1. Introduction
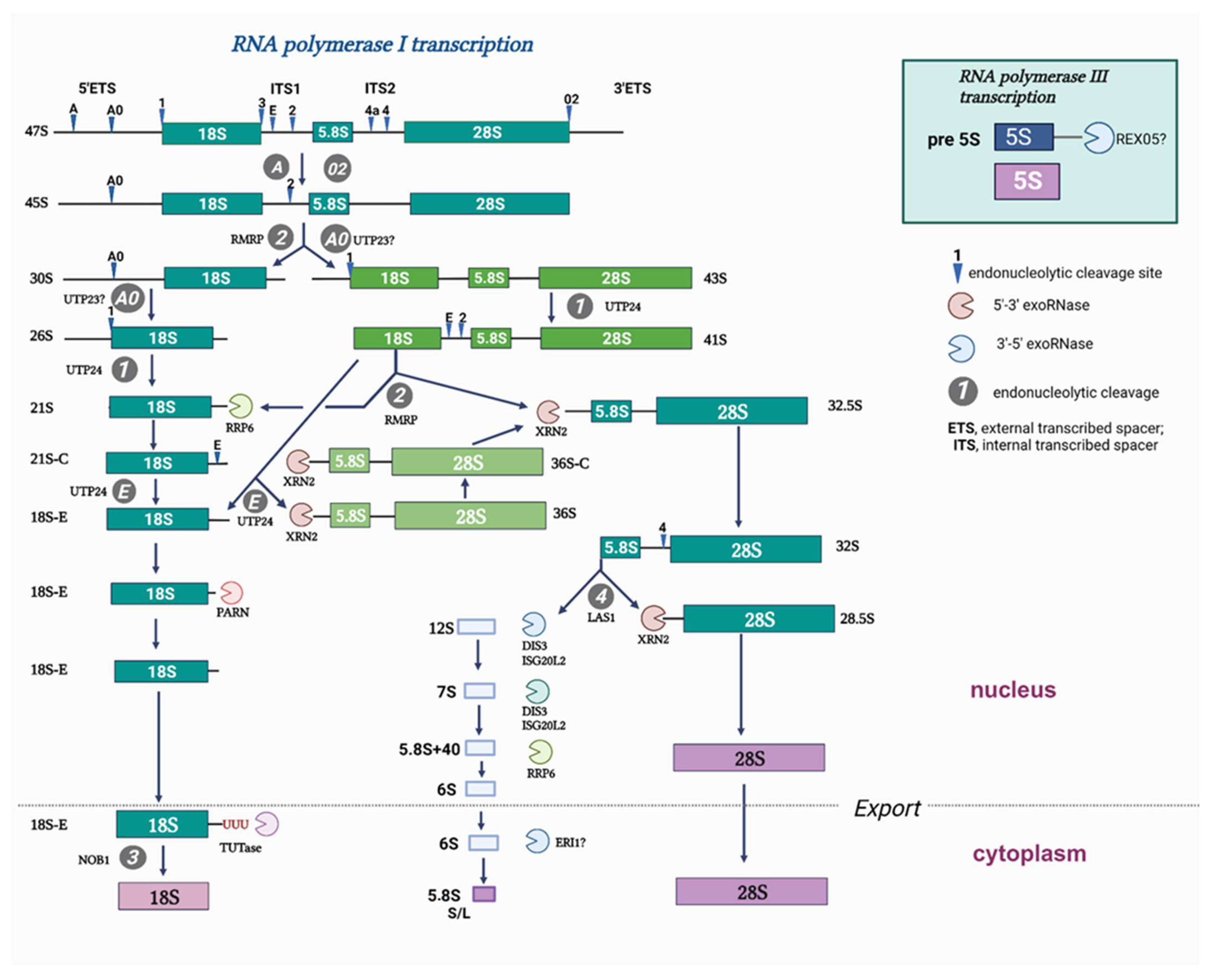
Ribosome biogenesis is a sophisticated time-ordered process, which adjusts the protein synthesis rate for the consumption of nutrients and external stimuli. It begins with the transcription of the ribosomal primary RNA precursor (35S/47S in
S. cerevisiae/H. sapiens). 47S pre-rRNA processing (
Figure 1) is coupled with the sequential recruitment of ribosome biogenesis factors and non-coding RNAs as well as the ordered coating of rRNA with ~80 ribosomal proteins during the formation of the functional 60S and 40S ribosomal subunits [
1]. Despite the considerable progress in understanding the ribosomal biogenesis in lower eukaryotes, in particular that in yeast
S. cerevisiae [
2,
3,
4,
5,
6,
7,
8], the complexity of the nucleolus and ribosomal precursors in higher eukaryotes hinders progress in obtaining structural and functional data on ribosome biogenesis [
9,
10,
11]. 47S pre-rRNA processing involves multiple human proteins that do not share sequence homology with
S. cerevisiae, thereby implying that the biogenesis of ribosomal subunits in humans is more complex and most likely involves steps that still remain poorly understood [
12].
Rpf1 and Esf1 are the nucleolar factors contributing to the biogenesis of 60S and 40S in
S. cerevisiae, respectively. Both have human homologs, which are RPF1 and ESF1, respectively [
13,
14]. A recent Cryo-EM study revealed the arrangement of RPF1 in the structure of human 60S subunit precursors. According to the structural data, RPF1 is present in the intermediates A1, B1, C1, and D1 and dissociates upon NVL2 recruiting and association [
12]. It is not clear whether it is required for the cleavage steps or provides a structure favoring the change in the pre-rRNA conformation that is mentioned above. Studying RPF1 in yeast uncovered both biochemical and structural details [
6,
8,
13]. RPF1-RNA co-immunoprecipitation experiments demonstrate its association with 35S, 27SA, and 27SB pre-ribosomal RNAs within the ITS1 and ITS2 sites. Rpf1 knockdown in yeast leads to the accumulation of the 27SA3 precursor and a decrease in the 7S precursor level, suggesting its possible interaction with the A3 site in the ITS1 and C2 sites in ITS2. Moreover, Rpf1 is thought to be a nucleation factor of the pre-66S complex in yeast [
13]. Less is known about the ESF1 protein. Esf1 knockdown in yeast leads to a dramatic decrease in 27SA2 and 20S pre-rRNAs, implying its role in the A2 site cleavage. Furthermore, the accumulation of the 35S and aberrant 23S pre-ribosomal RNAs has been observed, implicating the possible inhibition of A0 and A1 cleavage [
14]. Meanwhile, there are no biochemical or structural data concerning human ESF1 and its contribution to human ribosome biogenesis.
Here, we show that human RPF1 and ESF1 are directly involved in ribosomal subunits maturation. We evaluated the impact of RPF1 and ESF1 loss of function on their subcellular localization, pre-ribosomal RNA profiles, and association with the 60S and 40S precursors. We demonstrate that both RPF1 and ESF1 interact with the precursors of the large and small ribosomal subunits, respectively, and are thus involved in the early steps of their biogenesis.
2. Materials and Methods
2.1. DNA Constructs and Cloning
The shRNAs-coding sequences against RPF1 or ESF1 mRNAs or scramble control were selected from the Sigma MISSION® libraries and ordered as 21-mer top and bottom strands. Oligonucleotides were annealed and cloned into a pLKO.1-TRC vector (Addgene #10878) following the manufacturer’s protocol. The E. coli Stbl3 strain was used to prepare ultra-competent cells according to the Inoue method for routine cloning of pLKO.1-TRC-derived constructs to avoid any recombination events and increase the plasmid yield.
