

The Inflammasome-Dependent Dysfunction and Death of Retinal Ganglion Cells after Repetitive Intraocular Pressure Spikes

 , ,
, ,
Abstract
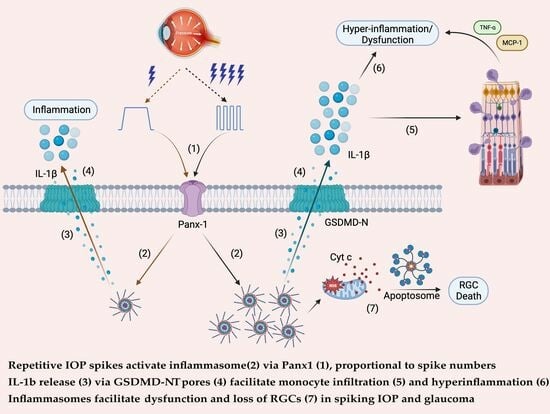
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Reagents
2.3. The Spiking OHT Injury Model
2.4. The Y437H-MYOC-Induced OHT Model
2.5. The Retinal Ischemia–Reperfusion Model
2.6. Intravitreal Cytokine Activity Assay
2.7. In Vivo Retinal Electrophysiology and Data Analysis
2.8. RGC Loss Assessment
2.9. Inflammasome Detection
2.10. Real-Time PCR
2.11. Immunohistochemistry
2.12. Statistical Analysis
3. Results
3.1. Acute Inflammasome Induction after spOHT
3.2. SpOHT Induce Functional and Structural Damage to RGCs
3.3. Inflammasome Activity Coordinates Retinal Neuroinflammation
3.4. Panx1 Is the Mechanosensitive Regulator of the Inflammasome
3.5. Inactivation of the Inflammasome Protects RGCs
3.6. Inner Retina Cell Types That Activate Inflammasome
3.7. Inflammasome Activation in Glaucoma
4. Discussion
4.1. Acute and Progressive Inflammasome Activation in the spOHT Model
4.2. Mechanosensor Signaling Drives Inflammasome Activation
4.3. The Neurotoxic Pathways Downstream of the Inflammasome
4.4. Pyroptosis vs. Apoptosis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Osborne, N.N.; Ugarte, M.; Chao, M.; Chidlow, G.; Bae, J.H.; Wood, J.P.; Nash, M.S. Neuroprotection in relation to retinal ischemia and relevance to glaucoma. Surv. Ophthalmol. 1999, 43 (Suppl. S1), S102–S128. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N. Mitochondria: Their role in ganglion cell death and survival in primary open-angle glaucoma. Exp. Eye Res. 2010, 90, 750–757. [Google Scholar] [CrossRef]
- Sappington, R.M.; Sidorova, T.; Long, D.J.; Calkins, D.J. TRPV1: Contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Investig. Ophthalmol. Vis. Sci. 2009, 50, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Pease, M.E.; McKinnon, S.J.; Quigley, H.A.; Kerrigan-Baumrind, L.A.; Zack, D.J. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2000, 41, 764–774. [Google Scholar]
- Calkins, D.J. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog. Retin. Eye Res. 2012, 31, 702–719. [Google Scholar] [CrossRef]
- Caprioli, J.; Coleman, A.L. Intraocular pressure fluctuation a risk factor for visual field progression at low intraocular pressures in the advanced glaucoma intervention study. Ophthalmology 2008, 115, 1123–1129.e3. [Google Scholar] [CrossRef] [PubMed]
- Asrani, S.; Zeimer, R.; Wilensky, J.; Gieser, D.; Vitale, S.; Lindenmuth, K. Large diurnal fluctuations in intraocular pressure are an independent risk factor in patients with glaucoma. J. Glaucoma 2000, 9, 134–142. [Google Scholar] [CrossRef]
- McMonnies, C.W. Intraocular pressure spikes in keratectasia, axial myopia, and glaucoma. Optom. Vis. Sci. 2008, 85, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Savastano, A.; Savastano, M.C.; Carlomusto, L.; Savastano, S. Bilateral Glaucomatous Optic Neuropathy Caused by Eye Rubbing. Case Rep. Ophthalmol. 2015, 6, 279–283. [Google Scholar] [CrossRef]
- Baskaran, M.; Raman, K.; Ramani, K.K.; Roy, J.; Vijaya, L.; Badrinath, S.S. Intraocular pressure changes and ocular biometry during Sirsasana (headstand posture) in yoga practitioners. Ophthalmology 2006, 113, 1327–1332. [Google Scholar] [CrossRef]
- Jasien, J.V.; Jonas, J.B.; de Moraes, C.G.; Ritch, R. Intraocular Pressure Rise in Subjects with and without Glaucoma during Four Common Yoga Positions. PLoS ONE 2015, 10, e0144505. [Google Scholar] [CrossRef]
