
PU.1 Expression Defines Distinct Functional Activities in the Phenotypic HSC Compartment of a Murine Inflammatory Stress Model

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Flow Cytometry
2.3. In Vivo Procedures
2.4. Cell Culture
2.5. Fluidigm qRT-PCR Analysis
2.6. Statistical Analysis
3. Results
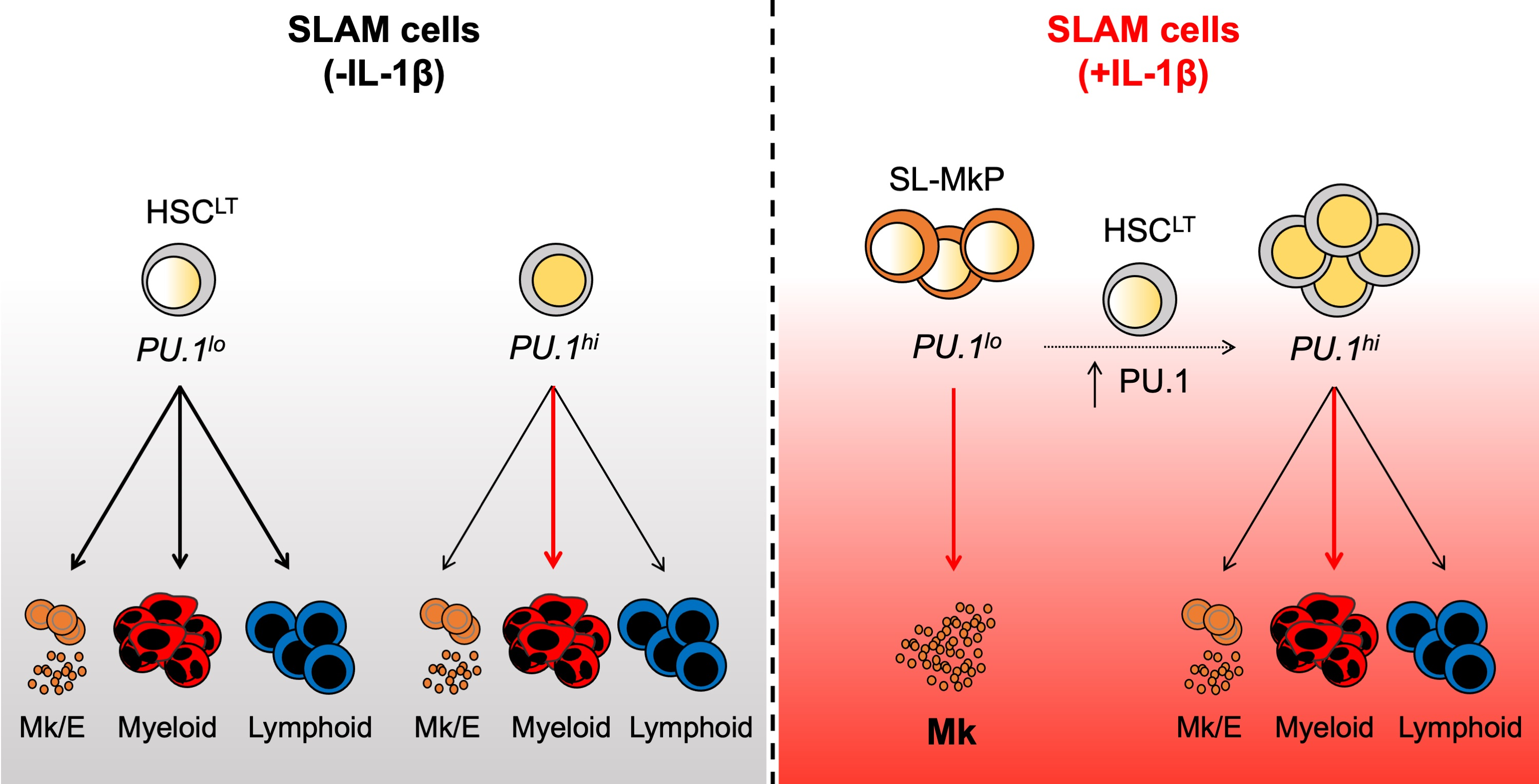
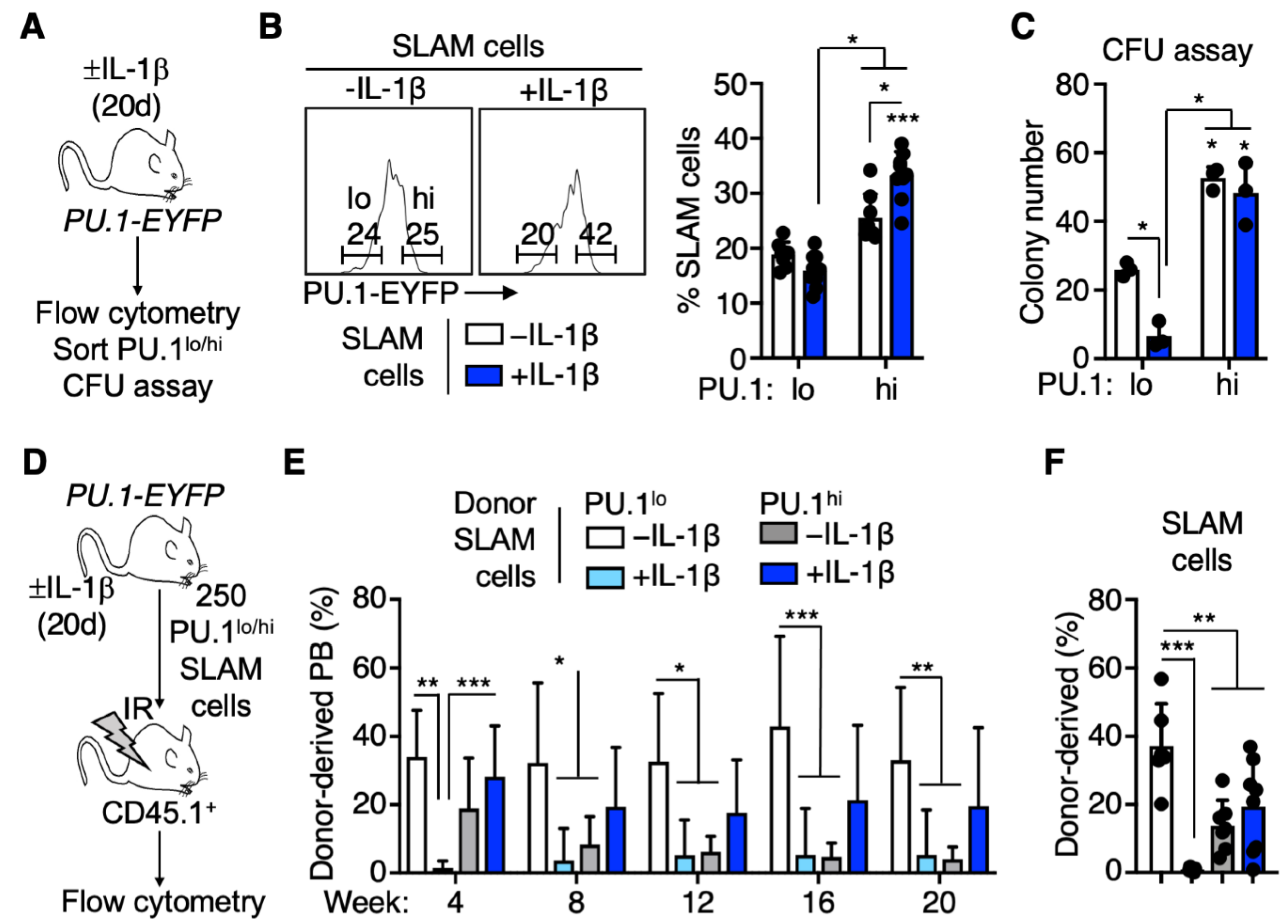
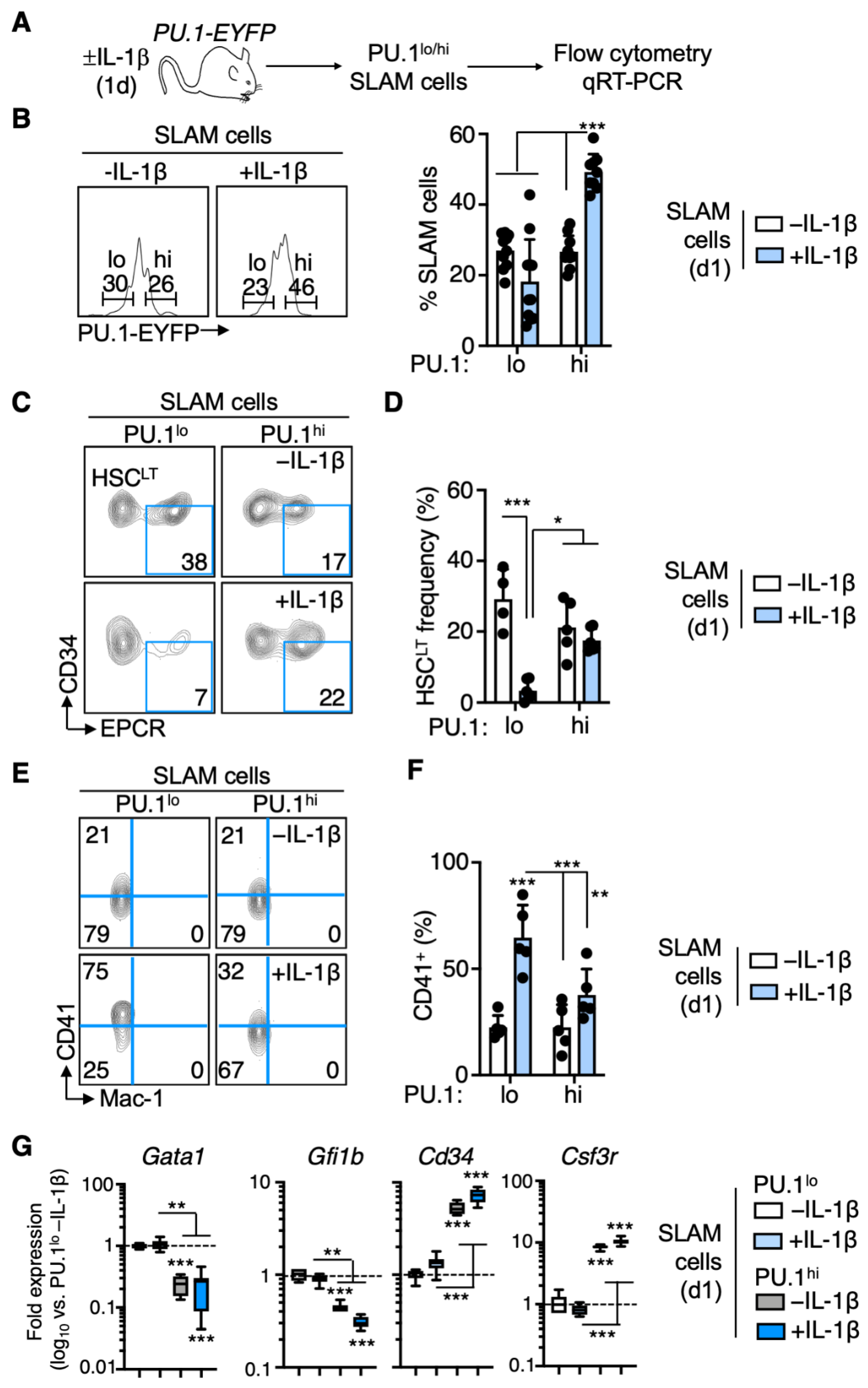
3.1. PU.1 Levels Identify Distinct Functional Activities in the SLAM Compartment following Chronic IL-1β Treatment
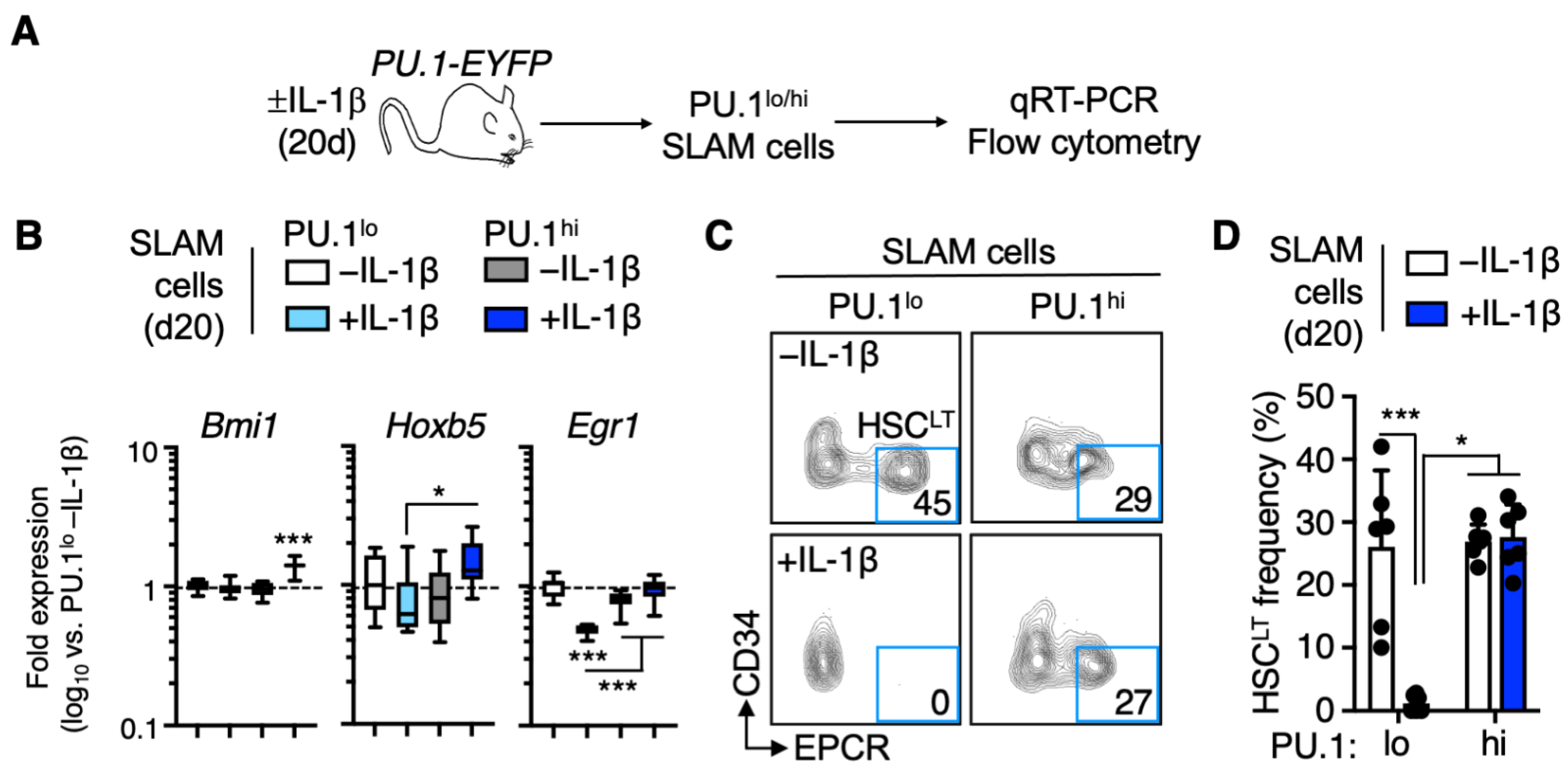
3.2. EPCR+ HSCLT Are Depleted from the PU.1lo SLAM Fraction following IL-1β Treatment
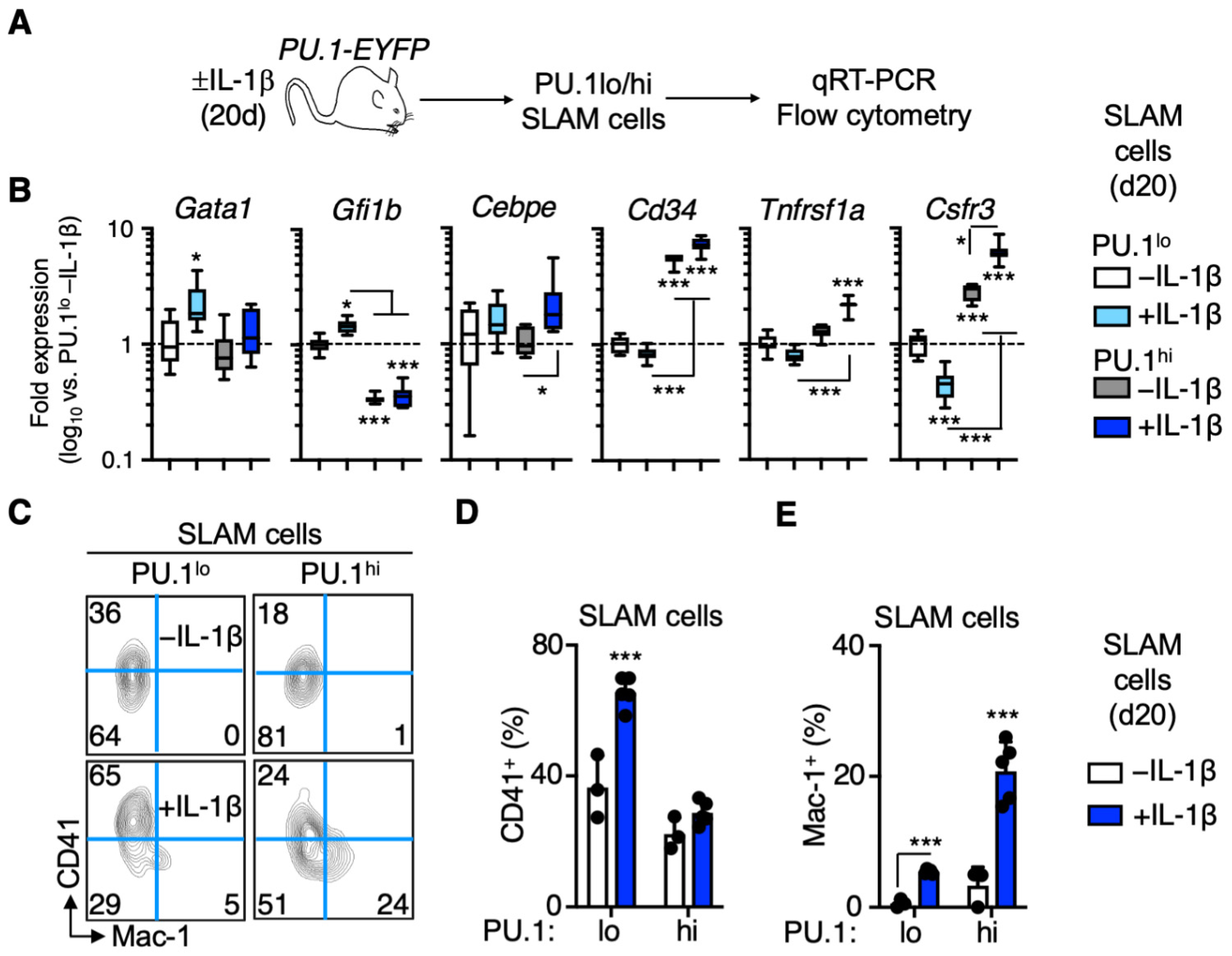
3.3. Distinct Patterns of Lineage Priming in the SLAM Compartment Associated with PU.1 Levels
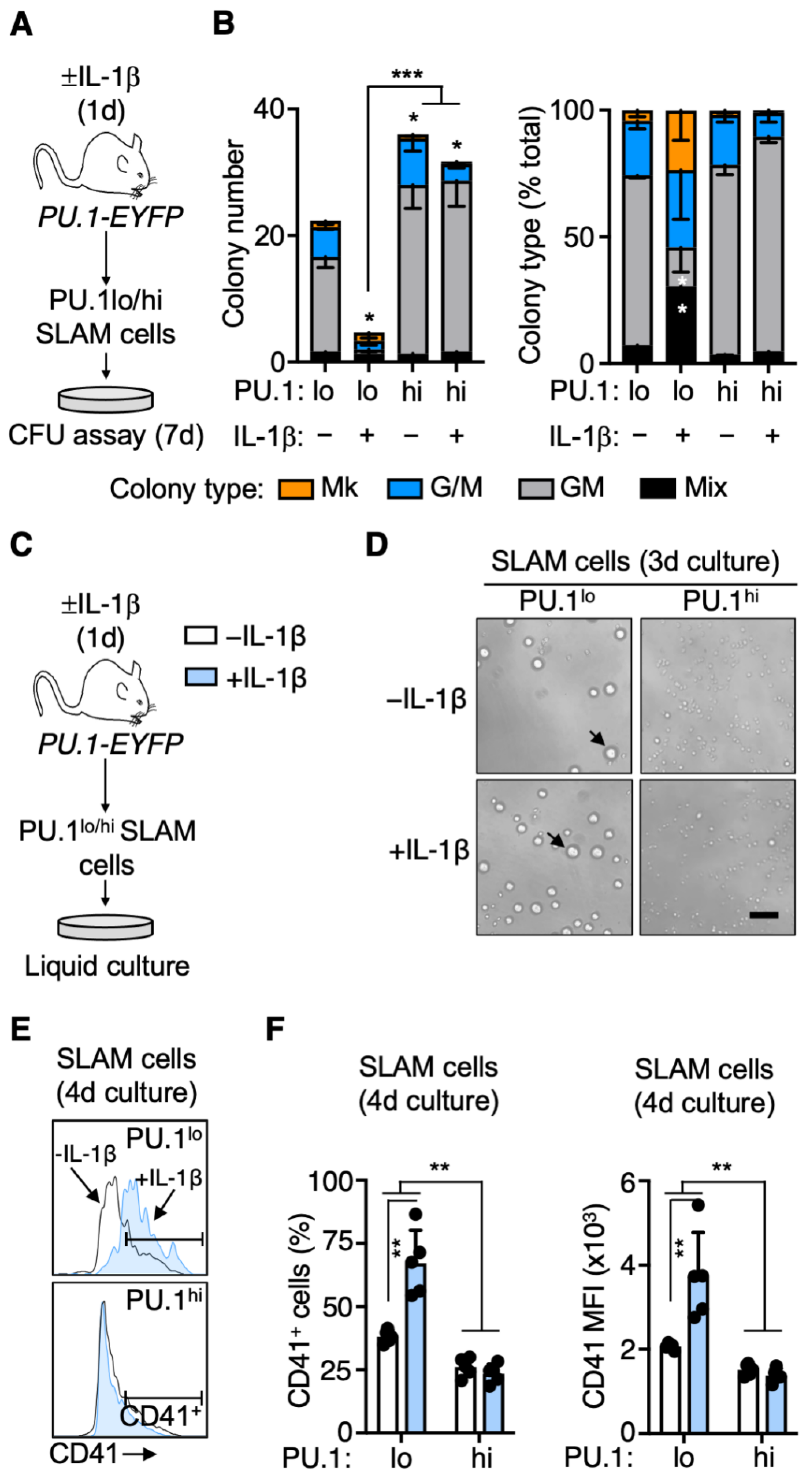
3.4. Rapid PU.1 Induction and Expansion of Lineage-Primed SLAM Cells in Response to IL-1β
3.5. PU.1lo SLAM Cells from IL-1β-Treated Mice Exhibit Characteristics of SL-MkP
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- King, K.Y.; Goodell, M.A. Inflammatory modulation of HSCs: Viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 2011, 11, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Caiado, F.; Pietras, E.M.; Manz, M.G. Inflammation as a regulator of hematopoietic stem cell function in disease, aging, and clonal selection. J. Exp. Med. 2021, 218, e20201541. [Google Scholar] [CrossRef]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.