2.2. Cell Culture, Lentivirus Particles Preparation, Viral Transduction, and siRNA Transfection
The HEK293 cell line was obtained from the Russian bank of cell lines (Institute of Cytology RAS, Saint Petersburg, Russia). Cells were grown in DMEM (PanEco, Moscow, Russia) and supplemented with 10% HyClone fetal bovine serum (Cytiva, Marlborough, MA, USA), 2 mM of L-glutamine, and penicillin/streptomycin (250 units/mL, all from PanEco, Moscow, Russia) at 37 °C and 5% CO
2. To produce lentivirus particles, HEK293 cells were co-transfected with plasmids encoding shRNA, GAG/Pol, Rev, and VSV-G using Lipofectamine 2000 (ThermoFisher, Wilmington, DE, USA) according to the protocol (
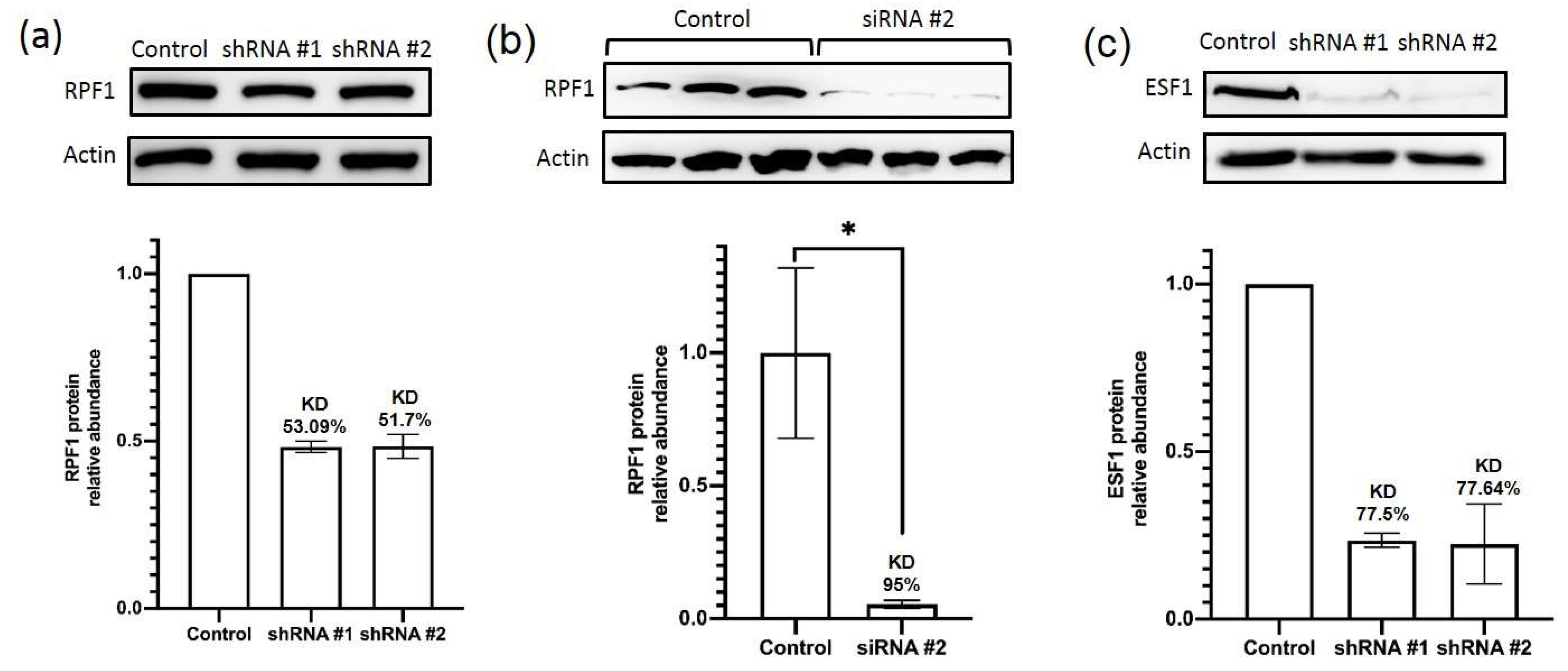
http://www.LentiGO-Vectors.de, accessed on 21 September 2022). The medium containing lentivirus particles was filtered through a 0.45 μm filter and used immediately or frozen and stored at −80 °C until use. Approximately 60% confluent wild-type HEK293 cells were transduced with the obtained lentivirus particles using Polybrene reagent added at a final concentration of 10 μg/mL (Miltenyi Biotec, Bergisch Gladbach, Germany). The pCDH-copGFP plasmid (10% of the total DNA amount) was added to a DNA mix to check the transduction efficiency. A total of 24 h after transduction, the medium was replaced with the fresh one without viral particles. Then, 3 days after transduction, puromycin was added to a final concentration of 10 μg/mL for cell selection. After selection, the knockdown efficiency was analyzed by RT-PCR and Western blotting.
Two siRNAs against RPF1 mRNA were designed and synthesized with additional modifications to improve their intracellular stability (GenTerra, Moscow, Russia). Approximately 70% confluent wild-type HEK293 cells were transfected by siRNAs individually or together at working concentrations of 5 nM and 15 nM according to Lipofectamine RNAiMAX (ThermoFisher, Wilmington, DE, USA) protocol. mRNA and protein level analysis as well as Northern blots were performed 3 days after transfection.
2.3. RT-PCR
Total RNA was isolated from the cells stably expressing shRNAs against RPF1 or ESF1 mRNAs or scramble control shRNA using TRIzol (ThermoFisher, Wilmington, DE, USA). RNA quality and concentration were assessed using the NanoDrop OneC spectrophotometer (ThermoFisher, Wilmington, DE, USA) at 260/280 nm wavelengths. cDNAs were prepared from 1 μg of RNA from each sample using SuperScript IV RT (ThermoFisher, Wilmington, DE, USA). The obtained cDNAs were diluted 10-fold and subjected to RT-PCR with primers to RPF1, ESF1, and GAPDH sequences spanning exon–exon junctions using 5X qPCRmix-HS SYBR (Evrogen, Moscow, Russia) according to the vendor’s protocol.
To prepare cDNA from ethynyl uridine-labeled RNA (see the corresponding sections in Materials and Methods), 50 μL of streptavidin magnetic beads with the bound biotin-RNA were incubated with a primer annealing solution (contains mix of primers, RNAse inhibitor, RNAse-free water) at 70 °C for 3 min. Then, the reverse transcriptase (RNAscribe RT, Biolabmix, Moscow, Russia) diluted in a reaction buffer was added, and the final mix was incubated following the manufacturer’s protocol. A total of 2.5 μL of the obtained cDNA were used for further RT-PCR with primers to 5′ETS or GAPDH. The primer sequences were as follows: 5′ETS-forward CGCGGGCCTGCTGTTCTCTC, 5′ETS-reverse CCCCGGGTGGGTCAGAGACC, GAPDH-forward GAAGGTGAAGGTCGGAGTCA and GAPDH-reverse TTGAGGTCAATGAAGGGGTC.