- Schuman, J.S.; Massicotte, E.C.; Connolly, S.; Hertzmark, E.; Mukherji, B.; Kunen, M.Z. Increased intraocular pressure and visual field defects in high resistance wind instrument players. Ophthalmology 2000, 107, 127–133. [Google Scholar] [CrossRef]
- Krist, D.; Cursiefen, C.; Junemann, A. Transitory intrathoracic and -abdominal pressure elevation in the history of 64 patients with normal pressure glaucoma. Klin. Monatsblätter Augenheilkd. 2001, 218, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Zhang, X.; Wang, N.; Li, B.; Tian, G.; Jonas, J.B. Cerebrospinal fluid pressure in ocular hypertension. Acta Ophthalmol. 2011, 89, e142–e148. [Google Scholar] [CrossRef] [PubMed]
- Barash, A.; Chui, T.Y.P.; Garcia, P.; Rosen, R.B. Acute Macular and Peripapillary Angiographic Changes with Intravitreal Injections. Retina 2020, 40, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Huang, B.B.; Ong, J.X.; Konopek, N.; Fawzi, A.A. Hemodynamic Effects of Anti-Vascular Endothelial Growth Factor Injections on Optical Coherence Tomography Angiography in Diabetic Macular Edema Eyes. Transl. Vis. Sci. Technol. 2022, 11, 5. [Google Scholar] [CrossRef]
- Soheilian, M.; Karimi, S.; Montahae, T.; Nikkhah, H.; Mosavi, S.A. Effects of intravitreal injection of bevacizumab with or without anterior chamber paracentesis on intraocular pressure and peripapillary retinal nerve fiber layer thickness: A prospective study. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 1705–1712. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Swaminathan, S.S.; Yang, J.; Barikian, A.; Shi, Y.; Shen, M.; Jiang, X.; Feuer, W.; Gregori, G.; Rosenfeld, P.J. Dose-Response Relationship between Intravitreal Injections and Retinal Nerve Fiber Layer Thinning in Age-Related Macular Degeneration. Ophthalmol. Retina 2021, 5, 648–654. [Google Scholar] [CrossRef]
- Pecora, L.; Sibony, P.; Fourman, S. Eye-rubbing optic neuropathy. Am. J. Ophthalmol. 2002, 134, 460–461. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Albalawi, F.; Beckel, J.M.; Lim, J.C.; Laties, A.M.; Mitchell, C.H. The P2X7 receptor links mechanical strain to cytokine IL-6 up-regulation and release in neurons and astrocytes. J. Neurochem. 2017, 141, 436–448. [Google Scholar] [CrossRef]
- Li, A.; Zhang, X.; Zheng, D.; Ge, J.; Laties, A.M.; Mitchell, C.H. Sustained elevation of extracellular ATP in aqueous humor from humans with primary chronic angle-closure glaucoma. Exp. Eye Res. 2011, 93, 528–533. [Google Scholar] [CrossRef]
- Xia, J.; Lim, J.C.; Lu, W.; Beckel, J.M.; Macarak, E.J.; Laties, A.M.; Mitchell, C.H. Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors. J. Physiol. 2012, 590, 2285–2304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, A.; Ge, J.; Reigada, D.; Laties, A.M.; Mitchell, C.H. Acute increase of intraocular pressure releases ATP into the anterior chamber. Exp. Eye Res. 2007, 85, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; Chen, H.; Li, F.; Zhu, Y.; Yin, W.; Zhuo, Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-kappaB pathway in acute glaucoma. J. Neuroinflam. 2015, 12, 137. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, X.; Luo, C.; Cai, J.; Powell, D.W. An astrocyte-specific proteomic approach to inflammatory responses in experimental rat glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4220–4233. [Google Scholar] [CrossRef]
- Markiewicz, L.; Pytel, D.; Mucha, B.; Szymanek, K.; Szaflik, J.; Szaflik, J.P.; Majsterek, I. Altered Expression Levels of MMP1, MMP9, MMP12, TIMP1, and IL-1beta as a Risk Factor for the Elevated IOP and Optic Nerve Head Damage in the Primary Open-Angle Glaucoma Patients. Biomed. Res. Int. 2015, 2015, 812503. [Google Scholar] [CrossRef]
