

Clinical Approaches for Mitochondrial Diseases

1
Korea Mouse Phenotyping Center, Seoul National University, Seoul 08826, Republic of Korea
2
Department of Medicine, Korea University College of Medicine, Seoul 02708, Republic of Korea
3
Laboratory Animal Resource and Research Center, Korea Research Institute of Bioscience and Biotechnology, Cheongju 28116, Republic of Korea
4
College of Veterinary Medicine and Research Institute of Veterinary Medicine, Chungbuk National University, Cheongju 28644, Republic of Korea
5
Department of Biomedical Sciences, Korea University College of Medicine, Seoul 02841, Republic of Korea
6
Department of Physiology, Korea University College of Medicine, Seoul 02841, Republic of Korea
*
Authors to whom correspondence should be addressed.
Cells 2023, 12(20), 2494; https://doi.org/10.3390/cells12202494
Submission received: 26 September 2023
/
Revised: 18 October 2023
/
Accepted: 19 October 2023
/
Published: 20 October 2023
(This article belongs to the Special Issue CRISPR-Based Genome Editing in Translational Research—Second Edition)
Abstract
:Mitochondria are subcontractors dedicated to energy production within cells. In human mitochondria, almost all mitochondrial proteins originate from the nucleus, except for 13 subunit proteins that make up the crucial system required to perform ‘oxidative phosphorylation (OX PHOS)’, which are expressed by the mitochondria’s self-contained DNA. Mitochondrial DNA (mtDNA) also encodes 2 rRNA and 22 tRNA species. Mitochondrial DNA replicates almost autonomously, independent of the nucleus, and its heredity follows a non-Mendelian pattern, exclusively passing from mother to children. Numerous studies have identified mtDNA mutation-related genetic diseases. The consequences of various types of mtDNA mutations, including insertions, deletions, and single base-pair mutations, are studied to reveal their relationship to mitochondrial diseases. Most mitochondrial diseases exhibit fatal symptoms, leading to ongoing therapeutic research with diverse approaches such as stimulating the defective OXPHOS system, mitochondrial replacement, and allotropic expression of defective enzymes. This review provides detailed information on two topics: (1) mitochondrial diseases caused by mtDNA mutations, and (2) the mechanisms of current treatments for mitochondrial diseases and clinical trials.
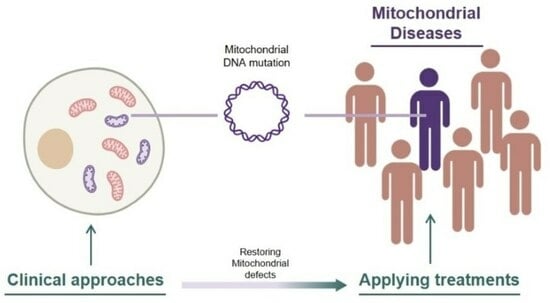
1. Introduction
Mitochondrial disease refers to conditions caused by mitochondrial dysfunction [1]. Due to the distribution of mitochondria in every cell of the body, organ defects and symptoms vary widely. However, there is one clear commonality: the inheritance of mutated DNA that encodes major components of oxidative phosphorylation (OXPHOS), leading to a loss of mitochondrial function [2]. Mitochondria are organelles composed of numerous nucleus-encoded proteins with various roles [3,4,5], as well as 13 self-encoded proteins crucial for OXPHOS, along with 2 self-encoded rRNAs and 22 tRNAs [6]. Because all mitochondrially encoded proteins play important roles in mitochondria, mutations in coding genes within mitochondria can directly lead to mitochondrial dysfunction [7].
Unlike genetic disorders associated with nuclear genomes, the replication of mutated mtDNA does not exhibit patterns of Mendelian inheritance due to the independent fusion-fission activity of mitochondria. Mitochondrial DNA-related mitochondrial disease does not require the 100% homoplasmy of mutated DNA in an organism. Despite the heteroplasmic distribution being diverse across individuals and types of diseases, there is considered to be a certain threshold of mtDNA mutation load required to exhibit symptoms of diseases [8,9]. Correlations between heteroplasmic levels and the expression of symptoms have also been reported in case studies involving a large amount of screening of mitochondrial disease patients [10,11]. DNA sequencing methods have rapidly advanced, making it easier to find disease-inducing DNA mutations and even distinguish every single mtDNA sequence in a cell [12]. Yet, due to the difficulty of isolating individual mitochondria, there is a greater demand for studies to reveal the ambiguous dynamics of the mitochondrial processing of mtDNA. One thing that is not ambiguous is the notable prevalence of mitochondrial diseases [13,14,15]. Therefore, we are facing the need to solve this threatening problem affecting our populations.
Because mitochondrial diseases involve different types of dysfunctions in mitochondria, approaches for the treatment of mitochondrial diseases also vary. These approaches include replacing the cytoplasm containing defective mitochondria with healthy mitochondria-containing cytoplasm through oocyte spindle transfer [16], targeting the underlying cause of mitochondrial disease by converting pathogenic point-mutated mtDNA to normal mtDNA [17,18,19], the use of chemical compounds to stimulate electron transfer chain in OXPHOS by bypassing the malfunctioning complex [20,21], and even researching phenolic compounds in our diet that have shown potential for reducing ROS through their antioxidant activity [22,23,24,25].
Mitochondrial defect also affects the closely associated protein, AMPK [26]. This, in turn, impacts the AMPK’s protein activity of regulating antitumor immunity [27,28]. In terms of this, research is being conducted to induce OXPHOS depression in mitochondria, which affects AMPK and further promotes cancer immunotherapy [29,30]. Studies to control the population of dysfunctional mitochondria by utilizing the involvement of AMPK protein in mitochondrial biogenesis are also in progress [31,32]. In the context of mitochondria-related autophagic processes, which involve the regulation of dysfunctional mitochondria populations, this approach holds the potential for treating mitochondrial diseases and is actively being investigated [33,34,35,36,37].
Countless studies are being conducted in the field of mitochondrial disease, but only a few studies have been completed with considerable efficiency in terms of restoring defective mitochondria, and an even more limited portion of groups have reached clinical trials with their treatments. This review focuses on treatments that are currently undergoing clinical trials for mitochondrial disease, with initial explanations of several mitochondrial diseases.
2. Features of Mitochondrial Disease
One notable aspect of mitochondrial disease is that, despite its diverse causes at the cellular level, symptoms are generally expressed through mitochondrial dysfunction. Consequently, although the main symptoms of mitochondrial disease vary depending on the type of disease, they appear to share common symptoms in the broader category of encephalomyopathy. Therefore, it is important to identify the defective mechanisms at the cellular level in order to understand the causes of mitochondrial disease.
Mitochondria are subcellular organelles known for producing ATP, which is used for cellular energy, through OXPHOS. Mitochondrial disease occurs when there are defects in the proteins involved in OXPHOS or other proteins related to mitochondrial function. Researchers have shown a correlation between mitochondrial performance and high-energy demanding cells, and mitochondrial proteome also varies depending on the tissue [38,39,40,41,42,43,44,45,46]. Therefore, mitochondrial dysfunction can be regarded as a deficiency in cellular energy, which leads to energy deficiency in nerve cells or myocytes, appearing as the main cause of mitochondrial disease. However, studies on patients with mitochondrial disease have reported that the main cause is often associated with lactic acidosis. Dysfunction in the electron transport chain leads to a decrease in ATP production, and low ATP level further increase glycolysis, resulting in an overproduction of pyruvate, which can be further reduced and converted to lactate [47,48,49].
Mitochondrial proteins can be encoded by both nuclear DNA and mtDNA, thus mitochondrial disease can also result from mutations in either or both types of DNA. The inheritance of mitochondrial diseases caused by nuclear DNA mutation can be easily identified by examining family histories, as the symptoms follow the rules of Mendelian inheritance. However, mitochondria have their own autonomous process of duplication and DNA replication. Therefore, mutated mtDNA can exist in a state of heteroplasmy, leading to cellular dysfunction and an increase in the expression of mitochondrial disease symptoms. The manifestation of these symptoms is expected to depend on a certain threshold of heteroplasmy, which varies depending on the type of disease and the individual carrying the mutated mtDNA [50,51,52]. Case studies have shown that the amount of mutated mtDNA can vary among patients with the same symptoms, and higher levels of heteroplasmy do not always correlate with the severity of symptoms. Even in the case of monozygotic twins with mitochondrial disease, different symptoms can be observed, suggesting that there are additional factors beyond DNA mutations that contribute to the expression of symptoms [53].
In case studies involving large amounts of patient data, a convincing correlation was found between heteroplasmy level and the age of onset [10,11], but no correlation was observed between symptoms. Due to the elusive nature of mitochondrial pathologies, it is important to understand the mechanisms and causes that trigger them.
3. Types of Mitochondrial Disease
Since mitochondrial diseases tend to have numerous causes beyond their prominent cause, this paper only focused on major types of disease-causing mutations. The description of mitochondrial disease with notable symptoms and case studies is given below (Table 1, Figure 1).
Leber hereditary optic neuropathy (LHON) is known to exhibit eye-related symptoms, such as sequential vision loss and dyschromatopsia. Patients with LHON also have a higher likelihood of experiencing neurological abnormalities, including peripheral neuropathy and myopathy, similar to other mitochondrial diseases, compared to the general population [80,81,82]. However, these symptoms do not manifest as a chain of events. Consequently, LHON is more commonly diagnosed based on optic neuropathy-related symptoms.
In LHON, up to 90% of clinical cases are associated with three mutations: m.G11778A (MT-ND4), m.A3460G (MT-ND1), and m.T14484C (MT-ND6). Among these mutations, m.G11778A is the most dominant [83]. ND1–6 are the components of the NADH-ubiquinone oxidoreductase complex (complex 1), and therefore, mutations associated with LHON cause defects in complex 1, which has the function of catalyzing reactions in the electron transport chain.
Patients with LHON have high levels of mutated DNA heteroplasmy, almost reaching the level of homoplasmy [83,84]. Also unaffected carriers of LHON-inducing mtDNA mutations can be easily found in the families with LHON patients [85,86,87,88], from which could be further inferred that LHON-inducing mtDNA mutations do not seem to be critical for the functioning of mitochondria. Therefore, several studies have been conducted to discover the triggers of symptom expression [89,90,91,92,93,94], but the underlying factors of LHON remain elusive.
The LHON mutation-derived mitochondrial defects mostly result in optic neuropathy, affecting optic nerve, axons, retinal ganglion cells, and photoreceptor cells [83,95]. Case studies have revealed affected retinal nerve fiber layers and impaired photoreceptor functions. Therefore, ocular symptoms are experienced, such as sequential loss of visual acuity and dyschromatopsia [54,55,56].
Myoclonic epilepsy with ragged red fiber (MERRF) is a multi-system disease characterized by progressive myoclonus and seizures [96]. One of the most common mutation among the patients with different types of mtDNA mutations is m.A8344G, which affects the mitochondrial tRNA lysine [97,98,99,100,101]. Another prominent mutation in the same gene is m.G8363A, along with the m.A3243G, m.G3255A, and m.T3291C mutations, which have also been reported to cause MERRF with a defects in tRNA leucine [57,58,59,99,102,103]. Mutations in tRNA-coding genes induce the global impairment of mtDNA-encoded proteins rather than affecting certain complexes or pathways [101,104,105]. This can result in the overall dysfunction of mitochondria. In case studies involving MERRF patients with tRNA lysine mutations, various symptoms were reported, such as unsteadiness of gait, optic atrophy, slurred speech, sensorineural hearing loss, and limb weakness [57,58,59].
One well-known mitochondrial disease is mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS). It is also caused by a mutation in the mitochondria-encoded tRNA leucine, specifically the m.A3243G mutation, which leads to mitochondrial dysfunction and similar effects as MERRF, affecting the synthesis of all mtDNA-encoded proteins [106,107,108,109].
MELAS is a multi-systemic disease; therefore, misdiagnosis was common until the utilization of DNA analysis on relatives or families of the patients. In one case study, a patient was initially diagnosed with diabetes at an early age, but the cause of the gradual development of diabetes remained elusive until the patient’s sister was diagnosed with maternally inherited diabetes and deafness (MIDD) [60]. Subsequent medical studies were then conducted, resulting in the diagnosis of MELAS. In total, it took 25 years from the initial diagnosis of diabetes for the patient to be diagnosed with MELAS. Stroke-like episodes are one of the main aspects used to diagnose MELAS [60,61,110], and symptoms of MELAS are commonly considered to begin before the age of 40 [111]. Recent studies have revealed that the disease can develop after the age of 40 in a small number of patients [112], and adult-onset cases of MELAS have also been reported [62,113,114,115,116]. In cases of late onset of the disease, most patients have low levels of heteroplasmy or significant variations in various tissues. Similar to other mitochondrial diseases, the improvement of symptoms is rare, and in most cases, the symptoms deteriorate rapidly.
Maternally inherited diabetes and deafness (MIDD) is caused by the same mutation as MELAS (m.A3243G). Since both diseases show a broad spectrum of symptoms, it is hard to differentiate between them. However, MIDD is characterized by sensorineural hearing loss and decreased insulin secretion [117,118,119]. A case study conducted with 161 patients with MIDD revealed a correlation between the age of onset of diabetes and the heteroplasmy level of the mutated mtDNA (m.A3243G).
There remains a question regarding the different consequences of MELAS and MIDD despite being associated with the same mutation. Previous case studies have shown that MIDD patients are likely to exhibit a low heteroplasmy of mutated DNA [120,121,122]; however, apart from this finding, no other direct evidence has been found to provide a clear explanation.
Leigh syndrome seems to be caused by a lack of energy in nerve cells [123], resulting in the failure of neuronal morphogenesis and maturation [124,125]. Numerous pathogenic mutations, more than 75 in mtDNA and nuclear DNA, have been reported [126,127]. Of the various causes, a characteristic mutation in mtDNA that is typically observed in Leigh syndrome is the mutation in the MT-ATP6 coding gene, and numerous types of mutations have been identified as pathogenic [128,129]. The defect in MT-ATP6 leads to the dysfunction of mitochondrial complex 5, which converts ADP to ATP, eventually resulting in a lack of energy.
Typical symptoms of Leigh syndrome include motor development delay, muscle disorder, and cardiac dysfunction. Case studies have shown that Leigh syndrome also exhibits a wide spectrum of mitochondrial disease-associated symptoms [130]. Although one of the main symptoms of Leigh syndrome is delayed development, some case studies have reported patients with a late onset of the first symptom [74,131,132].
Pearson syndrome, Kearns–Sayre syndrome (KSS), and chronic progressive external ophthalmoplegia (CPEO) are mtDNA deletion-induced mitochondrial diseases. Unlike other mitochondrial diseases, which show an increased distribution of mutated mtDNA in offspring, mtDNA deletion-related diseases appear to have less dependency on maternal inheritance [133,134,135]. Yet, the dynamics of mtDNA deletions are not fully understood, and there are potential hypotheses and supportive studies on the mechanisms [136,137].