2.4. SDS-PAGE and Western Blotting
A total of 1 × 106 HEK293-derived modified cells were lysed on ice in 200 μL of the lysis buffer (50 mM of Tris-HCl, 150 mM of NaCl, 10% glycerol, 1% Triton X-100, 1 mM of PMSF, 1 mM of DTT, at pH 7.5). Protein concentration in lysates was determined by Bradford reaction according to the standard protocol. Lysate samples or proteins extracted from sucrose were diluted with 5x Laemmli buffer and subjected to SDS-PAGE (25 μg per lane) in 12% gel prior to transfer to the nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA, USA). The membrane was blocked in 5% non-fat dry milk in TBST (20 mM of Tris-HCl, 150 mM of NaCl, 0.1% Tween 20, at pH 7.6) for 1 h at room temperature and incubated with rabbit anti-RPF1 (HPA024642, Sigma, Suffolk, UK, 1:1000 dilution), anti-ESF1 (SAB1411774, Sigma, Suffolk, UK, 1:1000 dilution), or anti-actin (A4700, Sigma, Suffolk, UK, 1:1000 dilution) primary antibodies in TBST supplied with 5% non-fat dry milk for 1 h at room temperature. After three 5 min washes in TBST, the membrane was incubated with the secondary anti-rabbit HRP-conjugated IgG (Sigma, Suffolk, UK, 1:20,000 in TBST supplied with 5% non-fat dry milk) for 1 h at room temperature. The membrane was then washed three times in TBST and incubated with ECL reagents (Cytiva, Marlborough, MA, USA) according to the vendor’s instruction and visualized using ChemiDoc imager (Bio-Rad Laboratories, Hercules, CA, USA). Densitometry analysis was performed with ImageLab 5.0 software.
2.5. Northern Blotting
Total RNA from HEK293-derived stable cell lines was isolated using TRIzol (ThermoFisher, Wilmington, DE, USA) following the standard protocol. RNA quality and concentration were measured using the NanoDrop OneC spectrophotometer (ThermoFisher, Wilmington, DE, USA) at 260/280 nm wavelengths. RNA was separated by gel electrophoresis in a 1.2% agarose gel containing 1.8 M of formaldehyde in 20 mM of MOPS/4 mM of NaAc/ 0.5 mM of EDTA (pH 7.0) used as a buffer. A total of 3 μg of total RNA were loaded to each lane and run at 2V/cm for 6 h. RNA was transferred to the BrightStar®-Plus positively charged nylon membrane (ThermoFisher, Wilmington, DE, USA) for 2 h