- Lin, C.; Wu, F.; Zheng, T.; Wang, X.; Chen, Y.; Wu, X. Kaempferol attenuates retinal ganglion cell death by suppressing NLRP1/NLRP3 inflammasomes and caspase-8 via JNK and NF-kappaB pathways in acute glaucoma. Eye 2019, 33, 777–784. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Sun, Q.; Xue, S.; Guan, H.; Ji, M. Activation of P2X7R-NLRP3 pathway in Retinal microglia contribute to Retinal Ganglion Cells death in chronic ocular hypertension (COH). Exp. Eye Res. 2019, 188, 107771. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Hu, H.; Sevigny, J.; Gabelt, B.T.; Kaufman, P.L.; Johnson, E.C.; Morrison, J.C.; Zode, G.S.; Sheffield, V.C.; Zhang, X.; et al. Rat, mouse, and primate models of chronic glaucoma show sustained elevation of extracellular ATP and altered purinergic signaling in the posterior eye. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3075–3083. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.R.; Macalinao, D.G.; Sousa, G.L.; Walden, M.; Soto, I.; Kneeland, S.C.; Barbay, J.M.; King, B.L.; Marchant, J.K.; Hibbs, M.; et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J. Clin. Investig. 2011, 121, 1429–1444. [Google Scholar] [CrossRef]
- Yang, X.; Luo, C.; Cai, J.; Powell, D.W.; Yu, D.; Kuehn, M.H.; Tezel, G. Neurodegenerative and inflammatory pathway components linked to TNF-alpha/TNFR1 signaling in the glaucomatous human retina. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8442–8454. [Google Scholar] [CrossRef]
- Soto, I.; Howell, G.R. The complex role of neuroinflammation in glaucoma. Cold Spring Harb. Perspect. Med. 2014, 4, a017269. [Google Scholar] [CrossRef]
- Dvoriantchikova, G.; Ivanov, D.; Barakat, D.; Grinberg, A.; Wen, R.; Slepak, V.Z.; Shestopalov, V.I. Genetic ablation of Pannexin1 protects retinal neurons from ischemic injury. PLoS ONE 2012, 7, e31991. [Google Scholar] [CrossRef] [PubMed]
- Pronin, A.; Pham, D.; An, W.; Dvoriantchikova, G.; Reshetnikova, G.; Qiao, J.; Kozhekbaeva, Z.; Reiser, A.E.; Slepak, V.Z.; Shestopalov, V.I. Inflammasome Activation Induces Pyroptosis in the Retina Exposed to Ocular Hypertension Injury. Front. Mol. Neurosci. 2019, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, J.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc. Natl. Acad. Sci. USA 2014, 111, 11181–11186. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Kocab, A.J.; Zacks, D.N.; Marshak-Rothstein, A.; Gregory-Ksander, M. A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma. J. Neuroinflam. 2019, 16, 184. [Google Scholar] [CrossRef]
- Yang, X.; Zeng, Q.; Tezel, G. Regulation of distinct caspase-8 functions in retinal ganglion cells and astroglia in experimental glaucoma. Neurobiol. Dis. 2021, 150, 105258. [Google Scholar] [CrossRef]
- Zhao, M.; Li, S.; Matsubara, J.A. Targeting Pyroptotic Cell Death Pathways in Retinal Disease. Front. Med. 2021, 8, 802063. [Google Scholar] [CrossRef]
- Krizaj, D.; Ryskamp, D.A.; Tian, N.; Tezel, G.; Mitchell, C.H.; Slepak, V.Z.; Shestopalov, V.I. From mechanosensitivity to inflammatory responses: New players in the pathology of glaucoma. Curr. Eye Res. 2014, 39, 105–119. [Google Scholar] [CrossRef]
- Evangelho, K.; Mogilevskaya, M.; Losada-Barragan, M.; Vargas-Sanchez, J.K. Pathophysiology of primary open-angle glaucoma from a neuroinflammatory and neurotoxicity perspective: A review of the literature. Int. Ophthalmol. 2019, 39, 259–271. [Google Scholar] [CrossRef]
