycelial cultures. J. Appl. Microbiol. 2005, 99, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Aguilar-Pontes, M.V.; de Vries, R.P.; Mäkelä, M.R. In silico analysis of putative sugar transporter genes in Aspergillus niger using phylogeny and comparative transcriptomics. Front Microbiol. 2018, 9, 1045. [Google Scholar] [CrossRef]
- Milner, D.S.; Attah, V.; Cook, E.; Maguire, F.; Savory, F.R.; Morrison, M.; Müller, C.A.; Foster, P.G.; Talbot, N.J.; Leonard, G.; et al. Environment-dependent fitness gains can be driven by horizontal gene transfer of transporter-encoding genes. Proc. Natl. Acad. Sci. USA 2019, 116, 5613–5622. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, K.M.V.; de Paula, R.G.; Antoniêto, A.C.C.; dos Reis, T.F.; Carraro, C.B.; Silva, A.C.; Almeida, F.; Rechia, C.G.V.; Goldman, G.H.; Silva, R.N. Characterization of a novel sugar transporter involved in sugarcane bagasse degradation in Trichoderma reesei. Biotechnol. Biofuels 2018, 11, 84. [Google Scholar] [CrossRef]
- Fang, W.; Leger, R.J. Mrt, a gene unique to fungi, encodes an oligosaccharide transporter and facilitates rhizosphere competency in Metarhizium robertsii. Plant. Physiol. 2010, 154, 1549–1557. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.X.; Ji, X.P.; Li, J.X.; Keyhani, N.O.; Feng, M.G.; Ying, S.H. A putative alpha-glucoside transporter gene BbAGT1 contributes to carbohydrate utilization, growth, conidiation and virulence of filamentous entomopathogenic fungus Beauveria bassiana. Res. Microbiol. 2013, 164, 480–489. [Google Scholar] [CrossRef]
- Zheng, P.; Xia, Y.; Xiao, G.; Xiong, C.; Hu, X.; Zhang, S.; Zheng, H.; Huang, Y.; Zhou, Y.; Wang, S.; et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011, 12, R116. [Google Scholar] [CrossRef] [Green Version]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef]
- Saier, M.H., Jr.; Reddy, V.S.; Tsu, B.V.; Ahmed, M.S.; Li, C.; Moreno-Hagelsieb, G. The Transporter Classification Database (TCDB): Recent advances. Nucleic Acids Res. 2016, 44, D372–D379. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium, The Gene Ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2018, 47, D330–D338.
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [Green Version]
- Väremo, L.; Nielsen, J.; Nookaew, I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef]
- Patil, K.R.; Nielsen, J. Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc. Natl. Acad. Sci. USA. 2005, 102, 2685–2689. [Google Scholar] [CrossRef] [Green Version]
- Consortium, T.U. UniProt: A hub for protein information. Nucleic Acids Res. 2014, 43, D204–D212. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2014, 1137, 1–15. [Google Scholar]
- Khodade, P.; Prabhu, R.; Chandra, N.; Raha, S.; Govindarajan, R. Parallel implementation of AutoDock. J. Appl. Crystallogr. 2007, 40, 598–599. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on sixlipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem 2008, 29, 1859–1865.50. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Nose, S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P.; et al. CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. Plos Comput. Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef] [Green Version]
- Doncheva, N.T.; Klein, K.; Domingues, F.S.; Albrecht, M. Analyzing and visualizing residue networks of protein structures. Trends Biochem. Sci. 2011, 36, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_ mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem Inf Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meliciani, I.; Klenin, K.; Strunk, T.; Schmitz, K.; Wenzel, W. Probing hot spots on protein-protein interfaces with all-atom freeenergy simulation. J. Chem Phys. 2009, 131, 034114. [Google Scholar] [CrossRef] [PubMed]
- Young, E.M.; Tong, A.; Bui, H.; Spofford, C.; Alper, H.S. Rewiring yeast sugar transporter preference through modifying a conserved protein motif. Proc. Natl. Acad. Sci. USA 2014, 111, 131. [Google Scholar] [CrossRef] [Green Version]
- Young, E.M.; Comer, A.D.; Huang, H.; Alper, H.S. A molecular transporter engineering approach to improving xylose catabolism in Saccharomyces Cerevisiae. Metab. Eng. 2012, 14, 401–411. [Google Scholar] [CrossRef]
- Sloothaak, J.; Tamayo-Ramos, J.A.; Odoni, D.I.; Laothanachareon, T.; Derntl, C.; Mach-Aigner, A.R.; Martins Dos Santos, V.A.P.; Schaap, P.J. Identification and functional characterization of novel xylose transporters from the cell factories Aspergillus niger and Trichoderma reesei. Biotechnol. Biofuels 2016, 9, 148. [Google Scholar] [CrossRef]
- Tusnady, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef]