according to the alkali transfer protocol. A total of 1 M of NaCl/10 mM of NaOH was used as a transfer buffer. The membrane was rinsed in 2x SSC buffer (0.3 M of NaCl, 30 mM of sodium citrate, at pH 7.0) and baked at 80 °C for 15 min in an oven. Hybridization with the biotin-labeled probes (10 ng/mL final concentration) was performed in the ULTRAhyb®-Oligo buffer (ThermoFisher, Wilmington, DE, USA) according to the manufacturer’s instructions at 42 °C for 12–16 h. The probe sequences were as follows: 5′ITS2-biotin–GGGGCGATTGATCGGCAAGCGACGCTC, 5′ITS1-biotin–GGCCTCGCCCTCCGGGCTCCG, 7SK-biotin-GACGCACATGGAGCGGTGAGGGAG, 28S-biotin-CCTCTTCGGGGGACGCGCGCGTGGCCCCGA and 18S-biotin-ATCGGCCCGAGGTTACTCTAGAGTCACCAAA. After hybridization, the membrane was washed twice with 2× SSC buffer supplemented with 0.5% SDS for 30 min at 42 °C. Bands were visualized with the Chemiluminescent Nucleic Acid Detection Module (ThermoFisher, Wilmington, DE, USA) at room temperature according to the manufacturer’s protocol using the x-ray film or ChemiDoc Imaging System (Bio-Rad Laboratories, Hercules, CA, USA). Densitometry analysis was performed by ImageLab 5.0 software.
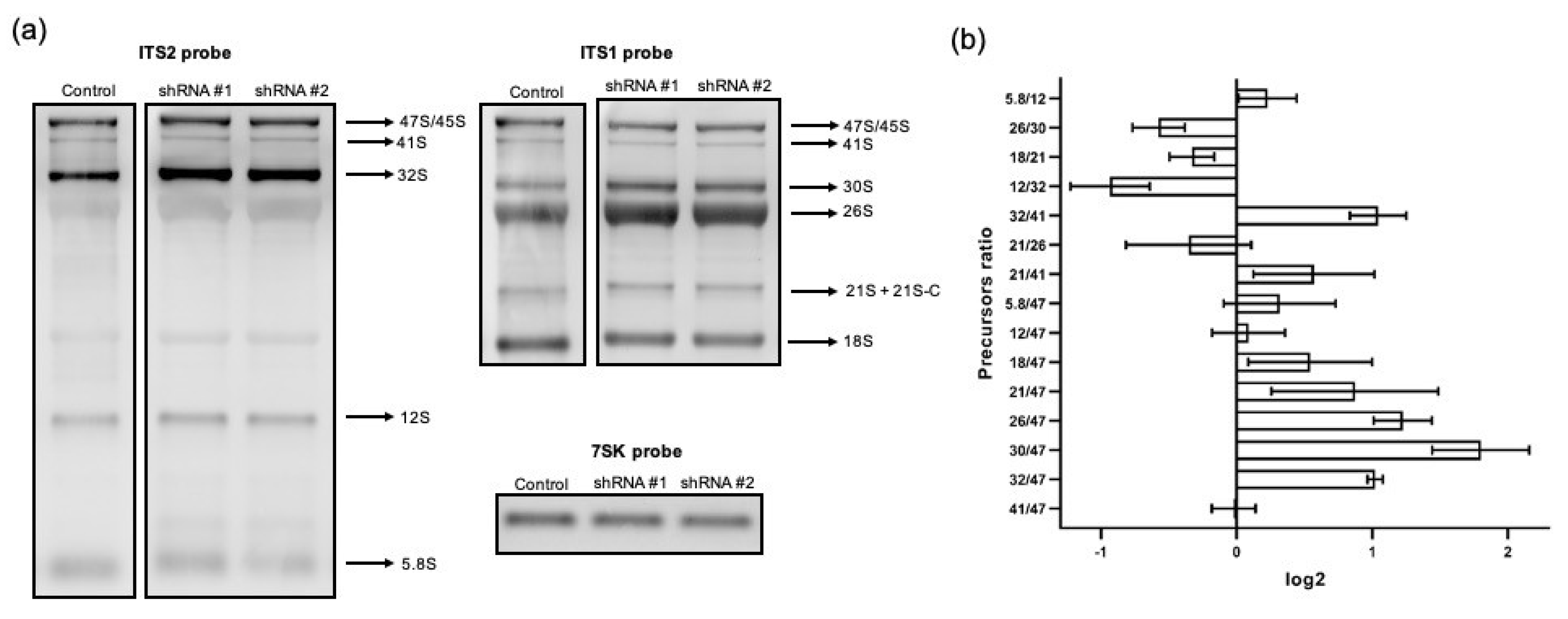
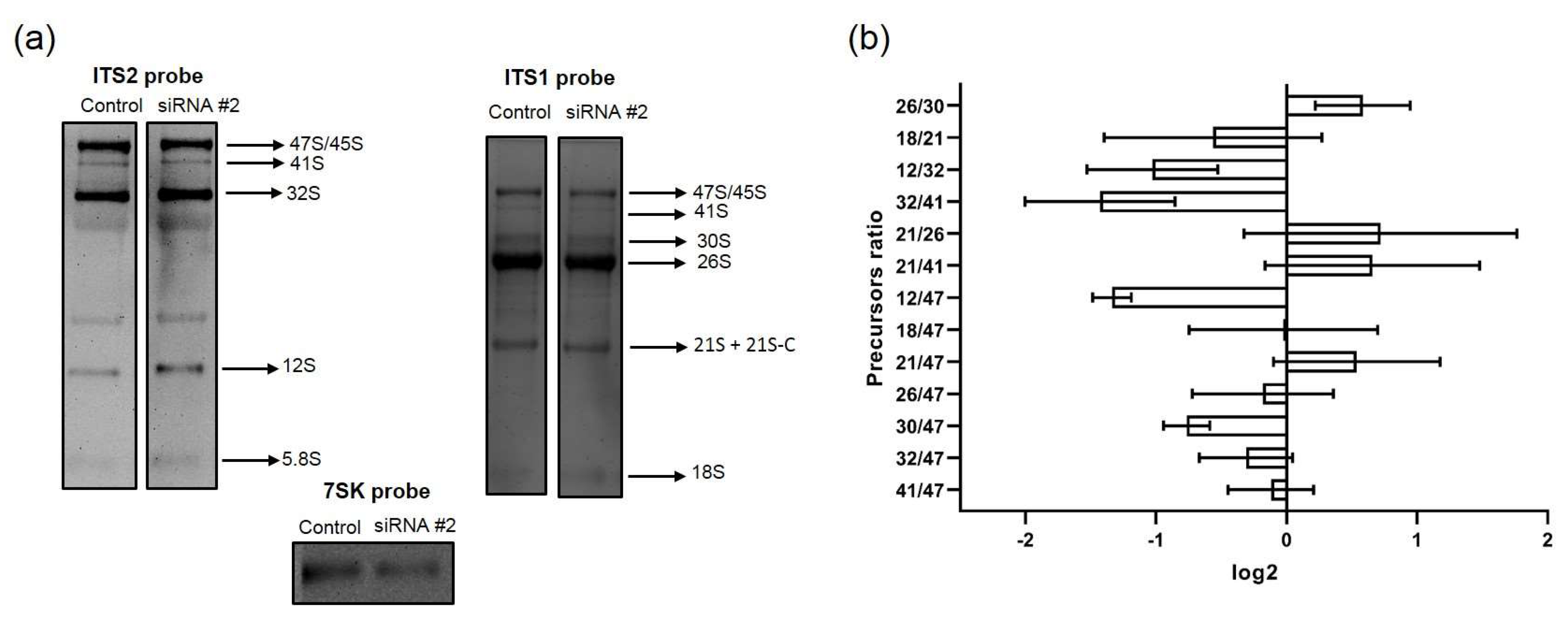
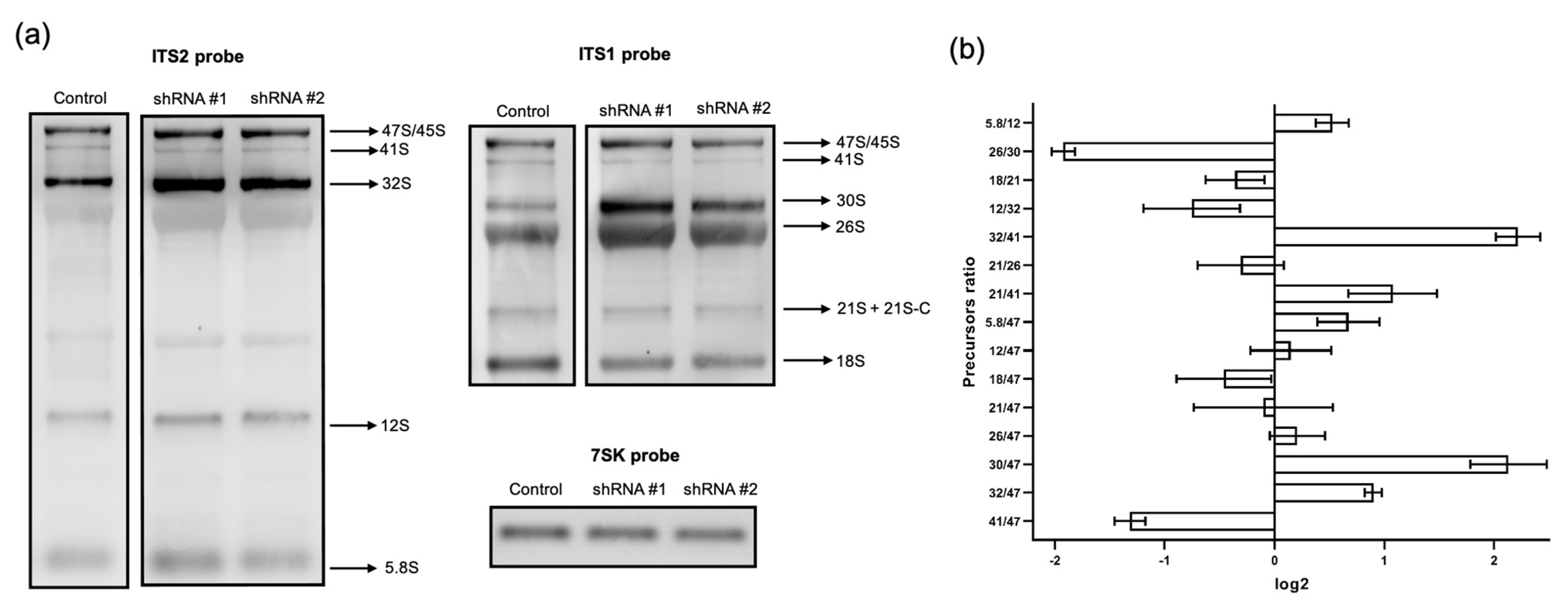
2.6. Analysis of rRNA Precursor Ratios
RAMP (Ratio Analysis of Multiple Precursors) profiles were generated according to [
15]; the density of rRNA precursor bands was determined as previously described. The ratio of precursors was calculated for each total RNA sample (pre-rRNA 41S/45-47S, 12S/32S, etc.) and was obtained from analyzing each individual lane following the hybridization with one probe. The log2 values of precursor ratios in the control samples were subtracted from log2 values of corresponding ratios in the samples from cells with either RPF1 or ESF1 knockdown. Resulting normalized log2 values were plotted on histograms and combined two different ratios into one RAMP profile. The first one represented the ratio of the corresponding pre-rRNA to the primary rRNA transcript, and the second represented the ratio between pre-rRNAs that formed substrate–product pairs, for example, 18SE/21S and 12S/32S.