- Tzeng, T.C.; Schattgen, S.; Monks, B.; Wang, D.; Cerny, A.; Latz, E.; Fitzgerald, K.; Golenbock, D.T. A Fluorescent Reporter Mouse for Inflammasome Assembly Demonstrates an Important Role for Cell-Bound and Free ASC Specks during In Vivo Infection. Cell Rep. 2016, 16, 571–582. [Google Scholar] [CrossRef]
- Mattson, D.L. Comparison of arterial blood pressure in different strains of mice. Am. J. Hypertens. 2001, 14, 405–408. [Google Scholar] [CrossRef]
- Morrison, J.C.; Cepurna, W.O.; Tehrani, S.; Choe, T.E.; Jayaram, H.; Lozano, D.C.; Fortune, B.; Johnson, E.C. A Period of Controlled Elevation of IOP (CEI) Produces the Specific Gene Expression Responses and Focal Injury Pattern of Experimental Rat Glaucoma. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6700–6711. [Google Scholar] [CrossRef]
- Bui, B.V.; Edmunds, B.; Cioffi, G.A.; Fortune, B. The gradient of retinal functional changes during acute intraocular pressure elevation. Investig. Ophthalmol. Vis. Sci. 2005, 46, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Rydz, C.; Nguyen Huu, V.A.; Rocha, L.; Palomino La Torre, C.; Lee, I.; Cho, W.; Jabari, M.; Donello, J.; Lyon, D.C.; et al. Stress induced aging in mouse eye. Aging Cell 2022, 21, e13737. [Google Scholar] [CrossRef] [PubMed]
- Grotegut, P.; Kuehn, S.; Meissner, W.; Dick, H.B.; Joachim, S.C. Intravitreal S100B Injection Triggers a Time-Dependent Microglia Response in a Pro-Inflammatory Manner in Retina and Optic Nerve. Mol. Neurobiol. 2020, 57, 1186–1202. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Dumitrescu, A.V.; Wadkins, D.; Elwood, B.W.; Gramlich, O.W.; Kuehn, M.H. Systemic Treatment with Pioglitazone Reverses Vision Loss in Preclinical Glaucoma Models. Biomolecules 2022, 12, 281. [Google Scholar] [CrossRef]
- Weaver, C.; Cyr, B.; de Rivero Vaccari, J.C.; de Rivero Vaccari, J.P. Inflammasome Proteins as Inflammatory Biomarkers of Age-Related Macular Degeneration. Transl. Vis. Sci. Technol. 2020, 9, 27. [Google Scholar] [CrossRef]
- Shrivastava, A.; Saxena, P.; Gupta, V.B. Spectrophotometric estimation of tamsulosin hydrochloride by acid-dye method. Pharm. Methods 2011, 2, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Porciatti, V. The mouse pattern electroretinogram. Doc. Ophthalmol. 2007, 115, 145–153. [Google Scholar] [CrossRef]
- Chou, T.H.; Romano, G.L.; Amato, R.; Porciatti, V. Nicotinamide-Rich Diet in DBA/2J Mice Preserves Retinal Ganglion Cell Metabolic Function as Assessed by PERG Adaptation to Flicker. Nutrients 2020, 12, 1910. [Google Scholar] [CrossRef] [PubMed]
- Porciatti, V.; Saleh, M.; Nagaraju, M. The pattern electroretinogram as a tool to monitor progressive retinal ganglion cell dysfunction in the DBA/2J mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2007, 48, 745–751. [Google Scholar] [CrossRef]
- Scheiblich, H.; Schlutter, A.; Golenbock, D.T.; Latz, E.; Martinez-Martinez, P.; Heneka, M.T. Activation of the NLRP3 inflammasome in microglia: The role of ceramide. J. Neurochem. 2017, 143, 534–550. [Google Scholar] [CrossRef] [PubMed]
- Dvoriantchikova, G.; Pronin, A.; Kurtenbach, S.; Toychiev, A.; Chou, T.H.; Yee, C.W.; Prindeville, B.; Tayou, J.; Porciatti, V.; Sagdullaev, B.T.; et al. Pannexin 1 sustains the electrophysiological responsiveness of retinal ganglion cells. Sci. Rep. 2018, 8, 5797. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.P.; Lotocki, G.; Marcillo, A.E.; Dietrich, W.D.; Keane, R.W. A molecular platform in neurons regulates inflammation after spinal cord injury. J. Neurosci. 2008, 28, 3404–3414. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.P.; Lotocki, G.; Alonso, O.F.; Bramlett, H.M.; Dietrich, W.D.; Keane, R.W. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J. Cereb. Blood Flow. Metab. 2009, 29, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Heilig, R.; Dick, M.S.; Sborgi, L.; Meunier, E.; Hiller, S.; Broz, P. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur. J. Immunol. 2018, 48, 584–592. [Google Scholar] [CrossRef]