The positions of mtDNA deletions are random, and the deletions are usually large, leading to overall mitochondrial dysfunction [133,138,139]. Three conditions share similar phenotypes, but in the case of Pearson’s syndrome, severe symptoms usually begin in infancy and causing death before the age of four [140]. Children who survive develop Kearns–Sayre syndrome (KSS) eventually experience a reduced quality of life and sudden death [141]. KSS and CPEO can be referred to as KSS minus or CPEO plus based on the severity of symptoms [142]. KSS also exhibits symptoms of progressive external ophthalmoplegia (PEO), but if a patient expresses isolated symptoms of PEO, they are referred to as CPEO [142]. Ultimately, since both diseases are associated with the same mtDNA deletion, a patient diagnosed with CPEO could experience a spectrum of symptoms in a multi-systemic disease and therefore become a patient with KSS [143,144].
Previously described mitochondrial diseases are mainly caused by mutations in mtDNA. There are also diseases that can result from both nuclear and mitochondrial mutations, such as Leigh syndrome and CPEO. Barth syndrome and Friedreich’s ataxia are mitochondrial diseases caused by mutations in nuclear DNA. Mutations in both cases do not disrupt the mitochondrial electron transport chain complexes, as they do not affect the composing subunits. Instead, these mutations affect proteins regulating the overall dynamics of mitochondria. Hence, they also result in the same dysfunctions observed in other mitochondrial diseases.
Due to the lack of treatments for mitochondrial diseases, the clinical approach in case studies typically involves a combination of known antioxidants, coenzyme Q10, and vitamins. Hence, there have been no significant improvements in the severe symptoms, except in the specific cases of LHON, which predominantly exhibits symptoms related to optic neuropathy, and other mitochondrial diseases exhibiting similar symptoms in the spectrum of encephalomyopathy. Therefore, a single treatment can potentially be applicable to various diseases if the restoration of disordered mitochondria can be demonstrated.
4. Clinical Approach with Treatments Involving Chemical Compounds
Intracellular interactions of disease-targeting molecules often affect various cellular mechanisms in addition to their primary target pathway. As a result, diverse clinical approaches can be conducted using the same treatment. This review specifically focuses on treatments aimed at rescuing dysfunctional mitochondria that are currently undergoing trials (Table 2, Figure 2).
Until now, the most widely used chemical compound in diseases caused by mitochondrial dysfunction has been coenzyme Q10. Coenzyme Q10 is a self-generated resource and also an FDA-approved dietary supplement [177,178], which is well-known for its role as a powerful antioxidant in cells [146,147]. Self-generated coenzyme Q10 serves as a diffusible electron carrier in the mitochondria. Based on the known functions of coenzyme Q10, various research studies have analyzed and reported the potential of coenzyme Q10 for therapeutic application in mitochondrial diseases [179,180]. A phase 3 clinical trial for mitochondrial disease was conducted and completed in 2013 (NCT number, NCT00432744). However, coenzyme Q10 still lacks evidence of significant results in restoring mitochondrial dysfunction, and its therapeutic effect remains elusive. In a phase 3 clinical trial for Parkinson’s disease, which can also be regarded as a pathology related to mitochondrial dysfunction [181], no beneficial evidence was found for rescuing defective mitochondria. Due to its limited success in clinical approaches for diseases, coenzyme Q10 is not yet FDA-approved for medical treatment of disease [178,182,183]. Although coenzyme Q10 (CoQ10) cannot be used to treat any medical condition, there is no issue regarding its use as a nutrient supplement. Therefore, it is often used as part of a cocktail of nutritional supplements, mixed with other vitamins for mitochondrial diseases.
Based on the partially beneficial effects of coenzyme Q10, related chemical compounds are being developed for treatments. Idebenone is the most recognized drug for mitochondrial diseases. Idebenone is a short-chain hydrosoluble quinone [184], which overcomes the disadvantage of insoluble coenzyme Q10 and has demonstrated strong antioxidant activity. It also functions as an electron carrier in the mitochondrial electron transfer [148,149,150,151,183]. Further studies have found that Idebenone can overcome complex 1 deficiency in patients with LHON by directly transferring electrons to the complex 3, bypassing complex 1. This restores the function of the electron transfer chain and normalizes the production of cellular energy [148,149,150,151]. Considering that almost all mitochondrial diseases have defects in complex 1 or the overall electron transfer chain, Idebenone could be used for the direct treatment of complex 1 defect, and it also shows promise for treating electron transport chain dysfunction. Clinical studies for various mitochondrial diseases are currently ongoing [185]. For the medical treatment of LHON patients, a phase 4 clinical trial has been completed (NCT02774005) and Raxone (Idebenone) has been approved by the European Medicines Agency (EMA) for use with LHON patients (product number for EMA, EMEA/H/C/003834). However, it has not received approval from the FDA.
Vatiquinone is a quinone-based molecule that shares structural similarities with coenzyme Q10 and Idebenone but exhibits a higher level of protectant activity against oxidative stress [186]. Further studies have revealed that Vatiquinone modulates the inflammation process and depletes glutathione (GSH) at the cellular level by acting as an inhibitor of 15-lipoxygenase (15-LO) [152]. The reduction in ROS activity has also been demonstrated in human samples from patients with Leigh syndrome and healthy subjects. Vatiquinone increased GSH levels and decreased oxidized GSH levels, leading to a significant reduction in ROS levels [153,154,187].
With promising results in the performance of defected mitochondria, clinical trials have progressed. Several supporting results were found in a trial of patients considered to be within 90 days of end-of-life [188]. Visual improvement was confirmed in a young patient with Leigh syndrome [189]. Furthermore, a phase 2/3 clinical trial for mitochondrial disease with refractory epilepsy was conducted (NCT04378075), but it was announced to have failed to achieve its primary endpoint of reducing observable motor seizures. However, phase 3 clinical trials for the safety study of other symptoms are currently ongoing. There is a trial underway with Vatiquinone-exposed patients for the treatment of Friedreich ataxia (NCT05515536) and another trial for the broad aspects of mitochondrial diseases (NCT05218655).
NAD+ is an important factor in the energy production by the mitochondria. The study of NAD+ precursors has shown potential in enhancing mitochondrial functions. In addition, a decrease in NAD+ levels is observed in mitochondrial diseases, which is caused by deficiencies in the mitochondrial electron transfer chain. This decrease in NAD+ causes a disruption in redox homeostasis and energy metabolism, ultimately affecting various signaling pathways in cells [156,190]. Various studies have shown that an increase in intracellular NAD+ levels can lead to an improvement in mitochondrial number and oxidative capacities [156]. Nicotinamide riboside (NR), a precursor of NAD+, has been studied for its potential to enhance mitochondrial function. It was expected to improve adiposity but failed to do so [191,192]. However, a few short-term follow-up studies reported the possibility of NR supplementation leading to physical performance improvements and the alleviation of minor symptoms related to circulatory system [191,192]. NR research in mice has focused on its effects on mitochondria, showing that it increases mitochondrial function, extends the lifespan of mice, and helps retain neuropathy [155,157,158]. In a human trial, the number of mitochondria increased, and the broad upregulation of NAD+ metabolism resulted in various enhancements of disease symptoms [193]. Currently, a phase 2 clinical trial is ongoing for patients with mitochondrial myopathy disorders.
KL1333 is a compound that acts as a NAD+ precursor through interaction with NAD(P)H quinone oxidoreductase 1 (NQO1), which is a NADH-to-NAD+ conversion enzyme [159]. It increases NAD+ levels and leads to enhancements. Currently, a phase 2 clinical study for mitochondrial disease is in progress (NCT05650229).
Peroxisome proliferator-activated receptors (PPARs) are transcription factors located in the nucleus that modulate several pathways through gene expression. PPARδ is a subtype of PPARs that is known to be highly expressed in skeletal muscle cells compared to other PPAR subtypes. It has a preference for increasing fatty acid oxidation and enhancing mitochondrial biogenesis [161,162,163,164,194]. Thus, in cases of mitochondrial disease where symptoms are observed in skeletal muscle, the use of PPARδ agonists could be a therapeutic option, which may increase bioenergetics in skeletal muscle. For the primary mitochondrial myopathy, which exhibits mitochondrial dysfunction-related symptoms especially in skeletal muscles, bocidelpar (ASP0367, clinical trial phase 2/3, NCT04641962) and mavodelpar (REN001, clinical trial phase 2, NCT04535609) are currently undergoing clinical trials as PPARδ agonists.
Elamipretide (SBT-272) has a distinct activity in restoring defective mitochondria compared to other chemical compounds described previously. The target of Elamipretide is the cardiolipin of the mitochondrial inner membrane [165], which has a role in maintaining the architecture and morphology of the mitochondria [166]. The proper structure of mitochondria is important for the proper functioning of mitochondrial proteins. Therefore, a defect in cardiolipin could lead to the impairment of mitochondrial structure, resulting in neural diseases [195,196,197]. Few studies have reported evidence that the recovery of cardiolipin levels could improve neuropathy [195,197], and more beneficial effects for mitochondrial disorders and heart failure have been observed [198,199,200].
Barth syndrome is a mitochondrial disease known to be caused by a defect in the Tafazzin protein, which further leads to cardiolipin abnormality [201]. In a preclinical study of Elamipretide, the network of mitochondria cristae was remedied, resulting in the improvement of bioenergetics dysfunction in rats [202]. The phase 3 clinical trial of Elamipretide in Barth syndrome (NCT03098797) has been completed, and another approach for recovering mitochondrial dysfunction is currently underway for primary mitochondrial myopathy, which is now in phase 3 of clinical trials (NCT05162768).
The disorder of cells caused by an increase in ROS is a direct result of mitochondrial defects. Sonlicromanol (KH176) is a compound that reduces ROS levels. It is a derivative of Trolox (water soluble Vitamin E) and possesses enhanced antioxidant properties [203,204]. Sonlicromanol mediates redox biology through its interaction with one of the major cellular signaling systems involved in antioxidant and redox processes, namely the thioredoxin system [160].
The successful rescue of a neural network within a high heteroplasmy load has been shown using patient-induced stem cells by modulating the neuronal transcriptome [205]. A clinical trial was conducted to rescue MELAS in a patient with the m.A3243G mutation (NCT04165239), and phase 2 of the trial has been completed.
Omaveloxolone (SKYCLARYS) is the only treatment with FDA approval for use in patients with mitochondrial disease (Friedreich’s ataxia). Friedreich’s ataxia is a disease characterized by defective Frataxin expression caused by a pathogenic repeat in the Frataxin coding gene, resulting in iron accumulation in mitochondria and increased oxidative stress in cells [167,168]. Omaveloxolone, which is a synthetic derivative of oleanolic acid, binds to Kelch-like ECH-associated protein 1 (Keap1), preventing the degradation of nuclear factor erythroid-2-related factor 2 (Nrf2). Nrf2 is a transcription factor that mediates antioxidant gene expression, represses pro-inflammatory gene expression, and enhances mitochondrial biogenesis. Thus, the application of Omaveloxolone restores cellular defects by protecting Nrf2. Omaveloxolone has shown a restoring effect on the disorder in electron transfer chain complex 1, rescuing mitochondrial functions [206,207]
Omaveloxolone is currently being used to treat patients with Friedreich’s ataxia. The FDA’s approval of Omaveloxolone may seem to indicate a greater pharmacological efficacy compared to other drugs currently under development for mitochondrial diseases. Even Raxone (Idebenone), which has demonstrated therapeutic effects for LHON disease in numerous case studies, has not yet received FDA approval. While Omaveloxolone is the first FDA-approved treatment for mitochondrial disease, there are conflicting opinions on its clinical effects, and the strength of the supporting efficacy data is still debatable [208].
Due to the approaches of drug treatments for mitochondrial diseases, most of the clinical effects occur at the level of defected pathways and do not affect DNA mutations, which are the original cause. Therefore, efficacy can vary among individuals, and the application of treatment needs to be continued throughout a lifetime.
5. Clinical Approach Using Non-Chemical Treatments
In terms of non-chemical treatments for mitochondrial diseases, there is a tendency to address the pathology by transferring healthy mitochondria or normal mitochondrial gene into cells with dysfunctional mitochondria, rather than targeting mitochondrial pathways (Table 3, Figure 2).
In the field of gene therapy, the adeno-associated virus (AAV) is known as a vehicle that can transduce packaged DNA (~4.7 kb) and enable allotropic expression in organisms [170,209,210,211].
A clinical approach for LHON patients, who have dysfunctional MT-ND4 in complex 1, involved a pre-clinical study using allotropic expression of normal MT-ND4 through mRNA delivery. A rodent model of LHON showed enhanced visual deficits, and the rescue of ATP synthesis was observed in patient-derived fibroblasts [170,171,172]. Subsequently, gene delivery using AAV was performed to achieve similar results through allotropic expression, resulting in certain improvements in a mouse model of LHON [171,212].
In the history of LUMEVOQ for LHON (rAAV2/2-ND4, GS010), a preclinical study was conducted using recombinant AAV serotype 2, which is commonly employed for transducing to retinal ganglion cell nuclei and does not cause retinal injury [172]. Application in human patients was carried out through the intravitreal injection (IVT) of human wild-type ND4-packaged rAAV2. Currently, a phase 3 clinical trial is underway for LHON patients with the m.G11778A mutation (NCT03293524), and some of the preliminary results are already providing evidence of improved visual acuity [213]. Additionally, evidence of contralateral improvement was found in unilateral treatment. Accordingly, viral gene transfer approaches are under research [214]. A group, using the same method of treatment for LHON, is currently in their clinical trial phase 2/3 (NR082, NCT03153293). Another group, using a slightly different approach with self-complementary AAV for packaging the vehicle [173], is currently conducting a phase 1 clinical trial (ScAAV2-P1ND4v2, NCT02161380).
Mitochondrial augmentation (MAT) is a therapeutic method that involves transferring normal exogenous mitochondria into cells with disordered mitochondria. Mitochondrial transfer between cells to restore respiration capability has been demonstrated by using mtDNA-mutated cells (A549) with hMSC or skin fibroblasts [215]. Further research found more evidence of mitochondrial transfer [216,217,218]. In addition, experiments have successfully restored mitochondrial defects via the ex vivo augmentation of patient-derived hematopoietic stem and progenitor cells (HSPCs) with normal donor exogenous mitochondria [174]. MAT treatment has also been conducted in human patients using patient-derived autologous CD34+ hematopoietic cells reinserted through intravenous infusion. Based on previous MAT treatments, a decrease in mtDNA heteroplasmy level was observed in the peripheral blood of patients (4/6), and some patients showed improvement in some parameters of physical examination [219].
MAT is a therapy that replaces disordered mitochondria in cells with normal mitochondria and is not limited to correcting specific pathogenic mutations. MAT (NCT03384420) for Pearson syndrome, which is caused by large and random mtDNA deletions, is currently undergoing phase 1/2 clinical trials.