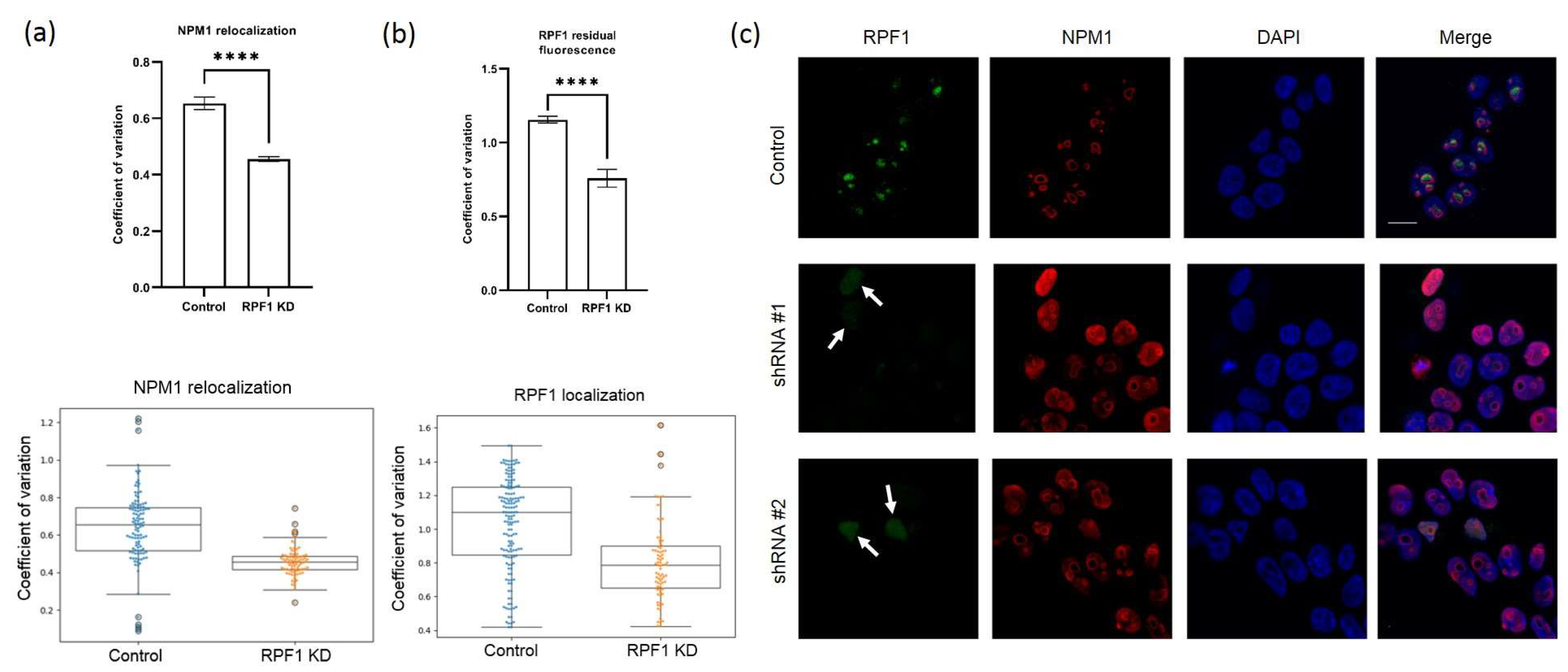
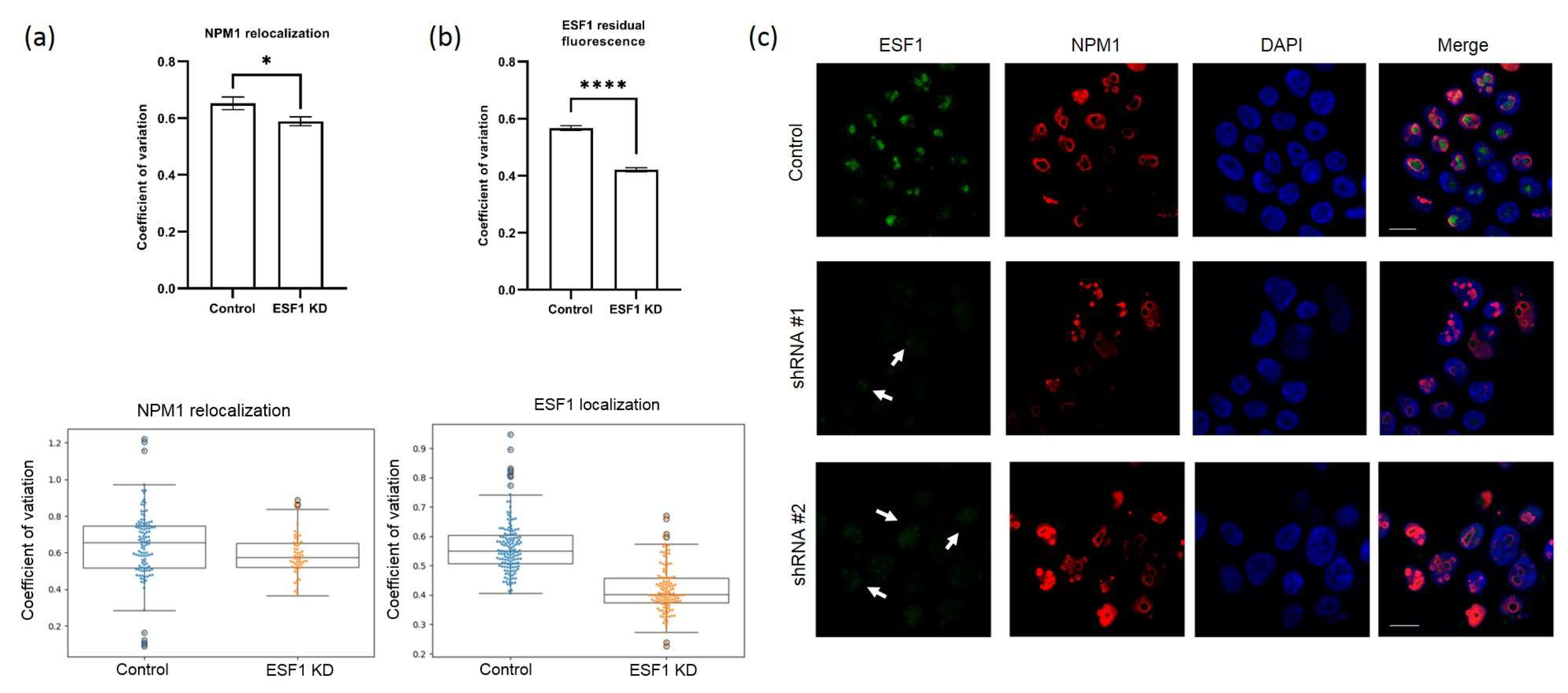
2.7. Immunocytochemistry
HEK293-derived stable cells were grown on cover slides. Cells were fixed in absolute acetone at −20 °C for 10 min and were incubated with a mixture of two antibodies as follows: rabbit anti-RPF1/anti-ESF1 (1:100 dilution) and either mouse anti-B23/nucleophosmin or mouse anti-SURF6 (1:200 dilution, Sigma, Suffolk, UK, B0556) for 1 h at room temperature. Cells were washed in PBS for 3 × 5 min and stained for 1 h at room temperature with a mixture of AlexaFluor 488 goat anti-mouse IgG (HCL) antibodies (ThermoFisher, Wilmington, DE, USA, A-11034, for detection of B23) and Alexa Fluor® 568 goat anti-rabbit IgG (H+L, ThermoFisher, Wilmington, DE, USA, A11004, for detection of RPF1/ESF1). To study co-localization with SURF6, cells were stained with a mixture of rabbit anti-RPF1/ESF1 and mouse anti-SURF6 antibodies, washed in PBS for 3x5 min, and incubated for 1 h at room temperature with a mixture of goat anti-mouse and goat anti-rabbit antibodies, as described above.
After incubation with secondary antibodies, the cells were washed with PBS for 3 × 5 min, stained with DNA-binding dye DAPI (1 mg/mL in PBS, 10 min) and fixed in Vectashield (Vector Laboratories, Newark, CA, USA). The preparations were analyzed using a DuoScan-Meta LSM510 confocal laser scanning microscope (Carl Zeiss Meditec AG, Jena, Germany) equipped with a Plan-Apochromat 63 × 1.40 (numerical aperture) oil Ph3 objective.