- Li, J.; Hao, J.H.; Yao, D.; Li, R.; Li, X.F.; Yu, Z.Y.; Luo, X.; Liu, X.H.; Wang, M.H.; Wang, W. Caspase-1 inhibition prevents neuronal death by targeting the canonical inflammasome pathway of pyroptosis in a murine model of cerebral ischemia. CNS Neurosci. Ther. 2020, 26, 925–939. [Google Scholar] [CrossRef]
- Bao, L.; Locovei, S.; Dahl, G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004, 572, 65–68. [Google Scholar] [CrossRef]
- Qu, Y.; Misaghi, S.; Newton, K.; Gilmour, L.L.; Louie, S.; Cupp, J.E.; Dubyak, G.R.; Hackos, D.; Dixit, V.M. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J. Immunol. 2011, 186, 6553–6561. [Google Scholar] [CrossRef]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 2019, 10, 1689. [Google Scholar] [CrossRef]
- Rogers, C.; Alnemri, E.S. Gasdermins: Novel mitochondrial pore-forming proteins. Mol. Cell Oncol. 2019, 6, e1621501. [Google Scholar] [CrossRef]
- Chen, H.; Deng, Y.; Gan, X.; Li, Y.; Huang, W.; Lu, L.; Wei, L.; Su, L.; Luo, J.; Zou, B.; et al. NLRP12 collaborates with NLRP3 and NLRC4 to promote pyroptosis inducing ganglion cell death of acute glaucoma. Mol. Neurodegener. 2020, 15, 26. [Google Scholar] [CrossRef] [PubMed]
- Adamczak, S.E.; de Rivero Vaccari, J.P.; Dale, G.; Brand, F.J., 3rd; Nonner, D.; Bullock, M.R.; Dahl, G.P.; Dietrich, W.D.; Keane, R.W. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J. Cereb. Blood Flow. Metab. 2014, 34, 621–629. [Google Scholar] [CrossRef]
- Jakobs, C.; Perner, S.; Hornung, V. AIM2 Drives Joint Inflammation in a Self-DNA Triggered Model of Chronic Polyarthritis. PLoS ONE 2015, 10, e0131702. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, Y.; Zhang, S.; Sun, X.; Wu, J. TRPV4-induced Muller cell gliosis and TNF-alpha elevation-mediated retinal ganglion cell apoptosis in glaucomatous rats via JAK2/STAT3/NF-kappaB pathway. J. Neuroinflam. 2021, 18, 271. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Xu, M.X.; Zhou, H.; Cheng, S.; Li, F.; Miao, Y.; Wang, Z. Tumor necrosis factor-alpha aggravates gliosis and inflammation of activated retinal Muller cells. Biochem. Biophys. Res. Commun. 2020, 531, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; He, Y.; Munoz-Planillo, R.; Liu, Q.; Nunez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.; Zetterqvist, A.V.; Wright, W.R.; Kirkby, N.S.; Mitchell, J.A.; Paul-Clark, M.J. Differential role of pannexin-1/ATP/P2X(7) axis in IL-1beta release by human monocytes. FASEB J. 2017, 31, 2439–2445. [Google Scholar] [CrossRef]
- Hung, S.C.; Choi, C.H.; Said-Sadier, N.; Johnson, L.; Atanasova, K.R.; Sellami, H.; Yilmaz, O.; Ojcius, D.M. P2X4 assembles with P2X7 and pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PLoS ONE 2013, 8, e70210. [Google Scholar] [CrossRef]
- El-Maadawy, W.H.; Hassan, M.; Badawy, M.H.; AbuSeada, A.; Hafiz, E. Probenecid induces the recovery of renal ischemia/reperfusion injury via the blockade of Pannexin 1/P2X7 receptor axis. Life Sci. 2022, 308, 120933. [Google Scholar] [CrossRef] [PubMed]
- Rusiecka, O.M.; Tournier, M.; Molica, F.; Kwak, B.R. Pannexin1 channels-a potential therapeutic target in inflammation. Front. Cell Dev. Biol. 2022, 10, 1020826. [Google Scholar] [CrossRef]