Another therapy option for the direct replacement of defective mitochondria is the use of patient-autologous mesoderm-derived stem cells, known as mesoangioblasts (MAB).
In a previous study, it was observed that MAB has low heteroplasmy loads despite having high loads of mutated mtDNA in skeletal muscle cells [176]. Mutated mtDNA seems to show a tendency to decrease in cultured satellite cells [175,220,221]. It has the advantage that when it is intra-arterially administrated to patients systemically and fused with damaged muscles, it contributes to inducing muscle regeneration due to its high myogenic potential [222,223]. In the first phase of the clinical trial, treatment with autologous MAB was conducted accompanied by ex vivo cultured patient-derived MAB. Currently, it is in a phase 2 clinical trial for the treatment of mitochondrial myopathy in patients with the m.A3243G mutation (NCT05962333).
For the treatment of unborn children whose parents have mitochondrial disease, mitochondrial replacement techniques (MRT, not listed in the table) are promising procedures, which can be used to replace almost all disordered mitochondria with healthy ones in human oocytes or zygotes [224,225]. Except for the fertilization process, various types of MRTs involve replacing the maternal nuclear DNA with the nuclear DNA in the donor’s cells. Despite the successful results of MRT operations [16], there have been debatable outcomes, such as the reversion of mutated DNA [226,227] and other ethical concerns [228]. Ultimately, the FDA has announced that performing MRT is not permitted in the USA.
Although clinical approaches using non-chemical treatments demonstrate their effect through numerous case studies, restoring efficiency remains elusive. Nevertheless, their approach towards achieving permanent effects, as opposed to chemical treatments, holds promise for a potential cure for mitochondrial diseases.
6. Targeted Genome Editing
In the field of clinical approaches for genetic diseases caused by nuclear DNA, there are exceptional genetic editing tools with CRISPR-Cas-based technology, which can target any sequence using target-complementary guide RNAs. In research on CRISPR-Cas-based base editors, which can edit single pathogenic base pairs with Cas protein and connected deaminase proteins [229], clinical trials are currently underway (NCT05456880, clinical study phase 1/2; NCT05885464, also phase 1/2).
In the case of mutations in mtDNA, the CRISPR-Cas system appears to be ineffective due to the mitochondrial import dynamics that prevent the transport of the guide RNA [230]. As a result, the current methods for targeted base editing of mutated mtDNA rely on approaches used prior to the development of the CRISPR-Cas system.
The transcription activator-like effector nuclease (TALEN) uses an array of TALE proteins to target specific sequences. Each TALE protein can recognize three nucleotides based on its RVD motifs [231,232,233]. Since TALE is formed by proteins, it can be transported to mitochondria using mitochondrial targeting signal amino acid sequences. Previous methods focused on TALEN for cutting DNA. Base editing with TALE array was not conducted before the emergence of CRISPR-Cas-related base editors (Figure 3).
The first application of base editing in mitochondria was conducted using a combination of bacterial cytidine deaminase toxin and TALE array [17]. In the progress of producing a mitochondrial cytosine base editor, the DddA toxin was divided in half to eliminate toxicity and become active after recruitment at the target site. Additionally, an uracil glycosylase inhibitor (UGI) was used to inhibit the repair mechanism of cytosine deamination. These modified DddA toxins are referred to as DddA-derived cytosine base editors (DdCBEs). After successfully applying targeted base conversion in mitochondria using DdCBEs, an in vivo mouse model with an induced pathogenic mutation of mtDNA was generated [234].
Although it was expected to be designed for the C-to-T base editor, the preference for base conversion only occurs at the 5′ TC motif. To overcome this bias, an improved editor called DddA11 with an expanded target range was developed [235]. Further development was conducted to enhance efficiency using a nuclear export signal, taking advantage of its preference to localize in the mitochondria rather than the nucleus [236].
The mitochondrial base editor is commonly used to induce pathogenic mutations in rodents to establish a mouse model of mitochondrial disease with disease phenotypes [234,236,237,238]. The use of DdCBEs in an in vivo model is usually conducted through microinjection at the zygote stage but cannot effectively target all mitochondria, resulting in a heteroplasmic distribution of mutated mtDNA. Since a high mutation load is required for the expression of mitochondrial diseases, there is a possibility of creating a mouse model with higher heteroplasmy levels through the continuous breeding of mutant mice [239]. In terms of effectiveness, improvements should focus on enhancing the overall impact on intracellular mtDNA.
After the demonstration of target-specific C-to-T base editing in mitochondria, an A-to-G base editor was also engineered using bacterial tRNA adenosine deaminase (TadA), which has shown its ability to mediate A-to-G conversion with the CRISPR-Cas system [240,241]. The first mitochondrial TALE-based A-to-G base editor, TALE-linked deaminases (TALED), uses an improved version of TadA, called TadA8e for base editing [18,242]. Since TadA lacks the ability to create a DNA bubble needed for its activity, TALED uses DddAtox splits to prepare single strands for TadA deamination [18]. Toxicity was the reason for splitting DddAtox in half. A monomeric form of TALED was also produced by inactivating the full DddAtox, but this change did not increase base editing efficiency.
One of the latest tools, mitochondrial DNA base editors (mitoBEs) uses mitochondria-derived nickase protein to create a suitable DNA structure for deaminase activity [19]. Both C-to-T and A-to-G conversions have been demonstrated using the deaminase protein rAPOBEC1 (for C-to-T) [229] and the TadA8e-V106W variant.
Given that researchers are discovering various editors using the CRISPR-Cas system [243,244,245], the development of a base editor based on CRISPR-Cas for mtDNA seems imminent. Improvements in mitochondrial DNA editing systems may lead to promising treatments and a permanent cure for mitochondrial diseases. However, many obstacles remain on the path to developing mitochondrial base editors for human clinical treatment. Diverse types of mitochondrial base editors have shown inconsistent performances with target-dependent efficiencies [17,18,19,235], as well as bystander editing within the target window. Genome-wide off-target effects have been observed in human cells [246] and mouse embryos [247], which is a major concern as it could lead to unexpected disorders. In addition, without the cytosine base editor DdCBE, other base editors still lack studies on in vivo models for further research. Nevertheless, mitochondrial base editing is still expected to be an attractive treatment method as it can correct mutations that cause mitochondrial disease and restore normal function.
7. Conclusions
Patients diagnosed with mitochondrial diseases are extremely rare, making it difficult to conduct sufficient case studies. Additionally, individuals with mitochondrial disease exhibit fatal symptoms, which poses a challenge for researchers attempting to study them. Therefore, it is a challenge to overcome these obstacles and develop effective treatments for mitochondrial disease.
In the past, it was impossible to recognize the underlying causes of mitochondrial disease. As a result, the approaches used to resolve it were also ambiguous, such as advising mtDNA mutation-harboring patients to exercise more to regenerate muscles, without considering their intolerance to physical activities [248,249]. While symptoms with a small portion of mutant mtDNA or age-related gradual mitochondrial defection can be managed with more nutritious diets or exercise, these methods cannot replace medical treatments.
Today, there are no obstacles in identifying DNA mutations in patients to find the underlying causes of mitochondrial diseases. Research on the dynamics of mitochondria continues, which leads us to aim for more specific pathways. Pharmacological advances are also leading to the discovery of new treatments for rare diseases.
There are several ongoing research studies focused on rescuing dysfunctional mitochondria that are not fully described in this review. For example, restoring mtDNA depletion by treating with deoxypyrimidine (deoxycytidine and deoxythymidine) to bypass the pyrimidine salvage pathway of mitochondria [169,250] is one clinical approach being used in human trials (NCT04802707, phase 2). OMT-28, a mitochondrial rescue agent involving Omega-3 fatty acid-related activity, is also under investigation (NCT05972954, Phase 2).
Although various drugs are currently undergoing human trials, many of them still seem to be ambiguous in terms of their effects on mitochondrial diseases, due to their indirect activities on the main causes. Mitochondrial diseases are inherited and permanent, and the known drugs for mitochondrial diseases provide only temporary relief rather than a cure. Therefore, a further approach to treatment should focus on addressing the underlying causes permanently.
MAT, or treatments with autologous MABs, could be regarded as one of the approaches that are the closest to human treatments involving the permanent replacement of defected mitochondria. While these therapies demonstrated notable effects in restoring defective mitochondria-harboring cells, no sufficient changes in the mitochondrial population that could completely cure mitochondrial diseases have been reported. AAV-mediated gene therapy could also be a potential approach for the treatment of mitochondrial diseases. Since spinal muscular atrophy, a genetic disease caused by nuclear DNA, has an FDA-approved AAV-mediated treatment, Zolgensma (Onasemnogene abeparvovec; FDA submission tracking number: 125694), and developing mitochondrial gene therapies using AAV seems to be promising approach. Many of these types of non-chemical treatments have been developed; however, they still have limitations in that their approach is more targeted towards specific tissues with direct symptoms rather than the entire body’s mitochondria. Mutated mitochondria are typically spread throughout the entire body in an individual, rather than being tissue-specific. Therefore, even if mitochondrial populations in one tissue are restored, it might lead to subsequent disorders in other tissues. Considering this, further research is required.
Expressed symptoms of mitochondrial diseases usually result in fatal conditions. However, current treatments for mitochondrial diseases are still considered remarkable if they show any effects on disease or improve some parameters. Despite the rarity of patients expressing severe symptoms, disease-inducing pathogenic mutations can be inherited without symptoms by many individuals. Without analyzing mtDNA sequences, there is a possibility that anyone could also be an unaffected carrier of mitochondrial diseases. Therefore, more attention is needed in the research of mitochondrial diseases and the clinical approaches to them.
Author Contributions
Discussion and writing, S.H., S.K., K.K., and H.L.; supervision, K.K. and H.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2023-00261905, RS-2023-00220894, NRF-2023R1A2C2004222).
Acknowledgments
Figures were constructed using BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schapira, A.H. Mitochondrial disease. Lancet 2006, 368, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Heusch, G.; Schulz, R. Nuclear-encoded mitochondrial proteins and their role in cardioprotection. Biochim. Biophys. Acta 2011, 1813, 1286–1294. [Google Scholar] [CrossRef]
- Monaghan, R.M.; Whitmarsh, A.J. Mitochondrial Proteins Moonlighting in the Nucleus. Trends Biochem. Sci. 2015, 40, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Lionaki, E.; Gkikas, I.; Tavernarakis, N. Differential Protein Distribution between the Nucleus and Mitochondria: Implications in Aging. Front. Genet. 2016, 7, 162. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Khotina, V.A.; Vinokurov, A.Y.; Bagheri Ekta, M.; Sukhorukov, V.N.; Orekhov, A.N. Creation of Mitochondrial Disease Models Using Mitochondrial DNA Editing. Biomedicines 2023, 11, 532. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef]
- Wong, L.-J.C. Diagnostic challenges of mitochondrial DNA disorders. Mitochondrion 2007, 7, 45–52. [Google Scholar] [CrossRef]
- Yang, M.; Xu, L.; Xu, C.; Cui, Y.; Jiang, S.; Dong, J.; Liao, L. The Mutations and Clinical Variability in Maternally Inherited Diabetes and Deafness: An Analysis of 161 Patients. Front. Endocrinol. 2021, 12, 728043. [Google Scholar] [CrossRef]