-M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Cain, D.W.; Kuraoka, M.; Kondo, M.; Kelsoe, G. IL-1R type I-dependent hemopoietic stem cell proliferation is necessary for inflammatory granulopoiesis and reactive neutrophilia. J. Immunol. 2009, 182, 6477–6484. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, G.; Mills, T.S.; Rabe, J.L.; Chavez, J.S.; Kuldanek, S.; Kirkpatrick, G.; Noetzli, L.; Jubair, W.K.; Zanche, M.; Meyers, J.R.; et al. Pro-inflammatory cytokine blockade attenuates myeloid expansion in a murine model of rheumatoid arthritis. Haematologica 2020, 105, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Rabe, J.L.; Hernandez, G.; Chavez, J.S.; Mills, T.S.; Nerlov, C.; Pietras, E.M. CD34 and EPCR coordinately enrich functional murine hematopoietic stem cells under normal and inflammatory conditions. Exp. Hematol. 2020, 81, 1–15.e16. [Google Scholar] [CrossRef]
- Chavez, J.S.; Rabe, J.L.; Loeffler, D.; Higa, K.C.; Hernandez, G.; Mills, T.S.; Ahmed, N.; Gessner, R.L.; Ke, Z.; Idler, B.M.; et al. PU.1 enforces quiescence and limits hematopoietic stem cell expansion during inflammatory stress. J. Exp. Med. 2021, 218, e20201169. [Google Scholar] [CrossRef]