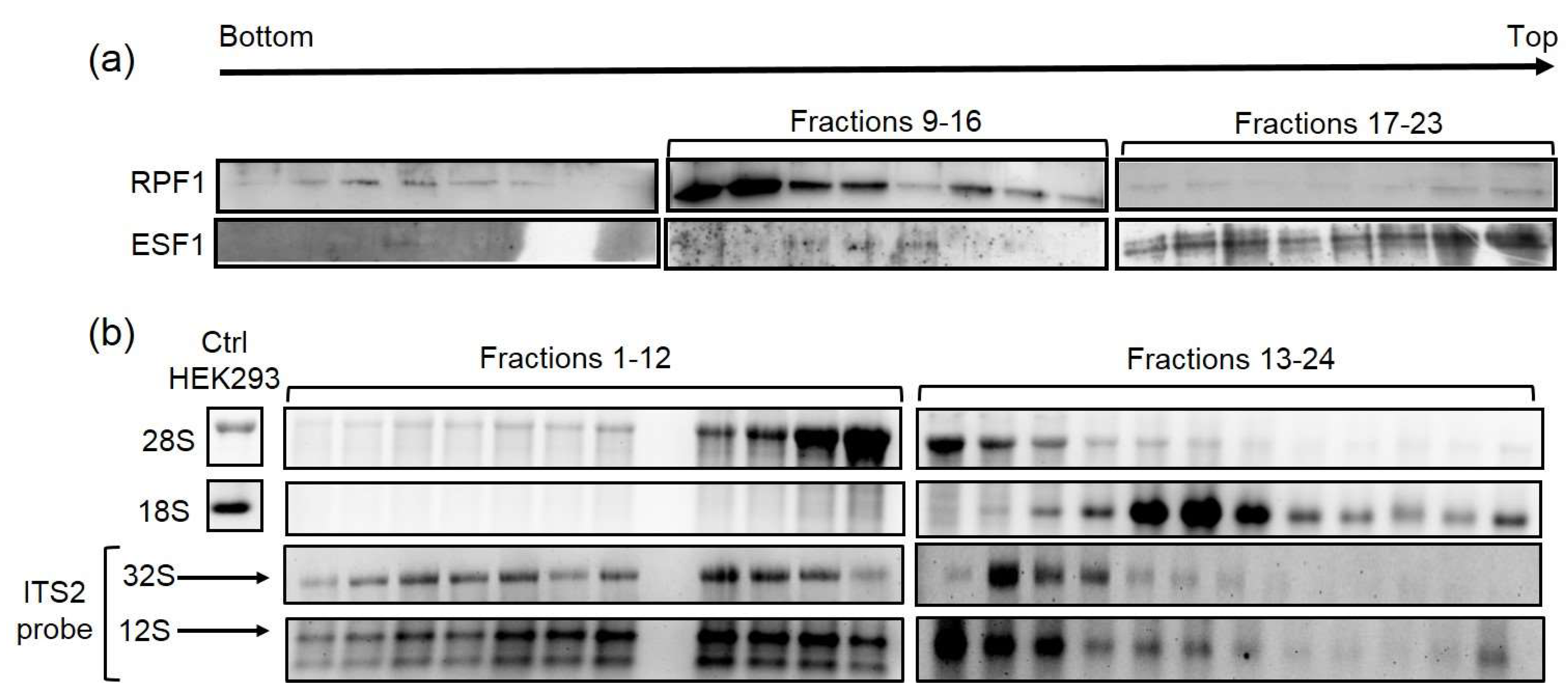
2.8. Sucrose Gradient
Ten T-75 flasks of wild-type HEK293 cells were cultured under standard conditions, harvested at a sub confluent density, frozen in liquid nitrogen, and stored at −80 °C until use. The cell pellet was thawed on ice and underwent nucleus extraction according to the protocol [
16]. Purified nuclei were treated with SN2 buffer supplemented with DNAse I to disrupt the nuclear membrane and destabilize the nucleolus following the PSE method [
17]. Lysates were centrifuged (12,300 ×
g, 10 min, 4 °C), and supernatants were loaded on linear 15–40% sucrose gradient containing 20 mM of HEPES-NaOH (pH 7.5), 200 mM of NaCl, 4 mM of EDTA, 0.1% Igepal CA-630, 0.1 mg/mL of heparin, and 1 mM of DTT. Ultracentrifugation was performed with a SW40Ti rotor (Beckman Coulter, Brea, CA, USA) at 36,000 × rpm for 6 h at 4 °C. Fractions (0.5 mL) were collected on a gradient collector system (Bio-Rad Laboratories, Hercules, CA, USA) and subsequently subjected to Western and Northern blot analyses. Samples were extracted through the phenol:chloroform:isoamyl alcohol mix. Aqueous phases were collected for RNA extraction, while organic phases were used for protein precipitation. Northern and Western blots were performed according to the corresponding section in Materials and Methods.
2.9. Polysome Profiling
Three T-75 flasks of each cell line with RPF1/ESF1 knockdown as well as control cells were cultured to obtain approximately 70–80% confluence. Cycloheximide was added to each flask to obtain the final concentration of 100 μg/mL, and cells were incubated under normal conditions for 30 min. After that, flasks were removed from the CO2 incubator and immediately placed on ice. Culturing media were completely aspirated, and cells were rinsed with 10 mL of ice-cold PBS+cycloheximide (100 μg/mL). A total of 500 μL of the polysome lysis buffer (20 mM of Tris-HCl, 250 mM of NaCl, 15 mM of MgCl2, 1 mM of DTT, 0.5% Triton X-100, cycloheximide at 100 μg/mL, 20 U/mL of DNAse I, RNAse inhibitor at 1 U/μL, at pH 7.5) were added to each flask and cells were scraped. The obtained lysates were transferred to 2 mL tubes, incubated for 20 min on ice, and centrifuged for 10 min at a maximum speed of 4 °C. Their concentration was measured, and 15 OD (approximately 350 μL) of each sample were loaded on a linear 10–60% sucrose gradient (the sucrose gradient buffer contains 20 mM of Tris-HCl, 250 mM of NaCl, 15 mM of MgCl2, at pH 7.5). Samples were centrifuged for 3 h at 35,000 rpm at 4 °C. After centrifugation, gradients were manually fractionated from the top to the bottom, and the optical density of each sample was measured at 260 nm using a NanoDrop OneC spectrophotometer (ThermoFisher, Wilmington, DE, USA).