- Ying, Y.; Liang, Y.; Luo, X.; Wei, M. Case Report: Clinical and Genetic Characteristics of Pearson Syndrome in a Chinese Boy and 139 Patients. Front. Genet. 2022, 13, 802402. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Wang, L.; Fan, Y.; Yuan, B.; Ramos-Mandujano, G.; Zhang, Y.; Alsolami, S.; Zhou, X.; Wang, J.; Shao, Y.; et al. Single-cell individual full-length mtDNA sequencing by iMiGseq uncovers unexpected heteroplasmy shifts in mtDNA editing. Nucleic Acids Res. 2023, 51, e48. [Google Scholar] [CrossRef]
- Thorburn, D.R. Mitochondrial disorders: Prevalence, myths and advances. J. Inherit. Metab. Dis. 2004, 27, 349–362. [Google Scholar] [CrossRef]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.S.; Belaramani, K.M.; Chan, C.K.; Chan, W.K.; Chan, W.L.; Chang, S.K.; Cheung, S.N.; Cheung, K.Y.; Cheung, Y.F.; Chong, S.J.; et al. Mitochondrial diseases in Hong Kong: Prevalence, clinical characteristics and genetic landscape. Orphanet J. Rare Dis. 2023, 18, 43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, H.; Luo, S.; Lu, Z.; Chavez-Badiola, A.; Liu, Z.; Yang, M.; Merhi, Z.; Silber, S.J.; Munne, S.; et al. Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod. Biomed. Online 2017, 34, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef]
- Cho, S.I.; Lee, S.; Mok, Y.G.; Lim, K.; Lee, J.; Lee, J.M.; Chung, E.; Kim, J.S. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell 2022, 185, 1764–1776.e12. [Google Scholar] [CrossRef]
- Yi, Z.; Zhang, X.; Tang, W.; Yu, Y.; Wei, X.; Zhang, X.; Wei, W. Strand-selective base editing of human mitochondrial DNA using mitoBEs. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef]
- Giorgio, V.; Petronilli, V.; Ghelli, A.; Carelli, V.; Rugolo, M.; Lenaz, G.; Bernardi, P. The effects of idebenone on mitochondrial bioenergetics. Biochim. Biophys. Acta 2012, 1817, 363–369. [Google Scholar] [CrossRef]
- Jaber, S.M.; Ge, S.X.; Milstein, J.L.; VanRyzin, J.W.; Waddell, J.; Polster, B.M. Idebenone Has Distinct Effects on Mitochondrial Respiration in Cortical Astrocytes Compared to Cortical Neurons Due to Differential NQO1 Activity. J. Neurosci. 2020, 40, 4609–4619. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Shen, W.; Yu, G.; Jia, H.; Li, X.; Feng, Z.; Wang, Y.; Weber, P.; Wertz, K.; Sharman, E.; et al. Hydroxytyrosol promotes mitochondrial biogenesis and mitochondrial function in 3T3-L1 adipocytes. J. Nutr. Biochem. 2010, 21, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Acuna, C.; Ferreira, J.; Speisky, H. Polyphenols and mitochondria: An update on their increasingly emerging ROS-scavenging independent actions. Arch. Biochem. Biophys. 2014, 559, 75–90. [Google Scholar] [CrossRef]
- Duan, J.; Li, Y.; Gao, H.; Yang, D.; He, X.; Fang, Y.; Zhou, G. Phenolic compound ellagic acid inhibits mitochondrial respiration and tumor growth in lung cancer. Food Funct. 2020, 11, 6332–6339. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Yu, M.; Wu, Y.; Xia, T.; Wang, L.; Song, K.; Zhang, C.; Lu, K.; Rahimnejad, S. Hydroxytyrosol Promotes the Mitochondrial Function through Activating Mitophagy. Antioxidants 2022, 11, 893. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Wang, J.; Li, Z.; Gao, L.; Qi, Y.; Zhu, H.; Qin, X. The regulation effect of AMPK in immune related diseases. Sci. China Life Sci. 2018, 61, 523–533. [Google Scholar] [CrossRef]
- Pokhrel, R.H.; Acharya, S.; Ahn, J.-H.; Gu, Y.; Pandit, M.; Kim, J.-O.; Park, Y.-Y.; Kang, B.; Ko, H.-J.; Chang, J.-H. AMPK promotes antitumor immunity by downregulating PD-1 in regulatory T cells via the HMGCR/p38 signaling pathway. Mol. Cancer 2021, 20, 133. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Z.; Hou, J.; Xiong, W.; Kim, H.; Chen, J.; Zheng, C.; Jiang, X.; Yoon, J.; Shen, J. Tumor Selective Metabolic Reprogramming as a Prospective PD-L1 Depression Strategy to Reactivate Immunotherapy. Adv. Mater. 2022, 34, e2206121. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, Z.; Hu, R.; Dong, M.; Zhou, X.; Ren, S.; Zhang, Y.; Chen, C.; Huang, R.; Zhu, M.; et al. Metabolic Intervention Liposome Boosted Lung Cancer Radio-Immunotherapy via Hypoxia Amelioration and PD-L1 Restraint. Adv. Sci. 2023, 10, e2207608. [Google Scholar] [CrossRef]
- Zhao, B.; Qiang, L.; Joseph, J.; Kalyanaraman, B.; Viollet, B.; He, Y.-Y. Mitochondrial dysfunction activates the AMPK signaling and autophagy to promote cell survival. Genes Dis. 2016, 3, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, A.I.; Ng, S.Y.; Mattina, S.R.; Ljubicic, V. AMPK is mitochondrial medicine for neuromuscular disorders. Trends Mol. Med. 2023, 29, 512–529. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Maraver, J.; Paz, M.V.; Cordero, M.D.; Bautista-Lorite, J.; Oropesa-Avila, M.; de la Mata, M.; Pavon, A.D.; de Lavera, I.; Alcocer-Gomez, E.; Galan, F.; et al. Critical role of AMP-activated protein kinase in the balance between mitophagy and mitochondrial biogenesis in MELAS disease. Biochim. Biophys. Acta 2015, 1852, 2535–2553. [Google Scholar] [CrossRef] [PubMed]
- Granatiero, V.; Giorgio, V.; Cali, T.; Patron, M.; Brini, M.; Bernardi, P.; Tiranti, V.; Zeviani, M.; Pallafacchina, G.; De Stefani, D.; et al. Reduced mitochondrial Ca(2+) transients stimulate autophagy in human fibroblasts carrying the 13514A>G mutation of the ND5 subunit of NADH dehydrogenase. Cell Death Differ. 2016, 23, 231–241. [Google Scholar] [CrossRef]
- Villanueva-Paz, M.; Povea-Cabello, S.; Villalon-Garcia, I.; Alvarez-Cordoba, M.; Suarez-Rivero, J.M.; Talaveron-Rey, M.; Jackson, S.; Falcon-Moya, R.; Rodriguez-Moreno, A.; Sanchez-Alcazar, J.A. Parkin-mediated mitophagy and autophagy flux disruption in cellular models of MERRF syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165726. [Google Scholar] [CrossRef]
- Shu, L.; Hu, C.; Xu, M.; Yu, J.; He, H.; Lin, J.; Sha, H.; Lu, B.; Engelender, S.; Guan, M.; et al. ATAD3B is a mitophagy receptor mediating clearance of oxidative stress-induced damaged mitochondrial DNA. EMBO J. 2021, 40, e106283. [Google Scholar] [CrossRef]
- Danese, A.; Patergnani, S.; Maresca, A.; Peron, C.; Raimondi, A.; Caporali, L.; Marchi, S.; La Morgia, C.; Del Dotto, V.; Zanna, C.; et al. Pathological mitophagy disrupts mitochondrial homeostasis in Leber’s hereditary optic neuropathy. Cell Rep. 2022, 40, 111124. [Google Scholar] [CrossRef]
- Zhang, J.; Merkle, H.; Hendrich, K.; Garwood, M.; From, A.H.; Ugurbil, K.; Bache, R.J. Bioenergetic abnormalities associated with severe left ventricular hypertrophy. J. Clin. Investig. 1993, 92, 993–1003. [Google Scholar] [CrossRef]
- Huss, J.M.; Kelly, D.P. Mitochondrial energy metabolism in heart failure: A question of balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, J.; Beard, D.A. Experimentally observed phenomena on cardiac energetics in heart failure emerge from simulations of cardiac metabolism. Proc. Natl. Acad. Sci. USA 2009, 106, 7143–7148. [Google Scholar] [CrossRef]
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Schautz, B.; Later, W.; Heymsfield, S.B.; Muller, M.J. Specific metabolic rates of major organs and tissues across adulthood: Evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr. 2010, 92, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. 2010, 5, 297–348. [Google Scholar] [CrossRef] [PubMed]
- Lotz, C.; Lin, A.J.; Black, C.M.; Zhang, J.; Lau, E.; Deng, N.; Wang, Y.; Zong, N.C.; Choi, J.H.; Xu, T.; et al. Characterization, design, and function of the mitochondrial proteome: From organs to organisms. J. Proteome Res. 2014, 13, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Thome, T.; Coleman, M.D.; Ryan, T.E. Mitochondrial Bioenergetic and Proteomic Phenotyping Reveals Organ-Specific Consequences of Chronic Kidney Disease in Mice. Cells 2021, 10, 3282. [Google Scholar] [CrossRef]
- Faria-Pereira, A.; Morais, V.A. Synapses: The Brain’s Energy-Demanding Sites. Int. J. Mol. Sci. 2022, 23, 3627. [Google Scholar] [CrossRef]
- Trigo, D.; Avelar, C.; Fernandes, M.; Sa, J.; da Cruz, E.S.O. Mitochondria, energy, and metabolism in neuronal health and disease. FEBS Lett. 2022, 596, 1095–1110. [Google Scholar] [CrossRef]
- Jackson, M.J.; Schaefer, J.A.; Johnson, M.A.; Morris, A.A.; Turnbull, D.M.; Bindoff, L.A. Presentation and clinical investigation of mitochondrial respiratory chain disease. A study of 51 patients. Brain 1995, 118 Pt 2, 339–357. [Google Scholar] [CrossRef]
- Koenig, M.K. Presentation and diagnosis of mitochondrial disorders in children. Pediatr. Neurol. 2008, 38, 305–313. [Google Scholar] [CrossRef]
- Miao, J.; Qian, Q.; Zand, L. Long-Standing Hypokalemia and Lactic Acidosis as the Primary Presentation of Mitochondrial Myopathy. Kidney Int. Rep. 2020, 5, 742–745. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Bernardino Gomes, T.M.; Ng, Y.S.; Pickett, S.J.; Turnbull, D.M.; Vincent, A.E. Mitochondrial DNA disorders: From pathogenic variants to preventing transmission. Human Mol. Genet. 2021, 30, R245–R253. [Google Scholar] [CrossRef] [PubMed]
- Chuenkongkaew, W.L.; Chinkulkitnivat, B.; Lertrit, P.; Chirapapaisan, N.; Kaewsutthi, S.; Suktitipat, B.; Mitrpant, C. Clinical expression and mitochondrial deoxyribonucleic acid study in twins with 14484 Leber’s hereditary optic neuropathy: A case report. World J. Clin. Cases 2022, 10, 6944–6953. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, S.; Hwang, J.-M. Visual Prognosis of Leber’s Hereditary Optic Neuropathy with T14484C Mitochondrial DNA Mutation in Koreans. J. Korean Ophthalmol. Soc. 2012, 53, 151. [Google Scholar] [CrossRef]
- Iorga, R.E.; Mihailovici, R.; Ozturk, M.R.; Costin, D. Leber’s hereditary optic neuropathy—Case report. Rom. J. Ophthalmol. 2018, 62, 64–71. [Google Scholar] [CrossRef]
- Miao, Q.M.; Cheng, Y.F.; Zheng, H.M.; Yuan, J.J.; Chen, C.Z. Photoreceptor changes in Leber hereditary optic neuropathy with m.G11778A mutation. Int. J. Ophthalmol. 2023, 16, 928–932. [Google Scholar] [CrossRef]
- Chandra, S.R.; Issac, T.G.; Gayathri, N.; Gupta, N.; Abbas, M.M. A typical case of myoclonic epilepsy with ragged red fibers (MERRF) and the lessons learned. J. Postgrad. Med. 2015, 61, 200–202. [Google Scholar] [CrossRef]
- Xu, H.L.; Lian, Y.J.; Chen, X. Brain atrophy in a patient with mitochondrial DNA G8363A mutation. Chin. Med. J. 2019, 132, 2141–2142. [Google Scholar] [CrossRef]
- Liu, X.Q.; Shen, S.Q.; Yang, G.C.; Liu, Q. Mitochondrial A3243G mutation causes mitochondrial encephalomyopathy in a Chinese patient: Case report. Medicine 2019, 98, e15534. [Google Scholar] [CrossRef]
- Windpessl, M.; Muller, P.; Wallner, M. Truth is a daughter of time: A case of MELAS diagnosed 25 years after initial manifestation. Oxf. Med. Case Rep. 2014, 2014, 24–25. [Google Scholar] [CrossRef]
- Martens, E.; Demeestere, J.; Verhaaren, B. Microhemorrhages in MELAS Lesions: A Case Report. J. Belg. Soc. Radiol. 2022, 106, 88. [Google Scholar] [CrossRef] [PubMed]
- Diao, S.P.; Chen, S.F.; Liu, A.Q.; Zhou, Z.H.; Peng, Z.X.; Hong, M.F. Elderly onset of MELAS in a male: A case report. Front. Neurol. 2022, 13, 1018529. [Google Scholar] [CrossRef] [PubMed]
- Kanaumi, T.; Hirose, S.; Goto, Y.; Naitou, E.; Mitsudome, A. An infant with a mitochondrial A3243G mutation demonstrating the MELAS phenotype. Pediatr. Neurol. 2006, 34, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Dubey, S.; Bhuin, S.; Lahiri, D.; Ray, B.K.; Finsterer, J. MELAS with multiple stroke-like episodes due to the variant m.13513G>A in MT-ND5. Clin. Case Rep. 2022, 10, e05361. [Google Scholar] [CrossRef]
- Tanaka, K.; Ueno, T.; Yoshida, M.; Shimizu, Y.; Ogawa, T.; Nishisaka, T.; Kurashige, T.; Masaki, T. Chronic kidney disease caused by maternally inherited diabetes and deafness: A case report. CEN Case Rep. 2021, 10, 220–225. [Google Scholar] [CrossRef]
- Li, M.C.H.; Symmonds, M.; Pretorius, P.M.; Sheerin, F.; Nithi, K.; Hofer, M.; Poulton, J.; Sen, A. Very late-onset mitochondrial cytopathy featuring epilepsia partialis continua and bilateral deafness: A case report. Seizure 2020, 76, 153–155. [Google Scholar] [CrossRef]
- Shoeleh, C.; Donato, U.M.; Galligan, A.; Vitko, J. A Case Report on Pearson Syndrome With Emphasis on Genetic Screening in Patients Presenting With Sideroblastic Anemia and Lactic Acidosis. Cureus 2023, 15, e33963. [Google Scholar] [CrossRef]
- Zhu, Q.; Chen, C.; Yao, J. Kearns-Sayre syndrome with a novel large-scale deletion: A case report. BMC Ophthalmol. 2022, 22, 35. [Google Scholar] [CrossRef]
- Grigalioniene, K.; Burnyte, B.; Balkeliene, D.; Ambrozaityte, L.; Utkus, A. Kearns-Sayre syndrome case. Novel 5,9 kb mtDNA deletion. Mol. Genet. Genom. Med. 2023, 11, e2059. [Google Scholar] [CrossRef]
- Sano, M.; Shimada, T.; Sakurai, A.; Goto, Y.-i.; Tsunemi, T.; Hattori, N. A case of chronic progressive external ophthalmoplegia presenting with central neurogenic hyperventilation. Brain Disord. 2022, 8, 100057. [Google Scholar] [CrossRef]
- Sirbu, C.A.; Sîrbu, O.-M.; Sandu, A.-M. Chronic progressive external ophtalmoplegia (CPEO)—Case presentation. Rom. J. Neurol./Rev. Romana Neurol. 2014, 13, 150–152. [Google Scholar] [CrossRef]