- Bruzzone, R.; Hormuzdi, S.G.; Barbe, M.T.; Herb, A.; Monyer, H. Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13644–13649. [Google Scholar] [CrossRef] [PubMed]
- Bargiotas, P.; Krenz, A.; Hormuzdi, S.G.; Ridder, D.A.; Herb, A.; Barakat, W.; Penuela, S.; von Engelhardt, J.; Monyer, H.; Schwaninger, M. Pannexins in ischemia-induced neurodegeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 20772–20777. [Google Scholar] [CrossRef] [PubMed]
- Morozumi, W.; Inagaki, S.; Iwata, Y.; Nakamura, S.; Hara, H.; Shimazawa, M. Piezo channel plays a part in retinal ganglion cell damage. Exp. Eye Res. 2020, 191, 107900. [Google Scholar] [CrossRef]
- Ryskamp, D.A.; Witkovsky, P.; Barabas, P.; Huang, W.; Koehler, C.; Akimov, N.P.; Lee, S.H.; Chauhan, S.; Xing, W.; Renteria, R.C.; et al. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J. Neurosci. 2011, 31, 7089–7101. [Google Scholar] [CrossRef]
- Lakk, M.; Young, D.; Baumann, J.M.; Jo, A.O.; Hu, H.; Krizaj, D. Polymodal TRPV1 and TRPV4 Sensors Colocalize but Do Not Functionally Interact in a Subpopulation of Mouse Retinal Ganglion Cells. Front. Cell Neurosci. 2018, 12, 353. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; He, H.; Long, J.; Sui, X.; Yang, J.; Lin, G.; Wang, Q.; Wang, Y.; Luo, Y. TRPV4 Regulates Soman-Induced Status Epilepticus and Secondary Brain Injury via NMDA Receptor and NLRP3 Inflammasome. Neurosci. Bull. 2021, 37, 905–920. [Google Scholar] [CrossRef]
- Liu, Y.; Fan, H.; Li, X.; Liu, J.; Qu, X.; Wu, X.; Liu, M.; Liu, Z.; Yao, R. Trpv4 regulates Nlrp3 inflammasome via SIRT1/PGC-1alpha pathway in a cuprizone-induced mouse model of demyelination. Exp. Neurol. 2021, 337, 113593. [Google Scholar] [CrossRef]
- Nishinaka, A.; Tanaka, M.; Ohara, K.; Sugaru, E.; Shishido, Y.; Sugiura, A.; Moriguchi, Y.; Toui, A.; Nakamura, S.; Shimada, K.; et al. TRPV4 channels promote vascular permeability in retinal vascular disease. Exp. Eye Res. 2023, 228, 109405. [Google Scholar] [CrossRef]
- Wang, A.Y.; Lee, P.Y.; Bui, B.V.; Jobling, A.I.; Greferath, U.; Brandli, A.; Dixon, M.A.; Findlay, Q.; Fletcher, E.L.; Vessey, K.A. Potential mechanisms of retinal ganglion cell type-specific vulnerability in glaucoma. Clin. Exp. Optom. 2020, 103, 562–571. [Google Scholar] [CrossRef]
- Desplat, A.; Penalba, V.; Gros, E.; Parpaite, T.; Coste, B.; Delmas, P. Piezo1-Pannexin1 complex couples force detection to ATP secretion in cholangiocytes. J. Gen. Physiol. 2021, 153, e202112871. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44.e6. [Google Scholar] [CrossRef]
- Volchuk, A.; Ye, A.; Chi, L.; Steinberg, B.E.; Goldenberg, N.M. Indirect regulation of HMGB1 release by gasdermin D. Nat. Commun. 2020, 11, 4561. [Google Scholar] [CrossRef] [PubMed]
- Broz, P. Unconventional protein secretion by gasdermin pores. Semin. Immunol. 2023, 69, 101811. [Google Scholar] [CrossRef]
- Chen, H.; Cho, K.S.; Vu, T.H.K.; Shen, C.H.; Kaur, M.; Chen, G.; Mathew, R.; McHam, M.L.; Fazelat, A.; Lashkari, K.; et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat. Commun. 2018, 9, 3209. [Google Scholar] [CrossRef]
- Williams, P.A.; Braine, C.E.; Kizhatil, K.; Foxworth, N.E.; Tolman, N.G.; Harder, J.M.; Scott, R.A.; Sousa, G.L.; Panitch, A.; Howell, G.R.; et al. Inhibition of monocyte-like cell extravasation protects from neurodegeneration in DBA/2J glaucoma. Mol. Neurodegener. 2019, 14, 6. [Google Scholar] [CrossRef]
- Gramlich, O.W.; Godwin, C.R.; Heuss, N.D.; Gregerson, D.S.; Kuehn, M.H. T and B Lymphocyte Deficiency in Rag1−/− Mice Reduces Retinal Ganglion Cell Loss in Experimental Glaucoma. Investig. Ophthalmol. Vis. Sci. 2020, 61, 18. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Tang, Y.; Yi, I.; Chen, D.F. The role of commensal microflora-induced T cell responses in glaucoma neurodegeneration. Prog. Brain Res. 2020, 256, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef]