- Etzrodt, M.; Ahmed, N.; Hoppe, P.S.; Loeffler, D.; Skylaki, S.; Hilsenbeck, O.; Kokkaliaris, K.D.; Kaltenbach, H.-M.; Stelling, J.; Nerlov, C.; et al. Inflammatory signals directly instruct PU.1 in HSCs via TNF. Blood 2019, 133, 816–819. [Google Scholar] [CrossRef]
- Ahmed, N.; Etzrodt, M.; Dettinger, P.; Kull, T.; Loeffler, D.; Hoppe, P.S.; Chavez, J.S.; Zhang, Y.; Ortega, G.C.; Hilsenbeck, O.; et al. Blood stem cell PU.1 upregulation is a consequence of differentiation without fast autoregulation. J. Exp. Med. 2022, 219, e20202490. [Google Scholar] [CrossRef]
- Koschmieder, S.; Rosenbauer, F.; Steidl, U.; Owens, B.M.; Tenen, D.G. Role of transcription factors C/EBPalpha and PU.1 in normal hematopoiesis and leukemia. Int. J. Hematol. 2005, 81, 368–377. [Google Scholar] [CrossRef]
- Pang, S.H.M.; De Graaf, C.A.; Hilton, D.J.; Huntington, N.; Carotta, S.; Wu, L.; Nutt, S.L. PU.1 Is Required for the Developmental Progression of Multipotent Progenitors to Common Lymphoid Progenitors. Front. Immunol. 2018, 9, 1264. [Google Scholar] [CrossRef] [PubMed]
- Nutt, S.L.; Metcalf, D.; D’Amico, A.; Polli, M.; Wu, L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J. Exp. Med. 2005, 201, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Staber, P.B.; Zhang, P.; Ye, M.; Welner, R.S.; Nombela-Arrieta, C.; Bach, C.; Kerenyi, M.; Bartholdy, B.A.; Zhang, H.; Jorda, M.A.; et