- Eliyan, Y.; Rezania, K.; Gomez, C.M.; Seibert, K. Pontine stroke in a patient with Chronic Progressive External Ophthalmoplegia (CPEO): A case report. BMC Neurol. 2023, 23, 231. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gomez, C.; Camara, Y.; Hirano, M.; Marti, R. 232nd ENMC international workshop: Recommendations for treatment of mitochondrial DNA maintenance disorders. 16–18 June 2017, Heemskerk, The Netherlands. Neuromuscul. Disord. 2022, 32, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.M.; Xin, C.J.; Wang, G.L.; Wu, X.M. Late-onset Leigh syndrome without delayed development in China: A case report. World J. Clin. Cases 2021, 9, 7133–7138. [Google Scholar] [CrossRef] [PubMed]
- Maruo, Y.; Ueda, Y.; Murayama, K.; Takeda, A. A case report of Leigh syndrome diagnosed by endomyocardial biopsy. Eur. Heart J. Case Rep. 2021, 5, ytaa582. [Google Scholar] [CrossRef]
- Legro, N.R.; Kumar, A.; Aliu, E. Case report of atypical Leigh syndrome in an adolescent male with novel biallelic variants in NDUFAF5 and review of the natural history of NDUFAF5-related disorders. Am. J. Med. Genet. A 2022, 188, 896–899. [Google Scholar] [CrossRef]
- Takeda, A.; Ueki, M.; Abe, J.; Maeta, K.; Horiguchi, T.; Yamazawa, H.; Izumi, G.; Chida-Nagai, A.; Sasaki, D.; Tsujioka, T.; et al. A case of infantile Barth syndrome with severe heart failure: Importance of splicing variants in the TAZ gene. Mol. Genet. Genom. Med. 2023, 11, e2190. [Google Scholar] [CrossRef]
- Amini, O.; Lakziyan, R.; Abavisani, M.; Sarchahi, Z. The cardiomyopathy of Friedreich’s ataxia common in a family: A case report. Ann. Med. Surg. 2021, 66, 102408. [Google Scholar] [CrossRef] [PubMed]
- Damasio, J.; Sardoeira, A.; Araujo, M.; Carvalho, I.; Sequeiros, J.; Barros, J. Rare occurrence of severe blindness and deafness in Friedreich ataxia: A case report. Cerebellum Ataxias 2021, 8, 17. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Chinnery, P.F. Leber Hereditary Optic Neuropathy; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews®: Seattle, WA, USA, 1993. [Google Scholar]
- Finsterer, J.; Zarrouk-Mahjoub, S. Leber’s hereditary optic neuropathy is multiorgan not mono-organ. Clin. Ophthalmol. 2016, 10, 2187–2190. [Google Scholar] [CrossRef]
- Esmaeil, A.; Ali, A.; Behbehani, R. Leber’s hereditary optic neuropathy: Update on current diagnosis and treatment. Front. Ophthalmol. 2023, 2, 1077395. [Google Scholar] [CrossRef]
- Shemesh, A.; Sood, G.; Margolin, E. Leber Hereditary Optic Neuropathy (LHON); StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Wong, A.; Cortopassi, G. 14—Mitochondrial Genetic Diseases. In Neurobiology of Disease; Gilman, S., Ed.; Academic Press: Burlington, MA, USA, 2007; pp. 157–161. [Google Scholar]
- Jacobi, F.K.; Leo-Kottler, B.; Mittelviefhaus, K.; Zrenner, E.; Meyer, J.; Pusch, C.M.; Wissinger, B. Segregation patterns and heteroplasmy prevalence in Leber’s hereditary optic neuropathy. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1208–1214. [Google Scholar]
- Dai, Y.; Wang, C.; Nie, Z.; Han, J.; Chen, T.; Zhao, X.; Ai, C.; Ji, Y.; Gao, T.; Jiang, P. Mutation analysis of Leber’s hereditary optic neuropathy using a multi-gene panel. Biomed. Rep. 2018, 8, 51–58. [Google Scholar] [CrossRef]
- Bianco, A.; Valletti, A.; Longo, G.; Bisceglia, L.; Montoya, J.; Emperador, S.; Guerriero, S.; Petruzzella, V. Mitochondrial DNA copy number in affected and unaffected LHON mutation carriers. BMC Res. Notes 2018, 11, 911. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska-Nowak, A.; Krawczynski, M.R.; Kosior-Jarecka, E.; Ambroziak, A.M.; Korwin, M.; Oldak, M.; Tonska, K.; Bartnik, E. Mitochondrial genome variation in male LHON patients with the m.11778G > A mutation. Metab. Brain Dis. 2020, 35, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Iommarini, L.; Giordano, L.; Maresca, A.; Pisano, A.; Valentino, M.L.; Caporali, L.; Liguori, R.; Deceglie, S.; Roberti, M.; et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain 2014, 137, 335–353. [Google Scholar] [CrossRef]
- Giordano, L.; Deceglie, S.; d’Adamo, P.; Valentino, M.L.; La Morgia, C.; Fracasso, F.; Roberti, M.; Cappellari, M.; Petrosillo, G.; Ciaravolo, S.; et al. Cigarette toxicity triggers Leber’s hereditary optic neuropathy by affecting mtDNA copy number, oxidative phosphorylation and ROS detoxification pathways. Cell Death Dis. 2015, 6, e2021. [Google Scholar] [CrossRef]
- Dombi, E.; Diot, A.; Morten, K.; Carver, J.; Lodge, T.; Fratter, C.; Ng, Y.S.; Liao, C.; Muir, R.; Blakely, E.L.; et al. The m.13051G>A mitochondrial DNA mutation results in variable neurology and activated mitophagy. Neurology 2016, 86, 1921–1923. [Google Scholar] [CrossRef]
- Moura-Coelho, N.; Pinto Proenca, R.; Tavares Ferreira, J.; Cunha, J.P. Late-onset Leber’s hereditary optic neuropathy: The role of environmental factors in hereditary diseases. BMJ Case Rep. 2019, 12, e227977. [Google Scholar] [CrossRef]
- Finsterer, J. More likely than through head trauma: Is LHON triggered by mitochondrion-toxic drugs or oxidative stress. Doc. Ophthalmol. 2021, 142, 395–396. [Google Scholar] [CrossRef]
- Vela-Sebastian, A.; Lopez-Gallardo, E.; Emperador, S.; Hernandez-Ainsa, C.; Pacheu-Grau, D.; Blanco, I.; Ros, A.; Pascual-Benito, E.; Rabaneda-Lombarte, N.; Presas-Rodriguez, S.; et al. Toxic and nutritional factors trigger Leber hereditary optic neuropathy due to a mitochondrial tRNA mutation. Clin. Genet. 2022, 102, 339–344. [Google Scholar] [CrossRef]
- Riordan-Eva, P.; Harding, A.E. Leber’s hereditary optic neuropathy: The clinical relevance of different mitochondrial DNA mutations. J. Med. Genet. 1995, 32, 81–87. [Google Scholar] [CrossRef]
- Hameed, S.; Tadi, P. Myoclonic Epilepsy and Ragged Red Fibers; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Hammans, S.R.; Sweeney, M.G.; Brockington, M.; Lennox, G.G.; Lawton, N.F.; Kennedy, C.R.; Morgan-Hughes, J.A.; Harding, A.E. The mitochondrial DNA transfer RNA(Lys)A-->G(8344) mutation and the syndrome of myoclonic epilepsy with ragged red fibres (MERRF). Relationship of clinical phenotype to proportion of mutant mitochondrial DNA. Brain 1993, 116 Pt 3, 617–632. [Google Scholar] [CrossRef] [PubMed]
- Velez-Bartolomei, F.; Lee, C.; Enns, G. Merrf; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews®: Seattle, WA, USA, 1993. [Google Scholar]
- Ripolone, M.; Zanotti, S.; Napoli, L.; Ronchi, D.; Ciscato, P.; Comi, G.P.; Moggio, M.; Sciacco, M. MERRF Mutation A8344G in a Four-Generation Family without Central Nervous System Involvement: Clinical and Molecular Characterization. J. Pers. Med. 2023, 13, 147. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-T.; Tay, H.-Y.; Yang, J.-T.; Liao, H.-H.; Ma, Y.-S.; Wei, Y.-H. Mitochondrial impairment and synaptic dysfunction are associated with neurological defects in iPSCs-derived cortical neurons of MERRF patients. J. Biomed. Sci. 2023, 30, 70. [Google Scholar] [CrossRef]
- Goto, Y.; Tsugane, K.; Tanabe, Y.; Nonaka, I.; Horai, S. A new point mutation at nucleotide pair 3291 of the mitochondrial tRNA(Leu(UUR)) gene in a patient with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Biochem. Biophys. Res. Commun. 1994, 202, 1624–1630. [Google Scholar] [CrossRef]
- Nishigaki, Y.; Tadesse, S.; Bonilla, E.; Shungu, D.; Hersh, S.; Keats, B.J.; Berlin, C.I.; Goldberg, M.F.; Vockley, J.; DiMauro, S.; et al. A novel mitochondrial tRNA(Leu(UUR)) mutation in a patient with features of MERRF and Kearns-Sayre syndrome. Neuromuscul. Disord. 2003, 13, 334–340. [Google Scholar] [CrossRef]
- Chomyn, A.; Meola, G.; Bresolin, N.; Lai, S.T.; Scarlato, G.; Attardi, G. In vitro genetic transfer of protein synthesis and respiration defects to mitochondrial DNA-less cells with myopathy-patient mitochondria. Mol. Cell. Biol. 1991, 11, 2236–2244. [Google Scholar] [CrossRef]
- Chomyn, A.; Lai, S.T.; Shakeley, R.; Bresolin, N.; Scarlato, G.; Attardi, G. Platelet-mediated transformation of mtDNA-less human cells: Analysis of phenotypic variability among clones from normal individuals—And complementation behavior of the tRNALys mutation causing myoclonic epilepsy and ragged red fibers. Am. J. Hum. Genet. 1994, 54, 966–974. [Google Scholar]
- Yoneda, M.; Chomyn, A.; Martinuzzi, A.; Hurko, O.; Attardi, G. Marked replicative advantage of human mtDNA carrying a point mutation that causes the MELAS encephalomyopathy. Proc. Natl. Acad. Sci. USA 1992, 89, 11164–11168. [Google Scholar] [CrossRef] [PubMed]
- Chomyn, A.; Enriquez, J.A.; Micol, V.; Fernandez-Silva, P.; Attardi, G. The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J. Biol. Chem. 2000, 275, 19198–19209. [Google Scholar] [CrossRef] [PubMed]
- Janssen, G.M.; Hensbergen, P.J.; van Bussel, F.J.; Balog, C.I.; Maassen, J.A.; Deelder, A.M.; Raap, A.K. The A3243G tRNALeu(UUR) mutation induces mitochondrial dysfunction and variable disease expression without dominant negative acting translational defects in complex IV subunits at UUR codons. Hum. Mol. Genet. 2007, 16, 2472–2481. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Guan, M.X. Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol. Cell. Biol. 2010, 30, 2147–2154. [Google Scholar] [CrossRef]
- Goldstein, A.; Servidei, S. Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes (MELAS). In Diagnosis and Management of Mitochondrial Disorders; Mancuso, M., Klopstock, T., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 81–100. [Google Scholar]
- Hirano, M.; Ricci, E.; Koenigsberger, M.R.; Defendini, R.; Pavlakis, S.G.; DeVivo, D.C.; DiMauro, S.; Rowland, L.P. Melas: An original case and clinical criteria for diagnosis. Neuromuscul. Disord. 1992, 2, 125–135. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef]
- Aurangzeb, S.; Vale, T.; Tofaris, G.; Poulton, J.; Turner, M.R. Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) in the older adult. Pract. Neurol. 2014, 14, 432–436. [Google Scholar] [CrossRef]
- Sunde, K.; Blackburn, P.R.; Cheema, A.; Gass, J.; Jackson, J.; Macklin, S.; Atwal, P.S. Case report: 5 year follow-up of adult late-onset mitochondrial encephalomyopathy with lactic acid and stroke-like episodes (MELAS). Mol. Genet. Metab. Rep. 2016, 9, 94–97. [Google Scholar] [CrossRef]
- Mukai, M.; Nagata, E.; Mizuma, A.; Yamano, M.; Sugaya, K.; Nishino, I.; Goto, Y.I.; Takizawa, S. Adult-onset Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke (MELAS)-like Encephalopathy Diagnosed Based on the Complete Sequencing of Mitochondrial DNA Extracted from Biopsied Muscle without any Myopathic Changes. Intern. Med. 2017, 56, 95–99. [Google Scholar] [CrossRef]
- Yang, F.; Peng, S.; Peng, Q. Diagnosis of adult-onset MELAS with suspected recurrent strokes: A case report. Exp. Ther. Med. 2022, 24, 466. [Google Scholar] [CrossRef]
- Gebhart, S.S.; Shoffner, J.M.; Koontz, D.; Kaufman, A.; Wallace, D. Insulin resistance associated with maternally inherited diabetes and deafness. Metabolism 1996, 45, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Guillausseau, P.J.; Massin, P.; Dubois-LaForgue, D.; Timsit, J.; Virally, M.; Gin, H.; Bertin, E.; Blickle, J.F.; Bouhanick, B.; Cahen, J.; et al. Maternally inherited diabetes and deafness: A multicenter study. Ann. Intern. Med. 2001, 134, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Oka, Y.; Kadowaki, T.; Kanatsuka, A.; Kuzuya, T.; Kobayashi, M.; Sanke, T.; Seino, Y.; Nanjo, K. Clinical features of diabetes mellitus with the mitochondrial DNA 3243 (A-G) mutation in Japanese: Maternal inheritance and mitochondria-related complications. Diabetes Res. Clin. Pract. 2003, 59, 207–217. [Google Scholar] [CrossRef] [PubMed]
- de Wit, H.M.; Westeneng, H.J.; van Engelen, B.G.; Mudde, A.H. MIDD or MELAS: That’s not the question MIDD evolving into MELAS: A severe phenotype of the m.3243A>G mutation due to paternal co-inheritance of type 2 diabetes and a high heteroplasmy level. Neth. J. Med. 2012, 70, 460–462. [Google Scholar] [PubMed]
- Goel, H.; Szczepanczyk, K.; Mirza, F.S. Late-Onset Melas with Midd: An Uncommon Age of Presentation. AACE Clin. Case Rep. 2018, 4, 228–231. [Google Scholar] [CrossRef]
- Bukhari, K.; Pennant, M. Mitochondrial Diabetes: The Clinical Spectrum of MIDD and MELAS in Association With the A3243G Mutation. J. Endocr. Soc. 2021, 5, A392–A393. [Google Scholar] [CrossRef]
- Rahman, S.; Blok, R.B.; Dahl, H.H.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar] [CrossRef]
- Iwata, R.; Casimir, P.; Vanderhaeghen, P. Mitochondrial dynamics in postmitotic cells regulate neurogenesis. Science 2020, 369, 858–862. [Google Scholar] [CrossRef]
- Inak, G.; Rybak-Wolf, A.; Lisowski, P.; Pentimalli, T.M.; Juttner, R.; Glazar, P.; Uppal, K.; Bottani, E.; Brunetti, D.; Secker, C.; et al. Defective metabolic programming impairs early neuronal morphogenesis in neural cultures and an organoid model of Leigh syndrome. Nat. Commun. 2021, 12, 1929. [Google Scholar] [CrossRef]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef]
- Lee, J.S.; Yoo, T.; Lee, M.; Lee, Y.; Jeon, E.; Kim, S.Y.; Lim, B.C.; Kim, K.J.; Choi, M.; Chae, J.H. Genetic heterogeneity in Leigh syndrome: Highlighting treatable and novel genetic causes. Clin. Genet. 2020, 97, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Sofou, K.; de Coo, I.F.M.; Ostergaard, E.; Isohanni, P.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; Lonnqvist, T.; Bindoff, L.A.; et al. Phenotype-genotype correlations in Leigh syndrome: New insights from a multicentre study of 96 patients. J. Med. Genet. 2018, 55, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Ganetzky, R.D.; Stendel, C.; McCormick, E.M.; Zolkipli-Cunningham, Z.; Goldstein, A.C.; Klopstock, T.; Falk, M.J. MT-ATP6 mitochondrial disease variants: Phenotypic and biochemical features analysis in 218 published cases and cohort of 14 new cases. Hum. Mutat. 2019, 40, 499–515. [Google Scholar] [CrossRef]