- Hulse, J.; Bhaskar, K. Crosstalk Between the NLRP3 Inflammasome/ASC Speck and Amyloid Protein Aggregates Drives Disease Progression in Alzheimer’s and Parkinson’s Disease. Front. Mol. Neurosci. 2022, 15, 805169. [Google Scholar] [CrossRef] [PubMed]
- Shestopalov, V.I.; Spurlock, M.; Gramlich, O.W.; Kuehn, M.H. Immune Responses in the Glaucomatous Retina: Regulation and Dynamics. Cells 2021, 10, 1973. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.K.; Khan, M.A.; Singh, S.K. Constitutive Inflammatory Cytokine Storm: A Major Threat to Human Health. J. Interferon Cytokine Res. 2020, 40, 19–23. [Google Scholar] [CrossRef] [PubMed]
- de Torre-Minguela, C.; Gomez, A.I.; Couillin, I.; Pelegrin, P. Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. FASEB J. 2021, 35, e21757. [Google Scholar] [CrossRef]
- de Vasconcelos, N.M.; Van Opdenbosch, N.; Van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019, 26, 146–161. [Google Scholar] [CrossRef]
- Kondolf, H.C.; D’Orlando, D.A.; Dubyak, G.R.; Abbott, D.W. Protein engineering reveals that gasdermin A preferentially targets mitochondrial membranes over the plasma membrane during pyroptosis. J. Biol. Chem. 2023, 299, 102908. [Google Scholar] [CrossRef]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflam. 2018, 15, 242. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Cardozo, B.H.; Foxworth, N.E.; John, S.W.M. Nicotinamide treatment robustly protects from inherited mouse glaucoma. Commun. Integr. Biol. 2018, 11, e1356956. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; John, S.W.M. Glaucoma as a Metabolic Optic Neuropathy: Making the Case for Nicotinamide Treatment in Glaucoma. J. Glaucoma 2017, 26, 1161–1168. [Google Scholar] [CrossRef]
- Gramlich, O.W.; Teister, J.; Neumann, M.; Tao, X.; Beck, S.; von Pein, H.D.; Pfeiffer, N.; Grus, F.H. Immune response after intermittent minimally invasive intraocular pressure elevations in an experimental animal model of glaucoma. J. Neuroinflam. 2016, 13, 82. [Google Scholar] [CrossRef]
- Resta, V.; Novelli, E.; Vozzi, G.; Scarpa, C.; Caleo, M.; Ahluwalia, A.; Solini, A.; Santini, E.; Parisi, V.; Di Virgilio, F.; et al. Acute retinal ganglion cell injury caused by intraocular pressure spikes is mediated by endogenous extracellular ATP. Eur. J. Neurosci. 2007, 25, 2741–2754. [Google Scholar] [CrossRef]
- Khanh Vu, T.H.; Chen, H.; Pan, L.; Cho, K.S.; Doesburg, D.; Thee, E.F.; Wu, N.; Arlotti, E.; Jager, M.J.; Chen, D.F. CD4+ T-Cell Responses Mediate Progressive Neurodegeneration in Experimental Ischemic Retinopathy. Am. J. Pathol. 2020, 190, 1723–1734. [Google Scholar] [CrossRef] [PubMed]
- Daftarian, N.; Zandi, S.; Piryaie, G.; Nikougoftar Zarif, M.; Ranaei Pirmardan, E.; Yamaguchi, M.; Behzadian Nejad, Q.; Hasanpour, H.; Samiei, S.; Pfister, I.B.; et al. Peripheral blood CD163(+) monocytes and soluble CD163 in dry and neovascular age-related macular degeneration. FASEB J. 2020, 34, 8001–8011. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Xiu, W.; Chen, Q.; Peng, K.; Zhu, X.; Wang, Z.; Xu, X.; Chen, Y.; Zhang, G.; Fu, J.; et al. Gut-licensed β7+ CD4+ T cells contribute to progressive retinal ganglion cell damage in glaucoma. Sci. Transl. Med. 2023, 15, eadg1656. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Heilig, R.; Dilucca, M.; Boucher, D.; Chen, K.W.; Hancz, D.; Demarco, B.; Shkarina, K.; Broz, P. Caspase-1 cleaves Bid to release mitochondrial SMAC and drive secondary necrosis in the absence of GSDMD. Life Sci. Alliance 2020, 3, e202000735. [Google Scholar] [CrossRef]