- Paulsen, P.A.; Custodio, T.F.; Pedersen, B.P. Crystal structure of the plant symporter STP10 illuminates sugar uptake mechanism in monosaccharide transporter superfamily. Nat. Commun. 2019, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Stourac, J.; Vavra, O.; Kokkonen, P.; Filipovic, J.; Pinto, G.; Brezovsky, J.; Damborsky, J.; Bednar, D. Caver Web 1.0: Identification of tunnels and channels in proteins and analysis of ligand transport. Nucleic Acids Res. 2019, 47, W414–W422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doncheva, N.T.; Assenov, Y.; Domingues, F.S.; Albrecht, M. Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc. 2012, 7, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhou, J.; Sun, M.; Chen, J.; Hu, G.; Shen, B. The construction of an amino acid network for understanding protein structure and function. Amino Acids 2014, 46, 1419–1439. [Google Scholar] [CrossRef]
- Hu, G.; Yan, W.; Zhou, J.; Shen, B. Residue interaction network analysis of Dronpa and a DNA clamp. J. Theor. Biol. 2014, 348, 55–64. [Google Scholar] [CrossRef]
- Kapetis, D.; Sassone, J.; Yang, Y.; Galbardi, B.; Xenakis, M.N.; Westra, R.L.; Szklarczyk, R.; Lindsey, P.; Faber, C.G.; Gerrits, M. Network topology of NaV1.7 mutations in sodium channel-related painful disorders. Bmc Syst. Biol. 2017, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.B.; Qin, Y.L.; Qi, Z.Q.; Niu, Y.; Liu, Z.J.; Liu, W.X.; He, H.; Cao, Z.M.; Yang, Y. Genome-Wide Analysis, Expression Profile, and Characterization of the Acid Invertase Gene Family in Pepper. Int. J. Mol. Sci. 2019, 20, 15. [Google Scholar] [CrossRef] [Green Version]
- Madej, M.G.; Sun, L.; Yan, N.; Kaback, H.R. Functional architecture of MFS-glucose transporters. Proc. Natl. Acad. Sci. USA 2014, 111, E719–E727. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Consensus Features | Numbers of Sugar Transporters |
|---|---|
| Protein domain (PFAM) | 49 |
| PF00083: Sugar transporter | 49 |
| Protein families (InterPro) | 65 |
| IPR003663: Sugar/inositol transporter | 41 |
| IPR004853: Sugar phosphate transporter | 8 |
| IPR005828: Sugar transporter | 9 |
| IPR005829: Sugar transporter | 5 |
| IPR007271: Nucleotide-sugar transporter | 1 |
| IPR011701: Major facilitator superfamily | 1 |
| Eukaryotic orthologous groups (KOG) | 63 |
| KOG0254: Predicted transporter | 48 |
| KOG1444: Nucleotide-sugar transporter | 2 |
| KOG1441: Carbohydrate transporter | 6 |
| KOG2234: UDP-galactose transporter | 1 |
| KOG0252: Inorganic phosphate transporter | 3 |
| KOG0769: Mitochondrial carrier protein | 1 |
| KOG4332: Sugar transporter | 1 |
| KOG0569: Carbohydrate transporter | 1 |
| Transporter classification database (TCDB) | 75 |
| 2.A.1: Major facilitator superfamily | 58 |
| 2.A.2: Glycoside symporter family | 3 |
| 2.A.7: Drug/metabolite transporter superfamily | 9 |
| 2.A.16: Dicarboxylate transporter family | 1 |
| 2.A.29: Mitochondrial carrier superfamily | 1 |
| 2.A.50: Glycerol uptake transporter family | 1 |
| 2.A.96: Acetate uptake transporter family | 2 |
| Total of non-redundant sugar transporters | 85 |
| Tunnel | Location | Throughput 1 | Cost | Bottleneck Radius 2 | Length 3 | Curvature 4 | Bottleneck 5 Residue |
|---|---|---|---|---|---|---|---|
| 37 | Between the TM7a and TM8b | 0.039658 | 3.227465 | 0.855433 | 84.94969 | 1.64852 | Phe 38; Tyr85; Trp441; Asn445 |
| 39 | Between the TM7a and TM11 | 0.029718 | 3.515996 | 0.855433 | 85.94388 | 1.555505 | |
| 41 | Between the TM7a and TM10b | 0.024121 | 3.724671 | 0.855433 | 94.43239 | 1.576965 | |
| 42 | Between the TM2 and TM11 | 0.017327 | 4.055483 | 0.855433 | 98.90193 | 2.068183 |
| Residues | ΔEMM | ΔGpolar | ΔGSASA | ΔGbind |
|---|---|---|---|---|
| Trp418 | −4.377 | 2.3992 | −0.4297 | −2.4067 |
| Phe38 | −2.7748 | 1.1358 | −0.3484 | −1.9848 |
| Ile174 | −1.4312 | 0.2321 | −0.2701 | −1.4684 |
| Ile35 | −1.4563 | 0.3508 | −0.2033 | −1.3111 |
| Val177 | −0.9842 | 0.2892 | −0.1434 | −0.8382 |
| Ile306 | −1.2814 | 0.7336 | −0.1411 | −0.69 |
| Trp441 | −2.524 | 2.0924 | −0.2414 | −0.6728 |
| Pro150 | −0.81 | 0.3465 | −0.0892 | −0.5512 |
| Leu414 | −1.9582 | 1.6616 | −0.1454 | −0.4428 |
| Ala307 | −0.7957 | 0.5096 | −0.1578 | −0.442 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirithep, K.; Xiao, F.; Raethong, N.; Zhang, Y.; Laoteng, K.; Hu, G.; Vongsangnak, W. Probing Carbon Utilization of Cordyceps militaris by Sugar Transportome and Protein Structural Analysis. Cells 2020, 9, 401. https://doi.org/10.3390/cells9020401
Sirithep K, Xiao F, Raethong N, Zhang Y, Laoteng K, Hu G, Vongsangnak W. Probing Carbon Utilization of Cordyceps militaris by Sugar Transportome and Protein Structural Analysis. Cells. 2020; 9(2):401. https://doi.org/10.3390/cells9020401
Chicago/Turabian StyleSirithep, Kanokwadee, Fei Xiao, Nachon Raethong, Yuhan Zhang, Kobkul Laoteng, Guang Hu, and Wanwipa Vongsangnak. 2020. "Probing Carbon Utilization of Cordyceps militaris by Sugar Transportome and Protein Structural Analysis" Cells 9, no. 2: 401. https://doi.org/10.3390/cells9020401