- Baertling, F.; Rodenburg, R.J.; Schaper, J.; Smeitink, J.A.; Koopman, W.J.; Mayatepek, E.; Morava, E.; Distelmaier, F. A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry 2014, 85, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Sakushima, K.; Tsuji-Akimoto, S.; Niino, M.; Saitoh, S.; Yabe, I.; Sasaki, H. Adult Leigh disease without failure to thrive. Neurologist 2011, 17, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Jabeen, S.A.; Sandeep, G.; Mridula, K.R.; Meena, A.K.; Borgohain, R.; Sundaram, C. Adult-onset Leigh’s disease: A rare entity. Ann. Indian Acad. Neurol. 2016, 19, 140–142. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef]
- Chinnery, P.F.; DiMauro, S.; Shanske, S.; Schon, E.A.; Zeviani, M.; Mariotti, C.; Carrara, F.; Lombes, A.; Laforet, P.; Ogier, H.; et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet 2004, 364, 592–596. [Google Scholar] [CrossRef]
- Wei, W.; Chinnery, P.F. Inheritance of mitochondrial DNA in humans: Implications for rare and common diseases. J. Intern. Med. 2020, 287, 634–644. [Google Scholar] [CrossRef]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Lightowlers, R.N.; Turnbull, D.M. What causes mitochondrial DNA deletions in human cells? Nat. Genet. 2008, 40, 275–279. [Google Scholar] [CrossRef]
- Nissanka, N.; Minczuk, M.; Moraes, C.T. Mechanisms of Mitochondrial DNA Deletion Formation. Trends Genet. 2019, 35, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Lott, M.T.; Voljavec, A.S.; Soueidan, S.A.; Costigan, D.A.; Wallace, D.C. Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: A slip-replication model and metabolic therapy. Proc. Natl. Acad. Sci. USA 1989, 86, 7952–7956. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; He, J.; Huang, Y.; Zhao, W. The generation of mitochondrial DNA large-scale deletions in human cells. J. Human Genet. 2011, 56, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.F.; Lee, H.J.; Chi, C.S.; Tsai, C.R.; Chang, T.K.; Wang, C.J. The neurological evolution of Pearson syndrome: Case report and literature review. Eur. J. Paediatr. Neurol. 2007, 11, 208–214. [Google Scholar] [CrossRef]
- Wild, K.T.; Goldstein, A.C.; Muraresku, C.; Ganetzky, R.D. Broadening the phenotypic spectrum of Pearson syndrome: Five new cases and a review of the literature. Am. J. Med. Genet. A 2020, 182, 365–373. [Google Scholar] [CrossRef]
- Horga, A.; Pitceathly, R.D.; Blake, J.C.; Woodward, C.E.; Zapater, P.; Fratter, C.; Mudanohwo, E.E.; Plant, G.T.; Houlden, H.; Sweeney, M.G.; et al. Peripheral neuropathy predicts nuclear gene defect in patients with mitochondrial ophthalmoplegia. Brain 2014, 137, 3200–3212. [Google Scholar] [CrossRef]
- Karpati, G.; Hilton-Jones, D.; Griggs, R.C. Disorders of Voluntary Muscle; Cambridge University Press: Cambridge, UK, 2001. [Google Scholar]
- van Everdingen, J.A.M.; de Faber, J.-T. Chapter 109—Chronic Progressive External Ophthalmoplegia (CPEO) 359.1 (Abiotrophic Ophthalmoplegia, CPEO with Ragged Red Fibres, Oculocraniosomatic Neuromuscular Disease, Ocular Myopathy, Olson’s Disease, Kearns–Sayre–Daroff Syndrome, Kearns–Sayre Syndrome, Progressive External Ophthalmoplegia Plus). In Roy and Fraunfelder’s Current Ocular Therapy, 6th ed.; Roy, F.H., Fraunfelder, F.W., Fraunfelder, F.T., Tindall, R., Jensvold, B., Eds.; W.B. Saunders: Edinburgh, Scotland, 2008; pp. 207–210. [Google Scholar]
- Lenaz, G.; Fato, R.; Formiggini, G.; Genova, M.L. The role of Coenzyme Q in mitochondrial electron transport. Mitochondrion 2007, 7, S8–S33. [Google Scholar] [CrossRef]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta 1995, 1271, 195–204. [Google Scholar] [CrossRef]
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7, S41–S50. [Google Scholar] [CrossRef]
- Mordente, A.; Martorana, G.E.; Minotti, G.; Giardina, B. Antioxidant properties of 2,3-dimethoxy-5-methyl-6-(10-hydroxydecyl)-1,4-benzoquinone (idebenone). Chem. Res. Toxicol. 1998, 11, 54–63. [Google Scholar] [CrossRef]
- Haefeli, R.H.; Erb, M.; Gemperli, A.C.; Robay, D.; Courdier Fruh, I.; Anklin, C.; Dallmann, R.; Gueven, N. NQO1-dependent redox cycling of idebenone: Effects on cellular redox potential and energy levels. PLoS ONE 2011, 6, e17963. [Google Scholar] [CrossRef] [PubMed]
- Erb, M.; Hoffmann-Enger, B.; Deppe, H.; Soeberdt, M.; Haefeli, R.H.; Rummey, C.; Feurer, A.; Gueven, N. Features of idebenone and related short-chain quinones that rescue ATP levels under conditions of impaired mitochondrial complex I. PLoS ONE 2012, 7, e36153. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Idebenone: A Review in Leber’s Hereditary Optic Neuropathy. Drugs 2016, 76, 805–813. [Google Scholar] [CrossRef]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, F.G.; Kinsman, S.L.; Cohen, B.H.; Goris, M.L.; Spicer, K.M.; Perlman, S.L.; Krane, E.J.; Kheifets, V.; Thoolen, M.; Miller, G.; et al. Brain uptake of Tc99m-HMPAO correlates with clinical response to the novel redox modulating agent EPI-743 in patients with mitochondrial disease. Mol. Genet. Metab. 2012, 107, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Petrillo, S.; Tozzi, G.; Carrozzo, R.; Martinelli, D.; Dionisi-Vici, C.; Di Giovamberardino, G.; Ceravolo, F.; Klein, M.B.; Miller, G.; et al. Glutathione: A redox signature in monitoring EPI-743 therapy in children with mitochondrial encephalomyopathies. Mol. Genet. Metab. 2013, 109, 208–214. [Google Scholar] [CrossRef]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Trammell, S.A.; Weidemann, B.J.; Chadda, A.; Yorek, M.S.; Holmes, A.; Coppey, L.J.; Obrosov, A.; Kardon, R.H.; Yorek, M.A.; Brenner, C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Sci. Rep. 2016, 6, 26933. [Google Scholar] [CrossRef]
- Seo, K.S.; Kim, J.H.; Min, K.N.; Moon, J.A.; Roh, T.C.; Lee, M.J.; Lee, K.W.; Min, J.E.; Lee, Y.M. KL1333, a Novel NAD(+) Modulator, Improves Energy Metabolism and Mitochondrial Dysfunction in MELAS Fibroblasts. Front. Neurol. 2018, 9, 552. [Google Scholar] [CrossRef] [PubMed]
- Beyrath, J.; Pellegrini, M.; Renkema, H.; Houben, L.; Pecheritsyna, S.; van Zandvoort, P.; van den Broek, P.; Bekel, A.; Eftekhari, P.; Smeitink, J.A.M. KH176 Safeguards Mitochondrial Diseased Cells from Redox Stress-Induced Cell Death by Interacting with the Thioredoxin System/Peroxiredoxin Enzyme Machinery. Sci. Rep. 2018, 8, 6577. [Google Scholar] [CrossRef] [PubMed]
- Barish, G.D.; Narkar, V.A.; Evans, R.M. PPAR delta: A dagger in the heart of the metabolic syndrome. J. Clin. Investig. 2006, 116, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.Z.; Nikolic, N.; Bakke, S.S.; Boekschoten, M.V.; Kersten, S.; Kase, E.T.; Rustan, A.C.; Thoresen, G.H. PPARdelta activation in human myotubes increases mitochondrial fatty acid oxidative capacity and reduces glucose utilization by a switch in substrate preference. Arch. Physiol. Biochem. 2014, 120, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Shine, R.W.; Dwyer, P.; Olson, L.; Truong, J.; Fredenburg, R.; Goddeeris, M.; Stickens, D.; Tozzo, E. PPARdelta modulation rescues mitochondrial fatty acid oxidation defects in the mdx model of muscular dystrophy. Mitochondrion 2019, 46, 51–58. [Google Scholar] [CrossRef]
- Fedorova, L.V.; Sodhi, K.; Gatto-Weis, C.; Puri, N.; Hinds, T.D., Jr.; Shapiro, J.I.; Malhotra, D. Peroxisome proliferator-activated receptor delta agonist, HPP593, prevents renal necrosis under chronic ischemia. PLoS ONE 2013, 8, e64436. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Probst, B.L.; Trevino, I.; McCauley, L.; Bumeister, R.; Dulubova, I.; Wigley, W.C.; Ferguson, D.A. RTA 408, A Novel Synthetic Triterpenoid with Broad Anticancer and Anti-Inflammatory Activity. PLoS ONE 2015, 10, e0122942. [Google Scholar] [CrossRef]
- Zighan, M.; Arkadir, D.; Douiev, L.; Keller, G.; Miller, C.; Saada, A. Variable effects of omaveloxolone (RTA408) on primary fibroblasts with mitochondrial defects. Front. Mol. Biosci. 2022, 9, 890653. [Google Scholar] [CrossRef]
- Lopez-Gomez, C.; Levy, R.J.; Sanchez-Quintero, M.J.; Juanola-Falgarona, M.; Barca, E.; Garcia-Diaz, B.; Tadesse, S.; Garone, C.; Hirano, M. Deoxycytidine and Deoxythymidine Treatment for Thymidine Kinase 2 Deficiency. Ann. Neurol. 2017, 81, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Augustin, S.; Ellouze, S.; Benit, P.; Bouaita, A.; Rustin, P.; Sahel, J.A.; Corral-Debrinski, M. The optimized allotopic expression of ND1 or ND4 genes restores respiratory chain complex I activity in fibroblasts harboring mutations in these genes. Biochim. Biophys. Acta 2008, 1783, 1707–1717. [Google Scholar] [CrossRef]
- Yu, H.; Koilkonda, R.D.; Chou, T.H.; Porciatti, V.; Ozdemir, S.S.; Chiodo, V.; Boye, S.L.; Boye, S.E.; Hauswirth, W.W.; Lewin, A.S.; et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, E1238–E1247. [Google Scholar] [CrossRef]
- Cwerman-Thibault, H.; Augustin, S.; Lechauve, C.; Ayache, J.; Ellouze, S.; Sahel, J.A.; Corral-Debrinski, M. Nuclear expression of mitochondrial ND4 leads to the protein assembling in complex I and prevents optic atrophy and visual loss. Mol. Ther. Methods Clin. Dev. 2015, 2, 15003. [Google Scholar] [CrossRef] [PubMed]
- Koilkonda, R.; Yu, H.; Talla, V.; Porciatti, V.; Feuer, W.J.; Hauswirth, W.W.; Chiodo, V.; Erger, K.E.; Boye, S.L.; Lewin, A.S.; et al. LHON gene therapy vector prevents visual loss and optic neuropathy induced by G11778A mutant mitochondrial DNA: Biodistribution and toxicology profile. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7739–7753. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, E.; Ben Yakir-Blumkin, M.; Blumenfeld-Kan, S.; Brody, Y.; Meir, A.; Melamed-Book, N.; Napso, T.; Pozner, G.; Saadi, E.; Shabtay-Orbach, A.; et al. Mitochondrial augmentation of CD34(+) cells from healthy donors and patients with mitochondrial DNA disorders confers functional benefit. NPJ Regen. Med. 2021, 6, 58. [Google Scholar] [CrossRef]
- Spendiff, S.; Reza, M.; Murphy, J.L.; Gorman, G.; Blakely, E.L.; Taylor, R.W.; Horvath, R.; Campbell, G.; Newman, J.; Lochmuller, H.; et al. Mitochondrial DNA deletions in muscle satellite cells: Implications for therapies. Hum. Mol. Genet. 2013, 22, 4739–4747. [Google Scholar] [CrossRef] [PubMed]
- van Tienen, F.; Zelissen, R.; Timmer, E.; van Gisbergen, M.; Lindsey, P.; Quattrocelli, M.; Sampaolesi, M.; Mulder-den Hartog, E.; de Coo, I.; Smeets, H. Healthy, mtDNA-mutation free mesoangioblasts from mtDNA patients qualify for autologous therapy. Stem Cell Res. Ther. 2019, 10, 405. [Google Scholar] [CrossRef]
- Arenas-Jal, M.; Sune-Negre, J.M.; Garcia-Montoya, E. Coenzyme Q10 supplementation: Efficacy, safety, and formulation challenges. Compr. Rev. Food Sci. Food Saf. 2020, 19, 574–594. [Google Scholar] [CrossRef]
- Sood, B.; Keenaghan, M. Coenzyme Q10; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Quinzii, C.M.; Hirano, M. Coenzyme Q and mitochondrial disease. Dev. Disabil. Res. Rev. 2010, 16, 183–188. [Google Scholar] [CrossRef]
- Hargreaves, I.P. Coenzyme Q10 as a therapy for mitochondrial disease. Int. J. Biochem. Cell Biol. 2014, 49, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group QE3 Investigators; Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Tarnopolsky, M.A. The mitochondrial cocktail: Rationale for combined nutraceutical therapy in mitochondrial cytopathies. Adv. Drug Deliv. Rev. 2008, 60, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Tragni, V.; Primiano, G.; Tummolo, A.; Cafferati Beltrame, L.; La Piana, G.; Sgobba, M.N.; Cavalluzzi, M.M.; Paterno, G.; Gorgoglione, R.; Volpicella, M.; et al. Personalized Medicine in Mitochondrial Health and Disease: Molecular Basis of Therapeutic Approaches Based on Nutritional Supplements and Their Analogs. Molecules 2022, 27, 3494. [Google Scholar] [CrossRef]
- Geromel, V.; Darin, N.; Chretien, D.; Benit, P.; DeLonlay, P.; Rotig, A.; Munnich, A.; Rustin, P. Coenzyme Q(10) and idebenone in the therapy of respiratory chain diseases: Rationale and comparative benefits. Mol. Genet. Metab. 2002, 77, 21–30. [Google Scholar] [CrossRef]
- Singh, A.; Faccenda, D.; Campanella, M. Pharmacological advances in mitochondrial therapy. EBioMedicine 2021, 65, 103244. [Google Scholar] [CrossRef]
- Shrader, W.D.; Amagata, A.; Barnes, A.; Enns, G.M.; Hinman, A.; Jankowski, O.; Kheifets, V.; Komatsuzaki, R.; Lee, E.; Mollard, P.; et al. alpha-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. Bioorg. Med. Chem. Lett. 2011, 21, 3693–3698. [Google Scholar] [CrossRef]
- Baudouin-Cornu, P.; Lagniel, G.; Kumar, C.; Huang, M.E.; Labarre, J. Glutathione degradation is a key determinant of glutathione homeostasis. J. Biol. Chem. 2012, 287, 4552–4561. [Google Scholar] [CrossRef]
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol. Genet. Metab. 2012, 105, 91–102. [Google Scholar] [CrossRef]
- Kouga, T.; Takagi, M.; Miyauchi, A.; Shimbo, H.; Iai, M.; Yamashita, S.; Murayama, K.; Klein, M.B.; Miller, G.; Goto, T.; et al. Japanese Leigh syndrome case treated with EPI-743. Brain Dev. 2018, 40, 145–149. [Google Scholar] [CrossRef]