al. Sustained PU.1 levels balance cell-cycle regulators to prevent exhaustion of adult hematopoietic stem cells. Mol. Cell 2013, 49, 934–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, B.; Vogler, T.O.; Narayanagari, S.; Bartholdy, B.; Todorova, T.I.; Ferreira, M.D.S.; Chen, J.; Yu, Y.; Mayer, J.; Barreyro, L.; et al. Minimal PU.1 reduction induces a preleukemic state and promotes development of acute myeloid leukemia. Nat. Med. 2015, 21, 1172–1181. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, P.S.; Schwarzfischer, M.; Loeffler, D.; Kokkaliaris, K.; Hilsenbeck, O.; Moritz, N.; Endele, M.; Filipczyk, A.; Gambardella, A.; Ahmed, N.; et al. Early myeloid lineage choice is not initiated by random PU.1 to GATA1 protein ratios. Nature 2016, 535, 299–302. [Google Scholar] [CrossRef]
- Kirstetter, P.; Anderson, K.; Porse, B.; Jacobsen, S.E.W.; Nerlov, C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat. Immunol. 2006, 7, 1048–1056. [Google Scholar] [CrossRef]
- Strasser, M.K.; Hoppe, P.S.; Loeffler, D.; Kokkaliaris, K.D.; Schroeder, T.; Theis, F.J.; Marr, C. Lineage marker synchrony in hematopoietic genealogies refutes the PU.1/GATA1 toggle switch paradigm. Nat. Commun. 2018, 9, 2697. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Y.; Miyanishi, M.; Wang, S.; Yamazaki, S.; Sinha, R.; Kao, K.S.; Seita, J.; Sahoo, D.; Nakauchi, S.Y.H.; Weissman, I.L. Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature 2016, 530, 223–227. [Google Scholar] [CrossRef]
- Sakamaki, T.; Kao, K.S.; Nishi, K.; Chen, J.Y.; Sadaoka, K.; Fujii, M.; Takaori-Kondo, A.; Weissman, I.L.; Miyanishi, M. Hoxb5 defines the heterogeneity of self-renewal capacity in the hematopoietic stem cell compartment. Biochem. Biophys. Res. Commun. 2021, 539, 34–41. [Google Scholar] [CrossRef]
- Min, I.M.; Pietramaggiori, G.; Kim, F.S.; Passegué, E.; Stevenson, K.E.; Wagers, A.J. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell 2008, 2, 380–391. [Google Scholar] [CrossRef] [Green Version]
- Van Der Meer, L.T.; Jansen, J.H.; Van Der Reijden, B.A. Gfi1 and Gfi1b: Key regulators of hematopoiesis. Leukemia 2010, 24, 1834–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiel, M.J.; Yilmaz, Ö.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weksberg, D.C.; Chambers, S.M.; Boles, N.C.; Goodell, M.A. CD150- side population cells represent a functionally distinct population of long-term hematopoietic stem cells. Blood 2008, 111, 2444–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challen, G.A.; Boles, N.C.; Chambers, S.M.; Goodell, M.A. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell 2010, 6, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Morita, Y.; Ema, H.; Nakauchi, H. Heterogeneity and hierarchy within the most primitive hematopoietic stem cell compartment. J. Exp. Med. 2010, 207, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Haas, S.; Hansson, J.; Klimmeck, D.; Loeffler, D.; Velten, L.; Uckelmann, H.; Wurzer, S.; Prendergast, M.; Schnell, A.; Hexel, K.; et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 2015, 17, 422–434. [Google Scholar] [CrossRef] [Green Version]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Graf, T.; Enver, T. Forcing cells to change lineages. Nature 2009, 462, 587–594. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, X.; Iwama, A.; Yu, C.; Smith, K.A.; Mueller, B.U.; Mueller, B.U.; Narravula, S.; Torbett, B.E.; Orkin, S.H.; et al. PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood 2000, 96, 2641–2648. [Google Scholar] [CrossRef]
- Wheat, J.C.; Sella, Y.; Willcockson, M.; Skoultchi, A.I.; Bergman, A.; Singer, R.H.; Steidl, U. Single-molecule imaging of transcription dynamics in somatic stem cells. Nature 2020, 583, 431–436. [Google Scholar] [CrossRef]
- Gazit, R.; Mandal, P.; Ebina, W.; Ben-Zvi, A.; Nombela-Arrieta, C.; Silberstein, L.E.; Rossi, D.J. Fgd5 identifies hematopoietic stem cells in the murine bone marrow. J. Exp. Med. 2014, 211, 1315–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapple, R.H.; Tseng, Y.-J.; Hu, T.; Kitano, A.; Takeichi, M.; Hoegenauer, K.A.; Nakada, D. Lineage tracing of murine adult hematopoietic stem cells reveals active contribution to steady-state hematopoiesis. Blood Adv. 2018, 2, 1220–1228. [Google Scholar] [CrossRef]
- Busch, K.; Klapproth, K.; Barile, M.; Flossdorf, M.; Holland-Letz, T.; Schlenner, S.; Reth, M.; Höfer, T.; Rodewald, H.-R. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 2015, 518, 542–546. [Google Scholar] [CrossRef]
- Hidalgo, A.; Peired, A.J.; Weiss, L.A.; Katayama, Y.; Frenette, P.S. The integrin alphaMbeta2 anchors hematopoietic progenitors in the bone marrow during enforced mobilization. Blood 2004, 104, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Bernitz, J.; Kim, H.S.; MacArthur, B.; Sieburg, H.; Moore, K. Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell 2016, 167, 1296–1309.e1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pla, A.S.; Macaulay, I.; Jensen, C.T.; Woll, P.S.; Luis, T.C.; Mead, A.; Moore, S.; Carella, C.; Matsuoka, S.; Jones, T.B.; et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 2013, 502, 232–236. [Google Scholar] [CrossRef]