2.10. Ethynyl Uridine (EU) Pulse Labeling
Stable cell lines with shRNA #1/#2 against
RPF1 mRNA as well as control cells were grown to 70% confluence in 6-well plates. EU (Jena Bioscience, Jena, Germany) was added to the final concentration of 0.5 mM, and cells were incubated for 1 h. Total RNA was extracted using the TRIzol reagent (ThermoFisher, Wilmington, DE, USA). A total of 5 μg of RNA were incubated in the click reaction buffer (final volume of 50 μL; it contains biotin-azide (PEG4 carboxamide-6-Azidohexanyl Biotin, ThermoFisher, Wilmington, DE, USA), CuSO
4, THPTA, sodium ascorbate, RNAse inhibitor 40 U, MOPS, EDTA, DEPC-treated water, at pH 7) at 25 °C, with 600 rpm in a thermo-shaker for 45 min. Then, 1 μL of RNAse-free glycogen (20 μg/μL, New England Biolabs, Ipswich, MA, USA, B1564S), 50 μL of 3 M of sodium acetate (pH 5.2), and 700 μL of cold 96% ethanol were added to the reaction mix. Samples were incubated overnight at −80 °C. RNA was pelleted by centrifugation at 13,000 ×
g for 20 min at 4 °C. The supernatants were removed, and RNA pellets were rinsed with 75% ethanol twice by centrifuging at 13,000×
g for 5 min at 4 °C. Pellets were air-dried for 5 min at room temperature and resuspended in 10 μL of RNAse-free water. RNA integrity was checked in formaldehyde agarose gel (see
Supplementary Figure S9).
2.11. Extraction of Biotinylated RNA Using Streptavidin Magnetic Beads
A total of 1 μg of biotinylated RNA isolated following EU labeling was used for binding to 50 μL of streptavidin magnetic beads (ThermoFisher, Wilmington, DE, USA, 88816) equilibrated with the BW buffer prior to incubation with RNA (bind and wash buffer, and the wash step was repeated 3 times; the buffer composition was as follows: 10 mM of Tris-HCl, 1mM of EDTA, 2 mM of NaCl, at pH 7.5). Then, RNA was mixed with 125 μL of 2× bind and wash buffer, 2 μL of RNAse inhibitor, and RNAse-free water to reach a final volume of 250 μL. The mix was heated at 68 °C for 5 min and added to 50 μL of pre-equilibrated streptavidin magnetic beads. The suspension was incubated at 25 °C in a thermo-shaker for 45 min at 900 rpm. Then, the beads were sequentially washed 5 times with the 10× bead volume of the bind and wash buffer and 5 times with the 10× beads volume of the low-salt buffer (0.1 M of NaCl). Finally, the beads were resuspended in 50 μL of the low-salt buffer for cDNA synthesis.
2.12. Image Processing and Quantification
NPM1, RPF1, or SURF6 signals were quantified using a custom Python 3 script. Images were preprocessed with ImageJ to convert the DAPI and NPM1 channels into separate 16-bit grayscale images. A minimum of 56 cells were analyzed for each treatment group. Nuclei segmentation was determined in the DAPI images using Li thresholding functions in the Scikit-Image Python package. The CV for individual nuclei, defined as a standard deviation of pixel intensity divided by mean pixel intensity, was calculated from the NPM1 images using the SciPy Python package. All data were normalized to the scramble control in each experiment. Data are represented as box plots with whiskers generated by the Matplotlib and Seaborn Python packages.
2.13. AlamarBlue Assay
The stable cell lines were seeded in 96-well plates at 50% confluence. A total of 10% of the AlamarBlue reagent was added according to the manufacturer’s protocol (ThermoFisher, Wilmington, DE, USA) to a culture medium, and cells were incubated for 4 h at 37 °C, 5% CO2. Absorbance was measured at 570 nm using Thermo Scientific Multiskan EX ELISA reader (ThermoFisher, Wilmington, DE, USA). Experiments were performed in triplicates.
2.14. Statistical Analysis
Results are expressed as mean ± standard error of mean (SEM). Microsoft Excel and GraphPad Prism 9.0 software were used for statistical analysis of data.
4. Discussion
Ribosome biogenesis is a well-orchestrated process involving myriads of assembly factors and non-coding RNAs that modulate the pre-rRNA processing. Progressing along the pathway is tightly regulated by the extra- and intracellular conditions to maintain an optimal protein synthesis routine.
The formation of the mature 28S ribosomal RNA during ribosome biogenesis in higher eukaryotes requires two major events. First, the RNAse MRP mediates the cleavage of the 41S or 45S pre-rRNA at site 2 (corresponding to the A3 site in
S. cerevisiae) in the 21S and 32S pre-rRNA intermediates in pathways 1 and 2, respectively [
25]. LAS1 endonuclease in the Rixosome complex (NOL9, WDR18, LAS1L, MDN1, PELP1, TEX10, and SENP3) performs the second cleavage at site 4 (corresponding to site C2 in
S. cerevisiae [
19]). This promotes the formation of the 12S and 28S pre-rRNA intermediates in both of the biogenesis pathways leading to NOG2-NSA2 dissociation and PET maturation [
26,
27]. The formation of a favorable structural environment for rRNA domain I precedes the cleavage event at this site [
28]. The early nucleolar SSF1 particle interacts with the so-called WDR74-module (WDR74-NOP52-RPF1-MAK16, corresponding to the Nsa1-module in
S. cerevisiae) [
29]. This complex clamps down domains I and II, which mark out the future PET site, and forms the intermediate NSA1 nucleolar precursor. During the subsequent maturation steps, AAA-ATPase NLV2 (corresponding to Rix7 in
S. cerevisiae) is recruited by promoting WDR74-module release, which is coupled with or precedes the dissociation of the SSF1-RRP15-SURF6 complex [
30]. These events lead to the formation of the PET rim and vestibule [
12,
22,
23,
24,
25,
26,
27,
28]. This is followed by the association of the 60S precursor with the forming factors of domain III and the subsequent recruitment of AAA-ATPase MDN1 (corresponding to Rea1 in
S. cerevisiae), which stimulates the detachment of BOP1-WDR12-DDX18 (corresponding to Erb1, Ytm1, and Has1 in
S. cerevisiae, respectively) and related factors [
31]. These processes are coupled with the incorporation of the NOG2 and 5S RNP complex and the formation of the late nucleolar 60S precursor [
12,
28,
32,
33]. The NOG2 particle interacts with the Rixosome complex, which possesses nuclease activity; it mediates ITS2 cleavage, PET, and central protuberance formation after its dissociation [
12,
27,
28].