- Morrison, J.C.; Cepurna, W.O.; Johnson, E.C. Modeling glaucoma in rats by sclerosing aqueous outflow pathways to elevate intraocular pressure. Exp. Eye Res. 2015, 141, 23–32. [Google Scholar] [CrossRef]
- Gramlich, O.W.; Godwin, C.R.; Wadkins, D.; Elwood, B.W.; Kuehn, M.H. Early Functional Impairment in Experimental Glaucoma Is Accompanied by Disruption of the GABAergic System and Inceptive Neuroinflammation. Int. J. Mol. Sci. 2021, 22, 7581. [Google Scholar] [CrossRef] [PubMed]
- Sappington, R.M.; Carlson, B.J.; Crish, S.D.; Calkins, D.J. The microbead occlusion model: A paradigm for induced ocular hypertension in rats and mice. Investig. Ophthalmol. Vis. Sci. 2010, 51, 207–216. [Google Scholar] [CrossRef]
- Ruiz-Ederra, J.; Verkman, A.S. Mouse model of sustained elevation in intraocular pressure produced by episcleral vein occlusion. Exp. Eye Res. 2006, 82, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Bui, K.M.; Patel, K.H.; Kim, H.; Arruda, J.A.; Wilensky, J.T.; Vajaranant, T.S. Effect of hemodialysis on intraocular pressure and ocular perfusion pressure. JAMA Ophthalmol. 2013, 131, 1525–1531. [Google Scholar] [CrossRef]
- Esen, F.; Eraslan, M.; Cerman, E.; Celiker, H.; Kazokoglu, H. Diurnal Spikes of Intraocular Pressure in Uveitic Glaucoma: A 24-hour Intraocular Pressure Monitoring Study. Semin. Ophthalmol. 2020, 35, 246–251. [Google Scholar] [CrossRef]
- Demirel, S.; Yanik, O.; Batioglu, F.; Ozmert, E. Intraocular pressure changes related to intravitreal injections of ranibizumab: Analysis of pseudophakia and glaucoma subgroup. Int. Ophthalmol. 2015, 35, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Pollmann, A.S.; Zhang, A.; Shuba, L.M. Intraocular pressure fluctuations in a professional woodwind musician with advanced glaucoma. Can. J. Ophthalmol. 2022, 57, e184–e185. [Google Scholar] [CrossRef] [PubMed]
- Koz, O.G.; Turkcu, M.F.; Yarangumeli, A.; Koz, C.; Kural, G. Normotensive glaucoma and risk factors in normotensive eyes with pseudoexfoliation syndrome. J. Glaucoma 2009, 18, 684–688. [Google Scholar] [CrossRef]
- Kandarakis, A.; Soumplis, V.; Karampelas, M.; Panos, C.; Kyriakos, N.; Baxevanakis, A.; Karagiannis, D. Efficacy of brimonidine in preventing intraocular pressure spikes following phacoemulsification in glaucoma patients. Eur. J. Ophthalmol. 2010, 20, 994–999. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spurlock, M.; An, W.; Reshetnikova, G.; Wen, R.; Wang, H.; Braha, M.; Solis, G.; Kurtenbach, S.; Galindez, O.J.; de Rivero Vaccari, J.P.; et al. The Inflammasome-Dependent Dysfunction and Death of Retinal Ganglion Cells after Repetitive Intraocular Pressure Spikes. Cells 2023, 12, 2626. https://doi.org/10.3390/cells12222626
Spurlock M, An W, Reshetnikova G, Wen R, Wang H, Braha M, Solis G, Kurtenbach S, Galindez OJ, de Rivero Vaccari JP, et al. The Inflammasome-Dependent Dysfunction and Death of Retinal Ganglion Cells after Repetitive Intraocular Pressure Spikes. Cells. 2023; 12(22):2626. https://doi.org/10.3390/cells12222626
Chicago/Turabian StyleSpurlock, Markus, Weijun An, Galina Reshetnikova, Rong Wen, Hua Wang, Michelle Braha, Gabriela Solis, Stefan Kurtenbach, Orlando J. Galindez, Juan Pablo de Rivero Vaccari, and et al. 2023. "The Inflammasome-Dependent Dysfunction and Death of Retinal Ganglion Cells after Repetitive Intraocular Pressure Spikes" Cells 12, no. 22: 2626. https://doi.org/10.3390/cells12222626