- Noetzli, L.J.; French, S.L.; Machlus, K. New Insights Into the Differentiation of Megakaryocytes From Hematopoietic Progenitors. Arter. Thromb. Vasc. Biol. 2019, 39, 1288–1300. [Google Scholar] [CrossRef]
- Grover, A.; Pla, A.S.; Thongjuea, S.; Carrelha, J.; Giustacchini, A.; Gambardella, A.; Macaulay, I.; Mancini, E.; Luis, T.C.; Mead, A.; et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat. Commun. 2016, 7, 11075. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chavez, J.S.; Rabe, J.L.; Hernandez, G.; Mills, T.S.; Niño, K.E.; Davizon-Castillo, P.; Pietras, E.M. PU.1 Expression Defines Distinct Functional Activities in the Phenotypic HSC Compartment of a Murine Inflammatory Stress Model. Cells 2022, 11, 680. https://doi.org/10.3390/cells11040680
Chavez JS, Rabe JL, Hernandez G, Mills TS, Niño KE, Davizon-Castillo P, Pietras EM. PU.1 Expression Defines Distinct Functional Activities in the Phenotypic HSC Compartment of a Murine Inflammatory Stress Model. Cells. 2022; 11(4):680. https://doi.org/10.3390/cells11040680
Chicago/Turabian StyleChavez, James S., Jennifer L. Rabe, Giovanny Hernandez, Taylor S. Mills, Katia E. Niño, Pavel Davizon-Castillo, and Eric M. Pietras. 2022. "PU.1 Expression Defines Distinct Functional Activities in the Phenotypic HSC Compartment of a Murine Inflammatory Stress Model" Cells 11, no. 4: 680. https://doi.org/10.3390/cells11040680