- Srivastava, S. Emerging therapeutic roles for NAD(+) metabolism in mitochondrial and age-related disorders. Clin. Transl. Med. 2016, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Dolopikou, C.F.; Kourtzidis, I.A.; Margaritelis, N.V.; Vrabas, I.S.; Koidou, I.; Kyparos, A.; Theodorou, A.A.; Paschalis, V.; Nikolaidis, M.G. Acute nicotinamide riboside supplementation improves redox homeostasis and exercise performance in old individuals: A double-blind cross-over study. Eur. J. Nutr. 2020, 59, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Remie, C.M.E.; Roumans, K.H.M.; Moonen, M.P.B.; Connell, N.J.; Havekes, B.; Mevenkamp, J.; Lindeboom, L.; de Wit, V.H.W.; van de Weijer, T.; Aarts, S.; et al. Nicotinamide riboside supplementation alters body composition and skeletal muscle acetylcarnitine concentrations in healthy obese humans. Am. J. Clin. Nutr. 2020, 112, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Lapatto, H.A.K.; Kuusela, M.; Heikkinen, A.; Muniandy, M.; van der Kolk, B.W.; Gopalakrishnan, S.; Pollanen, N.; Sandvik, M.; Schmidt, M.S.; Heinonen, S.; et al. Nicotinamide riboside improves muscle mitochondrial biogenesis, satellite cell differentiation, and gut microbiota in a twin study. Sci. Adv. 2023, 9, eadd5163. [Google Scholar] [CrossRef]
- Feng, L.; Luo, H.; Xu, Z.; Yang, Z.; Du, G.; Zhang, Y.; Yu, L.; Hu, K.; Zhu, W.; Tong, Q.; et al. Bavachinin, as a novel natural pan-PPAR agonist, exhibits unique synergistic effects with synthetic PPAR-gamma and PPAR-alpha agonists on carbohydrate and lipid metabolism in db/db and diet-induced obese mice. Diabetologia 2016, 59, 1276–1286. [Google Scholar] [CrossRef]
- de Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2020, 92, 86–95. [Google Scholar] [CrossRef]
- Falabella, M.; Vernon, H.J.; Hanna, M.G.; Claypool, S.M.; Pitceathly, R.D.S. Cardiolipin, Mitochondria, and Neurological Disease. Trends Endocrinol. Metab. 2021, 32, 224–237. [Google Scholar] [CrossRef]
- Gautam, M.; Genc, B.; Helmold, B.; Ahrens, A.; Kuka, J.; Makrecka-Kuka, M.; Gunay, A.; Kocak, N.; Aguilar-Wickings, I.R.; Keefe, D.; et al. SBT-272 improves TDP-43 pathology in ALS upper motor neurons by modulating mitochondrial integrity, motility, and function. Neurobiol. Dis. 2023, 178, 106022. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Dai, D.F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail. 2013, 6, 1067–1076. [Google Scholar] [CrossRef]
- Machiraju, P.; Wang, X.; Sabouny, R.; Huang, J.; Zhao, T.; Iqbal, F.; King, M.; Prasher, D.; Lodha, A.; Jimenez-Tellez, N.; et al. SS-31 Peptide Reverses the Mitochondrial Fragmentation Present in Fibroblasts From Patients With DCMA, a Mitochondrial Cardiomyopathy. Front. Cardiovasc. Med. 2019, 6, 167. [Google Scholar] [CrossRef] [PubMed]
- Reid Thompson, W.; Hornby, B.; Manuel, R.; Bradley, E.; Laux, J.; Carr, J.; Vernon, H.J. A phase 2/3 randomized clinical trial followed by an open-label extension to evaluate the effectiveness of elamipretide in Barth syndrome, a genetic disorder of mitochondrial cardiolipin metabolism. Genet. Med. 2021, 23, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.E.; Pennington, E.R.; Perry, J.B.; Dadoo, S.; Makrecka-Kuka, M.; Dambrova, M.; Moukdar, F.; Patel, H.D.; Han, X.; Kidd, G.K.; et al. The cardiolipin-binding peptide elamipretide mitigates fragmentation of cristae networks following cardiac ischemia reperfusion in rats. Commun. Biol. 2020, 3, 389. [Google Scholar] [CrossRef] [PubMed]
- Distelmaier, F.; Visch, H.J.; Smeitink, J.A.; Mayatepek, E.; Koopman, W.J.; Willems, P.H. The antioxidant Trolox restores mitochondrial membrane potential and Ca2+ -stimulated ATP production in human complex I deficiency. J. Mol. Med. 2009, 87, 515–522. [Google Scholar] [CrossRef]
- Blanchet, L.; Smeitink, J.A.; van Emst-de Vries, S.E.; Vogels, C.; Pellegrini, M.; Jonckheere, A.I.; Rodenburg, R.J.; Buydens, L.M.; Beyrath, J.; Willems, P.H.; et al. Quantifying small molecule phenotypic effects using mitochondrial morpho-functional fingerprinting and machine learning. Sci. Rep. 2015, 5, 8035. [Google Scholar] [CrossRef] [PubMed]
- Klein Gunnewiek, T.M.; Verboven, A.H.A.; Pelgrim, I.; Hogeweg, M.; Schoenmaker, C.; Renkema, H.; Beyrath, J.; Smeitink, J.; de Vries, B.B.A.; Hoen, P.A.C.; et al. Sonlicromanol improves neuronal network dysfunction and transcriptome changes linked to m.3243A>G heteroplasmy in iPSC-derived neurons. Stem Cell Rep. 2021, 16, 2197–2212. [Google Scholar] [CrossRef]
- Abeti, R.; Uzun, E.; Renganathan, I.; Honda, T.; Pook, M.A.; Giunti, P. Targeting lipid peroxidation and mitochondrial imbalance in Friedreich’s ataxia. Pharmacol. Res. 2015, 99, 344–350. [Google Scholar] [CrossRef]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich’s Ataxia Models. Front. Cell. Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef]
- Tiberi, J.; Segatto, M.; Fiorenza, M.T.; La Rosa, P. Apparent Opportunities and Hidden Pitfalls: The Conflicting Results of Restoring NRF2-Regulated Redox Metabolism in Friedreich’s Ataxia Pre-Clinical Models and Clinical Trials. Biomedicines 2023, 11, 1293. [Google Scholar] [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., 3rd; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Ozdemir, S.S.; Koilkonda, R.D.; Chou, T.H.; Porciatti, V.; Chiodo, V.; Boye, S.L.; Hauswirth, W.W.; Lewin, A.S.; Guy, J. Mutant NADH dehydrogenase subunit 4 gene delivery to mitochondria by targeting sequence-modified adeno-associated virus induces visual loss and optic atrophy in mice. Mol. Vis. 2012, 18, 1668–1683. [Google Scholar] [PubMed]
- Newman, N.J.; Yu-Wai-Man, P.; Subramanian, P.S.; Moster, M.L.; Wang, A.G.; Donahue, S.P.; Leroy, B.P.; Carelli, V.; Biousse, V.; Vignal-Clermont, C.; et al. Randomized trial of bilateral gene therapy injection for m.11778G>A MT-ND4 Leber optic neuropathy. Brain 2023, 146, 1328–1341. [Google Scholar] [CrossRef]
- McGrady, N.R.; Boal, A.M.; Risner, M.L.; Taiel, M.; Sahel, J.A.; Calkins, D.J. Ocular stress enhances contralateral transfer of lenadogene nolparvovec gene therapy through astrocyte networks. Mol. Ther. 2023, 31, 2005–2013. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Dong, L.F.; Rohlena, J.; Zobalova, R.; Nahacka, Z.; Rodriguez, A.M.; Berridge, M.V.; Neuzil, J. Mitochondria on the move: Horizontal mitochondrial transfer in disease and health. J. Cell Biol. 2023, 222, e202211044. [Google Scholar] [CrossRef]
- Jacoby, E.; Bar-Yosef, O.; Gruber, N.; Lahav, E.; Varda-Bloom, N.; Bolkier, Y.; Bar, D.; Blumkin, M.B.; Barak, S.; Eisenstein, E.; et al. Mitochondrial augmentation of hematopoietic stem cells in children with single large-scale mitochondrial DNA deletion syndromes. Sci. Transl. Med. 2022, 14, eabo3724. [Google Scholar] [CrossRef]
- Sampaolesi, M.; Torrente, Y.; Innocenzi, A.; Tonlorenzi, R.; D’Antona, G.; Pellegrino, M.A.; Barresi, R.; Bresolin, N.; De Angelis, M.G.; Campbell, K.P.; et al. Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science 2003, 301, 487–492. [Google Scholar] [CrossRef]
- Roobrouck, V.D.; Clavel, C.; Jacobs, S.A.; Ulloa-Montoya, F.; Crippa, S.; Sohni, A.; Roberts, S.J.; Luyten, F.P.; Van Gool, S.W.; Sampaolesi, M.; et al. Differentiation potential of human postnatal mesenchymal stem cells, mesoangioblasts, and multipotent adult progenitor cells reflected in their transcriptome and partially influenced by the culture conditions. Stem Cells 2011, 29, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Dellavalle, A.; Sampaolesi, M.; Tonlorenzi, R.; Tagliafico, E.; Sacchetti, B.; Perani, L.; Innocenzi, A.; Galvez, B.G.; Messina, G.; Morosetti, R.; et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell Biol. 2007, 9, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Dellavalle, A.; Maroli, G.; Covarello, D.; Azzoni, E.; Innocenzi, A.; Perani, L.; Antonini, S.; Sambasivan, R.; Brunelli, S.; Tajbakhsh, S.; et al. Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat. Commun. 2011, 2, 499. [Google Scholar] [CrossRef]
- Cree, L.; Loi, P. Mitochondrial replacement: From basic research to assisted reproductive technology portfolio tool-technicalities and possible risks. Mol. Hum. Reprod. 2015, 21, 3–10. [Google Scholar] [CrossRef]
- Sharma, H.; Singh, D.; Mahant, A.; Sohal, S.K.; Kesavan, A.K.; Samiksha. Development of mitochondrial replacement therapy: A review. Heliyon 2020, 6, e04643. [Google Scholar] [CrossRef] [PubMed]
- Connor, S. When replacement becomes reversion. Nat. Biotechnol. 2017, 35, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Sato, S.; Ooka, R.; Akashi, K.; Nakamura, A.; Miyado, K.; Akutsu, H.; Tanaka, M. Mitochondrial replacement by genome transfer in human oocytes: Efficacy, concerns, and legality. Reprod. Med. Biol. 2021, 20, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Pompei, M.; Pompei, F. Overcoming bioethical, legal, and hereditary barriers to mitochondrial replacement therapy in the USA. J. Assist. Reprod. Genet. 2019, 36, 383–393. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Moscou, M.J.; Bogdanove, A.J. A Simple Cipher Governs DNA Recognition by TAL Effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, S.; Baek, G.; Kim, A.; Kang, B.C.; Seo, H.; Kim, J.S. Mitochondrial DNA editing in mice with DddA-TALE fusion deaminases. Nat. Commun. 2021, 12, 1190. [Google Scholar] [CrossRef]
- Mok, B.Y.; Kotrys, A.V.; Raguram, A.; Huang, T.P.; Mootha, V.K.; Liu, D.R. CRISPR-free base editors with enhanced activity and expanded targeting scope in mitochondrial and nuclear DNA. Nat. Biotechnol. 2022, 40, 1378–1387. [Google Scholar] [CrossRef]
- Lee, S.; Lee, H.; Baek, G.; Namgung, E.; Park, J.M.; Kim, S.; Hong, S.; Kim, J.S. Enhanced mitochondrial DNA editing in mice using nuclear-exported TALE-linked deaminases and nucleases. Genome Biol. 2022, 23, 211. [Google Scholar] [CrossRef]
- Guo, J.; Chen, X.; Liu, Z.; Sun, H.; Zhou, Y.; Dai, Y.; Ma, Y.E.; He, L.; Qian, X.; Wang, J.; et al. DdCBE mediates efficient and inheritable modifications in mouse mitochondrial genome. Mol. Ther.—Nucleic Acids 2022, 27, 73–80. [Google Scholar] [CrossRef]
- Tan, L.; Qi, X.; Kong, W.; Jin, J.; Lu, D.; Zhang, X.; Wang, Y.; Wang, S.; Dong, W.; Shi, X.; et al. A conditional knockout rat resource of mitochondrial protein-coding genes via a DdCBE-induced premature stop codon. Sci. Adv. 2023, 9, eadf2695. [Google Scholar] [CrossRef]
- Silva-Pinheiro, P.; Mutti, C.D.; Van Haute, L.; Powell, C.A.; Nash, P.A.; Turner, K.; Minczuk, M. A library of base editors for the precise ablation of all protein-coding genes in the mouse mitochondrial genome. Nat. Biomed. Eng. 2023, 7, 692–703. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Thuronyi, B.W.; Koblan, L.W.; Levy, J.M.; Yeh, W.-H.; Zheng, C.; Newby, G.A.; Wilson, C.; Bhaumik, M.; Shubina-Oleinik, O.; Holt, J.R.; et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol. 2019, 37, 1070–1079. [Google Scholar] [CrossRef]
- Zhao, D.; Li, J.; Li, S.; Xin, X.; Hu, M.; Price, M.A.; Rosser, S.J.; Bi, C.; Zhang, X. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat. Biotechnol. 2021, 39, 35–40. [Google Scholar] [CrossRef]
- Kurt, I.C.; Zhou, R.; Iyer, S.; Garcia, S.P.; Miller, B.R.; Langner, L.M.; Grünewald, J.; Joung, J.K. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 2021, 39, 41–46. [Google Scholar] [CrossRef]
- Chen, L.; Hong, M.; Luan, C.; Gao, H.; Ru, G.; Guo, X.; Zhang, D.; Zhang, S.; Li, C.; Wu, J.; et al. Adenine transversion editors enable precise, efficient A•T-to-C•G base editing in mammalian cells and embryos. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Meng, H.; Liu, L.; Zhao, H.; Rao, X.; Yan, Y.; Wu, H.; Liu, M.; He, A.; Yi, C. Mitochondrial base editor induces substantial nuclear off-target mutations. Nature 2022, 606, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, Z.; Xu, K.; Feng, H.; Xie, L.; Li, D.; Zuo, Z.; Zhang, M.; Xu, C.; Yang, H.; et al. Mitochondrial base editor DdCBE causes substantial DNA off-target editing in nuclear genome of embryos. Cell Discov. 2022, 8, 27. [Google Scholar] [CrossRef]
- Jeppesen, T.D.; Schwartz, M.; Olsen, D.B.; Wibrand, F.; Krag, T.; Duno, M.; Hauerslev, S.; Vissing, J. Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain 2006, 129, 3402–3412. [Google Scholar] [CrossRef]
- Murphy, J.L.; Blakely, E.L.; Schaefer, A.M.; He, L.; Wyrick, P.; Haller, R.G.; Taylor, R.W.; Turnbull, D.M.; Taivassalo, T. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain 2008, 131, 2832–2840. [Google Scholar] [CrossRef]
- Lopez-Gomez, C.; Sanchez-Quintero, M.J.; Lee, E.J.; Kleiner, G.; Tadesse, S.; Xie, J.; Akman, H.O.; Gao, G.; Hirano, M. Synergistic Deoxynucleoside and Gene Therapies for Thymidine Kinase 2 Deficiency. Ann. Neurol. 2021, 90, 640–652. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Symptoms and causes of mitochondrial diseases. The upper side of the figure displays commonly diagnosed symptoms, with symptoms affecting the entire body placed on the left and organ-specific symptoms on the right. As mitochondrial diseases have various causes, only well-known pathogenic mtDNA mutations are listed. A line and brief illustration indicate a correlation between pathogenic mutations and the affected sites. Details can be found in Table 1, Section 3.