How do our functional and biochemical data on RPF1 fit into this complex picture? Although RPF1 depletion downmodulated site 4 cleavage, the effect of RPF1 knockdown was indirect. This idea is supported by the fact that the Rixosome complex did not contain RPF1. Cryo-EM data suggest that RPF1 is no longer associated with the 60S precursor at the time of site 4 cleavage. It is likely that the structural support of domains I and II provided by the WRD74 module was not sufficient due to PRF1 absence in the complexes formed prior to site 4 cleavage. We suggest that the lack of RPF1 might negatively affect the formation of Rixosome- or LAS1-binding sites and results in the lowered efficiency of 32S pre-rRNA cleavage at site 4 within the ITS2 region. Nevertheless, this defect in ITS2 grounding and cleavage is insufficient for the significant disruption of the ribosome biogenesis and cell proliferation rate, which is supported by the results of AlamarBlue assay (
Supplementary Figure S6a). The same results have been observed for
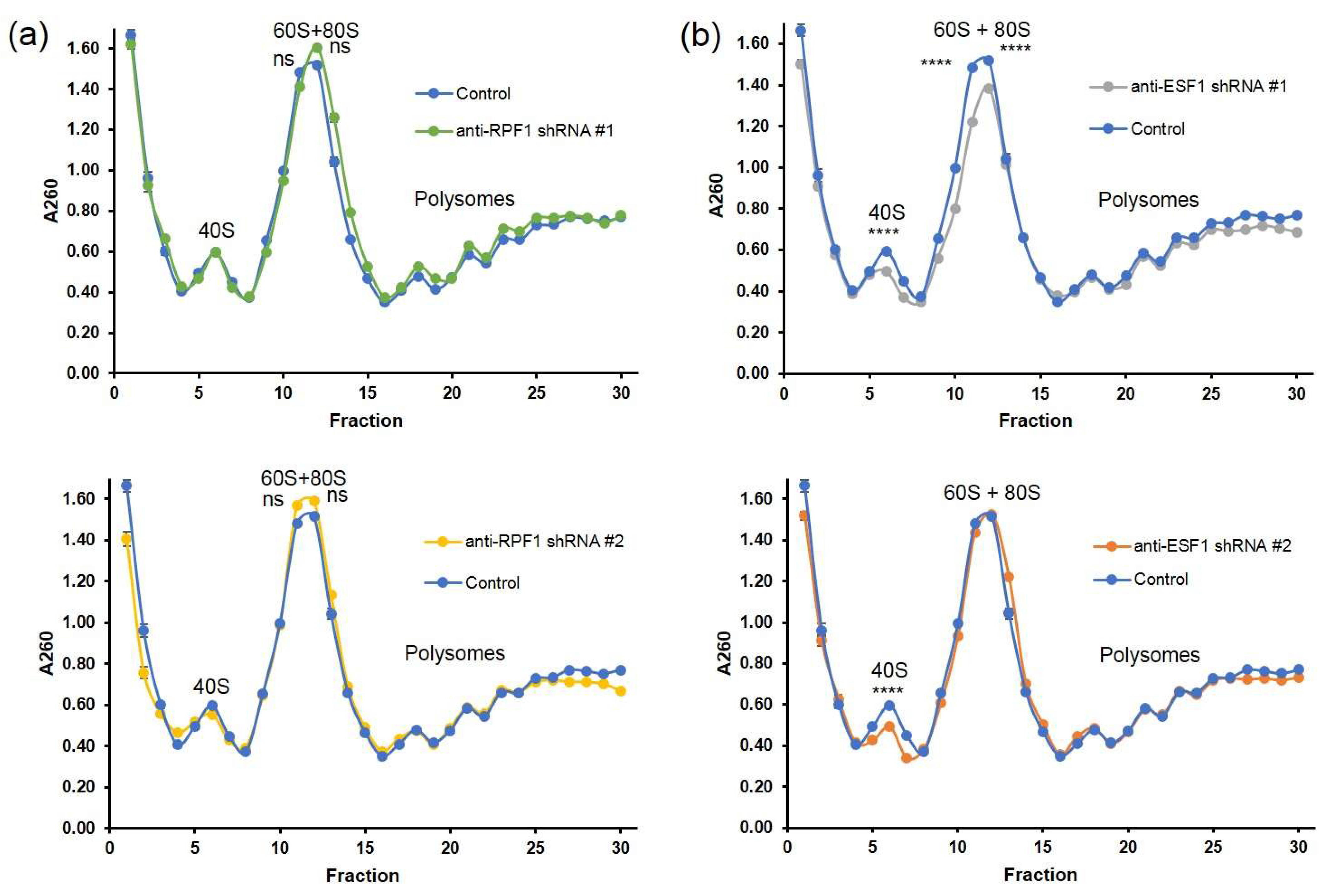
S. cerevisiae. Pre-60S particles can bypass the nuclear quality control mechanism, enter the following maturation steps in the cytoplasm, and interact with the mature 40S subunit. Indeed, these aberrant 80S ribosomes can be found in the polysome fraction. These observations are supported by our polysome profiling data that show that 60S-80S and polysome fractions are not depleted in RPF1-deficient cells. However, the mechanisms of the quality control evasion of the ITS2-containing particles and the translation properties of these abnormal ribosomes still remain elusive [
34].
Of note, 5′ETS augmentation in cells with shRNA-induced RPF1 knockdown suggests either an increase in the Pol I transcription rate or inhibited 5′ETS cleavage. According to the published data, NPM1 absence in the nucleolus may lead to a decrease in H3K9 methylation at the promoters of the rDNA genes [
21]. We observed the disruption of the 41S–32S–21S axis in the stable cell lines with RPF1 knockdown. An elevated level of the 5′ETS precursor also indicated that RPF1 might contribute to the early steps of ribosome biogenesis, and its absence resulted in the deregulation of A0 processing. This assumption is in line with the data obtained from yeast, which revealed that RPF1 co-immunoprecipitated with an early 35S rRNA-containing precursor. Eventually, an elevated 5′ETS level might be caused by either rDNA promoter demethylation or decreased 5′ETS processing as well as both these factors.
18S rRNA maturation in humans requires more advanced re-modeling and processing steps in the nucleoplasm compared with the 18S formation in yeast [
28,
35,
36]. The human 47S primary transcript contains a 5′ETS region that is six times longer [
37]. The early stages of human pre-ribosomal RNA processing appear to begin with the association of a nascent transcript with UTP-A, UTP-B, and U3 snoRNA (similar to yeast) to stabilize the 5′ETS segment, form the 5′ETS particle, and provide the favorable structural conditions for the docking of the other assembly factors [
28,
38]. The modular assembly of the 5′-, central, 3′-major, and 3′-minor domains of the 18S rRNA segment starts only when it becomes accessible. The proper domain organization and association with the assembled 5′ETS complex leads to cleavage at the A0 site by the endonuclease UTP24 both in yeast and in humans. Subsequent complex re-modeling is accomplished by the association of new assembly factors and SSU processome formation [
5,
28]. The simplicity of yeast nucleolar organization allowed for isolating and describing the stepwise domain formation by Cryo-EM. These studies resolved a series of 5′ETS structures in different states [
2,
3,
4,
5,
7]. The 5′ETS formation in humans remains a mystery due to a lack of structural data [
39]. The ESF1 protein and its yeast homolog Esf1 are the earliest assembly factors. They interact with the 5′ domain of the 18S rRNA segment during SSU processome formation according to the biochemical data [
5,
7]. Despite significant progress in the structural characterization of pre-SSU, 3D models of yeast particles do not provide any information regarding the 5′ domain of 18S with the attached Esf1 protein and its partners. Earlier, it was shown that while each 18S rRNA domain predominantly grows as an independent module, the presence of these domains in favorable spatial organization leads to A0 site cleavage. This is accompanied through the almost immediate dissociation of the earliest factors and the compaction of the precursor into SSU [
5]. According to our data, ESF1 knockdown resulted in the 41S/47S and 26S/30S ratio decline, thereby strongly demonstrating a reduced efficiency in the A0 site cleavage. Reduced A0 site processing resulted in an elevation in the 32S/47S and 30S/47S ratios. This finding may serve as solid evidence for pathway 2 activation, which starts with site 2 cleavage instead of that of A0. The 26S/30S ratio was drastically reduced; however, the net level of matured 18S did not significantly change. This implies that the A0 site cleavage is most likely skipped during rRNA maturation along pathway 2.
Together with our previous data [
18], these results demonstrate that human cells utilize ribosome biogenesis pathways extremely effectively to maintain a bulk protein synthesis level. Knockdown of the pre-ribosome assembly factors, especially those that exert accessory functions and are not directly involved in cleavage events, did not drastically drop the rate of ribosomal subunit maturation but rather fine-tuned the maturation events and the re-direction of the pre-rRNA processing paths. These assumptions are supported by our polysome profiling data. Finally, it should be noted that for our study, we utilized transformed cells that likely possess altered control mechanisms, which might result in increased plasticity and the relaxed control of ribosome biogenesis.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