Figure 1.
Symptoms and causes of mitochondrial diseases. The upper side of the figure displays commonly diagnosed symptoms, with symptoms affecting the entire body placed on the left and organ-specific symptoms on the right. As mitochondrial diseases have various causes, only well-known pathogenic mtDNA mutations are listed. A line and brief illustration indicate a correlation between pathogenic mutations and the affected sites. Details can be found in Table 1, Section 3.

Figure 2.
Interactions of treatments for mitochondrial diseases. Targeting points of drugs are indicated with arrows with brief illustrations. The treatments are displayed using their known names and features, and further detailed mechanisms for each treatment can be found in Section 4 and Section 5 and Table 2 and Table 3.
Figure 2.
Interactions of treatments for mitochondrial diseases. Targeting points of drugs are indicated with arrows with brief illustrations. The treatments are displayed using their known names and features, and further detailed mechanisms for each treatment can be found in Section 4 and Section 5 and Table 2 and Table 3.

Figure 3.
Currently developed mitochondrial base editor. Identical proteins are displayed with the same shape and color. Target and edited nucleotides are marked using boxes, with the edited nucleotides shown in orange. An arrow indicates another form of the same base editor with different deaminase activity. Further details can be found in Section 6.
Figure 3.
Currently developed mitochondrial base editor. Identical proteins are displayed with the same shape and color. Target and edited nucleotides are marked using boxes, with the edited nucleotides shown in orange. An arrow indicates another form of the same base editor with different deaminase activity. Further details can be found in Section 6.


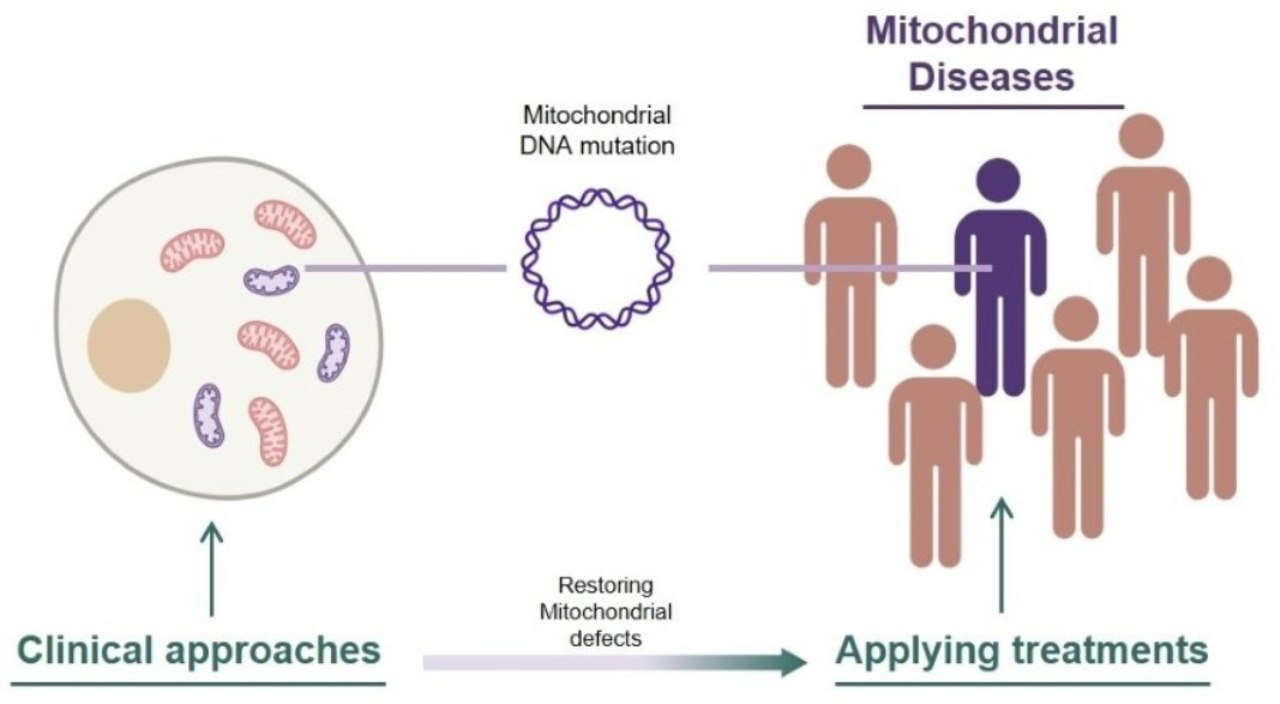{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Known diseases with mitochondrial malfunction.
| Disease | Main Symptoms | Case Reports References | Cause |
|---|---|---|---|
| LHON (Leber hereditary optic neuropathy) | Vision loss (retinal ganglion cells and axon loss) | Significant decrease in visual acuity, dyschromatopsia [54,55,56] | m.T14484C (Complex1 ND6) |
| m.G3460A (Complex1 ND1) | |||
| m.G11778A (Complex1 ND4) | |||
| MERRF (Myoclonic epilepsy with ragged red fiber) | Myoclonus, generalized epilepsy, ataxia, myopathy, exercise intolerance | Bilateral primary optic atrophy, sensorineural hearing loss, gait unsteadiness [57] | m.A8344G (tRNA lysine) |
| Slurred speech, fatigue, hearing loss, limb weakness [58] | m.G8363A (tRNA lysine) | ||
| Dysphonia, memory loss, stroke-like episode [59] | m.A3243G (tRNA leucine) | ||
| MELAS (Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes) | Muscle weakness in whole body, dementia, aphasia, myoclonus, ataxia | Developing diabetes, hearing loss, progressively deteriorating functional status [60] | m.A3243G (tRNA leucine) |
| Aphasia, subtle loss of muscle strength in right arm [61] | |||
| Migraine-like headaches, muscle damage [62] | |||
| disturbances of consciousness, ventilatory failure [63] | |||
| Aeizures, transient sensory disturbances, weakness, visual impairment, cognitive impairment [64] | m.G13513A (Complex1 ND5) | ||
| Maternally inherited diabetes and deafness (MIDD) | Chronic kidney disease, deafness, loss of oral sensation, neuropathy, hearing loss, myopathy | Proteinuria, glomerular abnormality progressive renal failuare [65] | m.A3243G (tRNA leucine) |
| Hearing loss, loss of oral sensation, dysarthria, drolling [66] | |||
| Hearing loss, central nervous system diseases, myopathy, cardiac disease, nephropathy, underweight [10] | |||
| Pearson syndrome | Failure to thrive, malabsorption | Refractory anemia, digestive system failuare, bone marrow failure, metabolic disorders, gastrointestinal symptoms, renal disorders, pancreatic exocrine insufficiency [11] | Large deletion of mtDNA |
| Malabsorption, lactic acidemia, sideroblasts on bone marrow evaluation, microcephaly [67] | |||
| Kearns–Sayre syndrome (KSS) | Heart block, growth retardation, external ophthalmoplegia, vestibular dysfunction | Vision loss, progressive external ophthalmoplegia, retinitis pigmentosa, heart block, vestibular dysfunction, growth retardation [68] | Large deletion of mtDNA |
| Photophobia, nystagmus, sensorineural hearing loss, tremor, progressive cerebellar ataxia [69] | |||
| Chronic progressive external ophthalmoplegia (CPEO) | Loosing control of eyelids and eye movements, ptosis | Central neurogenic hyperventilation, restriction of eye movement and ptosis [70] | Multiple deletion of mtDNA |
| Low birth weight and congenital deafness [71] | |||
| Hemifacial weakness, dysarthria, mental retardation, sensorineural hearing loss [72] | mtDNA mutation with mitochondrial protein-encoding nuclear DNA mutations [73] | ||
| Ophthalmoplegia, strabismus, loosing control of extraocular muscles [71] | |||
| Leigh syndrome | Neurological symptoms, cardiac dysfunction, dyspraxia | Blepharoptosis, ptosis, inability to walk, sensory deficits, fever [74] | m.T9176C (MT-ATP6) |
| Delay of psychomotor development, cardiomyopathy [75] | m.A14453G (Complex1 ND6) | ||
| Bilateral exotropia, nystagmus bilateral, childhood-onset neuromuscular regression [76] | Nuclear NDUFAF5 gene mutation, (Complex 1 NDUFAF5) | ||
| Barth syndrome | Heart failure | Left ventricular hypertrophy, heart failure with metabolic crisis [77] | Nuclear TAZ gene C640T (mitochondrial acyl chain composition remodeling enzyme) |
| Friedreich’s ataxia | Ataxia, gait unsteadiness, cardiomyopathy | Chest pain, dyspnea, palpitation, left ventricular hypertrophy [78] | Nuclear FAZ gene (mitochondrial iron metabolism related enzyme) |
| Blindness, sensorineural deafness [79], |
Table 2.
Drugs for clinical trials for treating mitochondrial diseases.
| Treatment | Mechanisms | Target Disease | Clinical Trial /NCT Number |
|---|---|---|---|
| Coenzyme Q10 | Diffusible electron carrier of the mitochondrial respiratory chain [145], lipid peroxidation interfering antioxidants [146,147] | Mitochondrial diseases | Phase 3 (Completed) /NCT00432744 |
| RAXONE (Idebenone) | Transfer electrons directly to complex III by bypassing malfunctional complex I [148,149,150,151] | LHON (Leber hereditary optic neuropathy) | Phase 4 (Completed) /NCT02774005 |
| Vatiquinone (EPI-743, PTC-743) | Inhibiting 15-lipoxygenase (15-LO) [152] leads to increased GSH levels and decreased oxidized GSH [153,154] | Friedreich ataxia | Phase 3 (Active) /NCT05515536 |
| Mitochondrial disease with refractory epilepsy | Phase 2/3 (Active) /NCT04378075 | ||
| Mitochondrial respiratory chain diseases | Phase 2 (Active) /NCT01370447 | ||
| Nicotinamide Riboside (NR) | Precursor of Nicotinamide adenine dinucleotide (NAD+); increasing intracellular NAD+ level increases mitochondrial function and mitochondrial number [155,156,157,158] | Mitochondrial myopathy disorder | Phase 2 (Active) /NCT05590468 |
| KL1333 | Interacts with NQO1 and acts as precursor of NAD+, recovers deficiency of mitochondrial respiratory chain [159] | Mitochondrial disease | Phase 2 (Active) /NCT05650229 |
| Sonlicromanol (KH176) | ROS-redox modulator through interaction with the Thioredoxin System, which is a major antioxidant and redox signaling cellular system [160] | m.A3243G causing mitochondrial diseases | Phase 2 (Completed) /NCT04165239 |
| Bocidelpar (ASP0367) | Agonist of PPARδ, enhances fatty acid oxidation, mitochondrial respiration and oxidative metabolism, which further leads to increment of skeletal muscle genes expression [161,162,163] | Primary mitochondrial myopathy | Phase 2/3 (Active) /NCT04641962 |
| Mavodelpar (REN001, HPP593) | Agonist of PPARδ upregulates oxidative stress defense genes, contributes to attenuating oxidative stress [164] | Primary mitochondrial myopathy | Phase 2 (Active) /NCT04535609 |
| Elamipretide (SBT-272) | Binding mitochondrial inner membrane cardiolipin; rescues dysmorphology of mitochondria [165,166] | Primary mitochondrial myopathy | Phase 3 (Active) /NCT05162768 |
| Barth syndrome | Phase 3 (Completed) /NCT03098797 | ||
| Mitochondrial dysfunction in age-related macular degeneration | Phase 2 (Completed) /NCT03891875 | ||
| Friedreich Ataxia | Phase 1/2 (Active) /NCT05168774 | ||
| Skyclarys (Omaveloxolone) | Nrf2 degradation inhibitor; upregulates the expression of antioxidant gene, downregulates the expression of pro-inflammatory genes, and enhances mitochondrial biogenesis [167,168] | Friedreich’s ataxia | FDA approved |
| Deoxynucleosides Pyrimidines (Deoxycytidine dC and Deoxythymidine dT) | Enhances mtDNA maintenance by bypassing malfunctional mitochondrial pyrimidine salvage pathway [169] | Mitochondrial depletion syndromes with neurological phenotypes dysfunction | Phase 2 (Active) /NCT04802707 |
Table 3.
Non-chemical clinical treatments for mitochondrial diseases.
| Treatment | Mechanisms | Target disease | Clinical trials |
|---|---|---|---|
| LUMEVOQ (GS010, rAAV2/2-ND4), | MT-ND4 deficiency rescue via allotopic expression of normal MT-ND4 using recombinant AAV [170,171,172] | LHON (Leber hereditary optic neuropathy) | Phase 3 (Active) /NCT03293524 |
| NR082 (rAAV2-ND4), | Phase 2/3 (Active) /NCT03153293 | ||
| ScAAV2-P1ND4v2 | MT-ND4 deficiency rescue via allotopic expression of normal MT-ND4 using self-complementary AAV [173] | Phase 1 (Active) /NCT02161380 | |
| Mitochondrial augmentation | Replacement of dysfunctional mitochondria with healthy-exogenous donor mitochondria using in vitro uptake [174] | mtDNA depletion disease (Pearson syndrome) | Phase 1/2 (Active) /NCT03384420 |
| Mesoangioblasts (MABs) | Intra-arterial injection of in vitro cultured patient-autologous mesoangioblasts, which harbor far fewer mtDNA mutations despite a much higher mutation load in patient [175,176] | Mitochondrial myopathy with m.A3243G mutation | Phase 2 (Active) /NCT05962333 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hong, S.; Kim, S.; Kim, K.; Lee, H. Clinical Approaches for Mitochondrial Diseases. Cells 2023, 12, 2494. https://doi.org/10.3390/cells12202494
AMA Style
Hong S, Kim S, Kim K, Lee H. Clinical Approaches for Mitochondrial Diseases. Cells. 2023; 12(20):2494. https://doi.org/10.3390/cells12202494
Chicago/Turabian StyleHong, Seongho, Sanghun Kim, Kyoungmi Kim, and Hyunji Lee. 2023. "Clinical Approaches for Mitochondrial Diseases" Cells 12, no. 20: 2494. https://doi.org/10.3390/cells12202494
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.