
Pleiotropic Effects of the Protease-Activated Receptor 1 (PAR1) Inhibitor, Vorapaxar, on Atherosclerosis and Vascular Inflammation

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patient Studies
2.2. Experimental Mouse Study
2.3. Materials
2.4. Histology, Immunostaining, and Confocal Microscopy
2.5. qPCR
2.6. Isolation of Endothelial Cells
2.7. Human Monocytic Cells (THP-1) Cells
2.8. ELISA
2.9. Stimulation of Platelet-Rich Plasma and Platelet-Poor Plasma with Aortic Plaque Material
2.10. Stimulation of Human PRP with PPP from ApoEko Mice
2.11. Statistical Analysis
3. Results
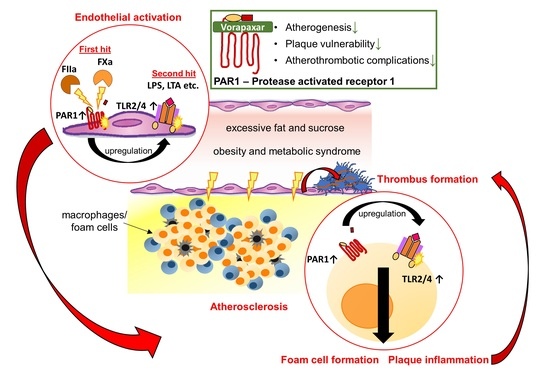
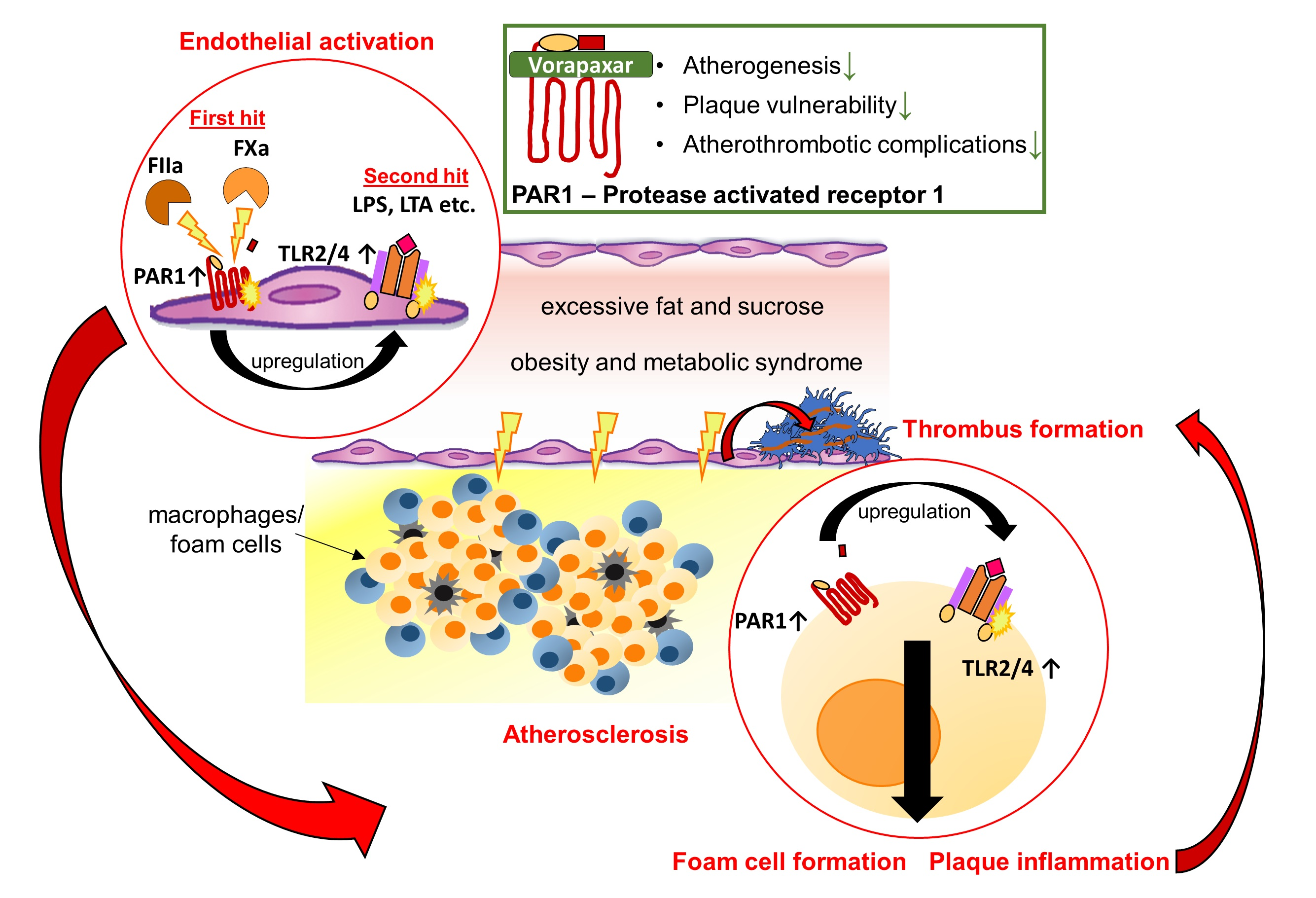
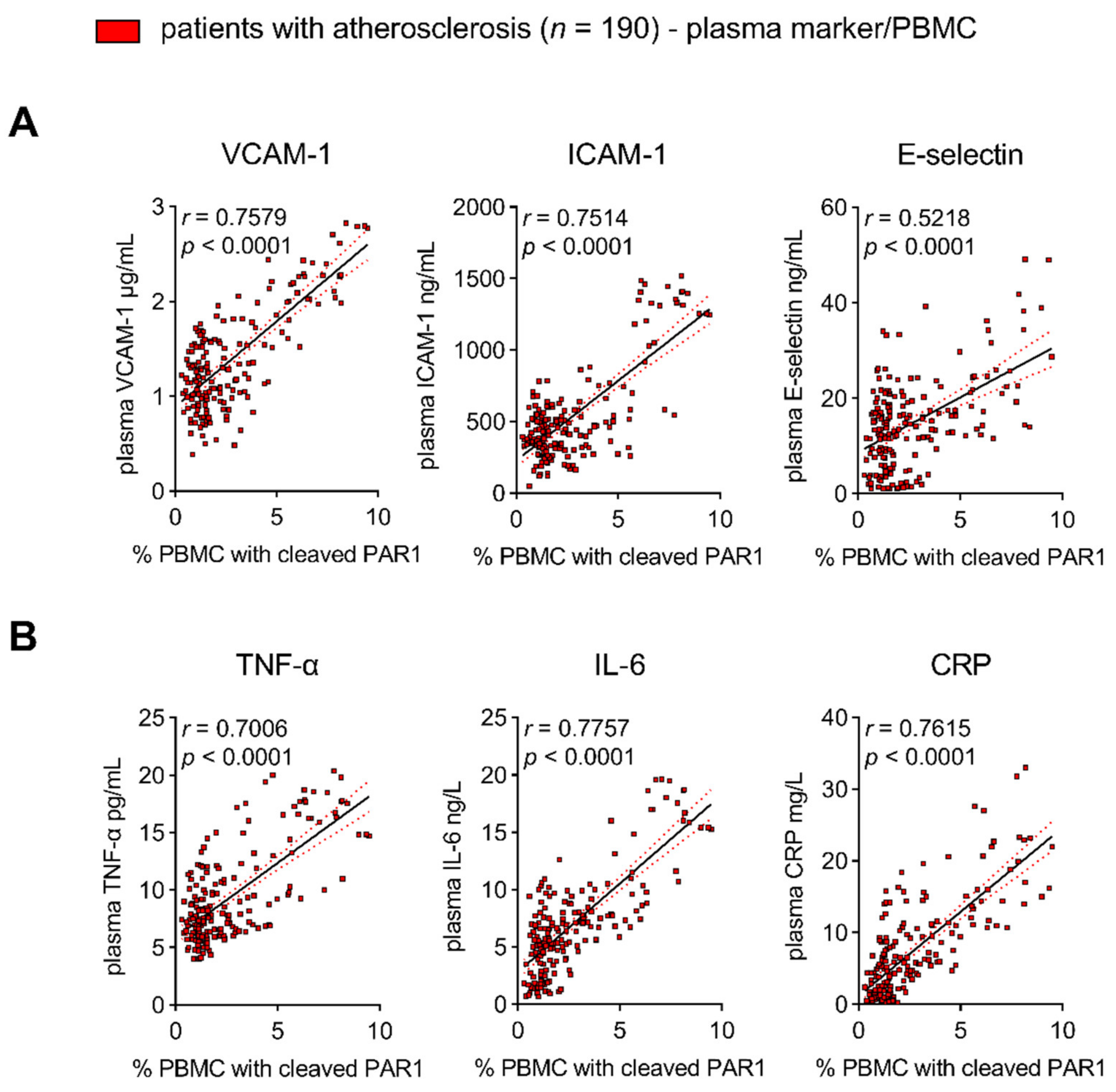
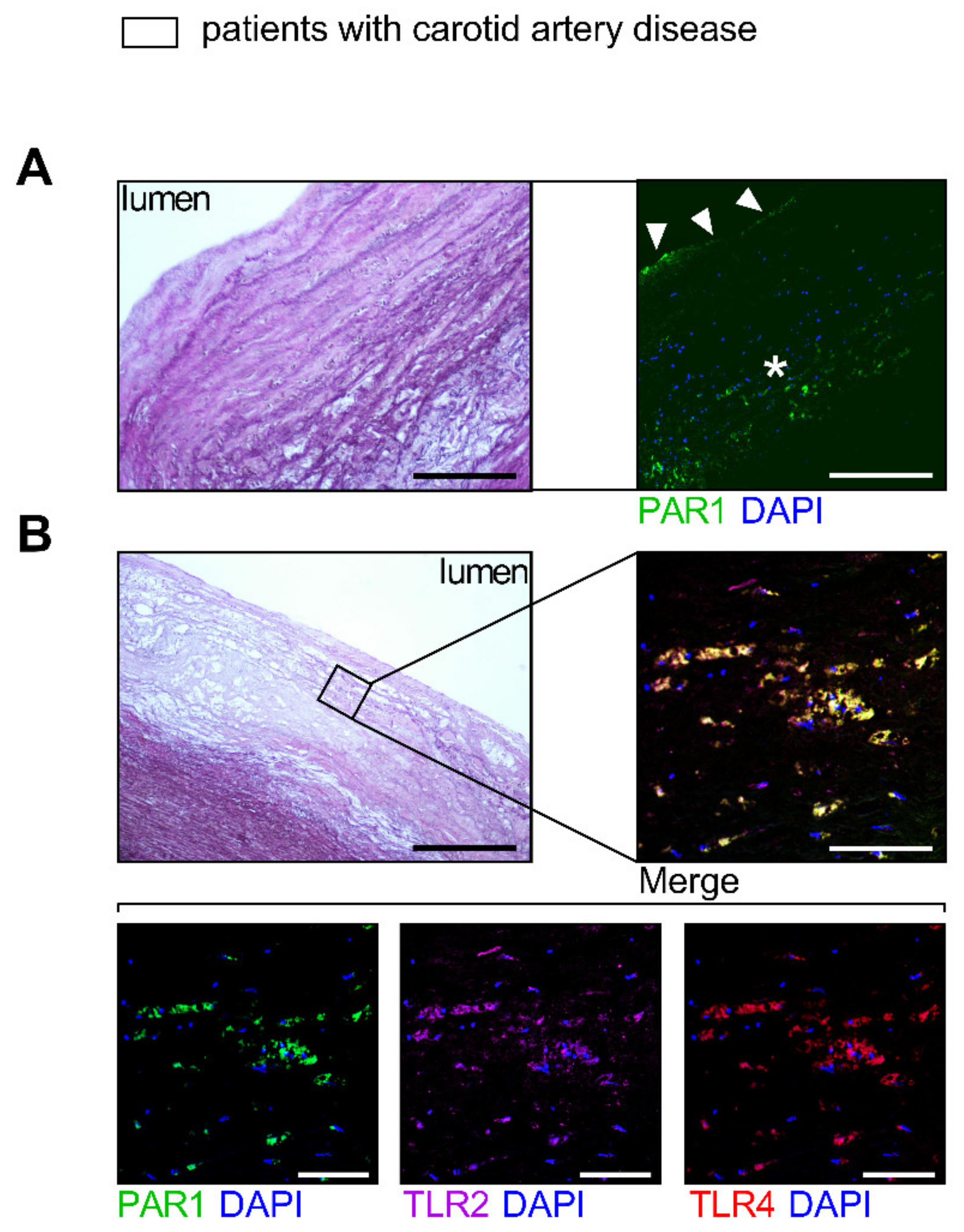
3.1. Target Identification of PAR1-Mediated Thrombo-Inflammation in Patients with Atherosclerotic Disease
3.1.1. PAR1 Activation Corresponds to Endothelial Activation and Vascular Inflammation
3.1.2. PAR1 Colocalizes with TLR2 and TLR4 in Human Atherosclerotic Disease
3.2. Evaluation of Pleiotropic, Vasoprotective Effects of the PAR1 Inhibitor Vorapaxar
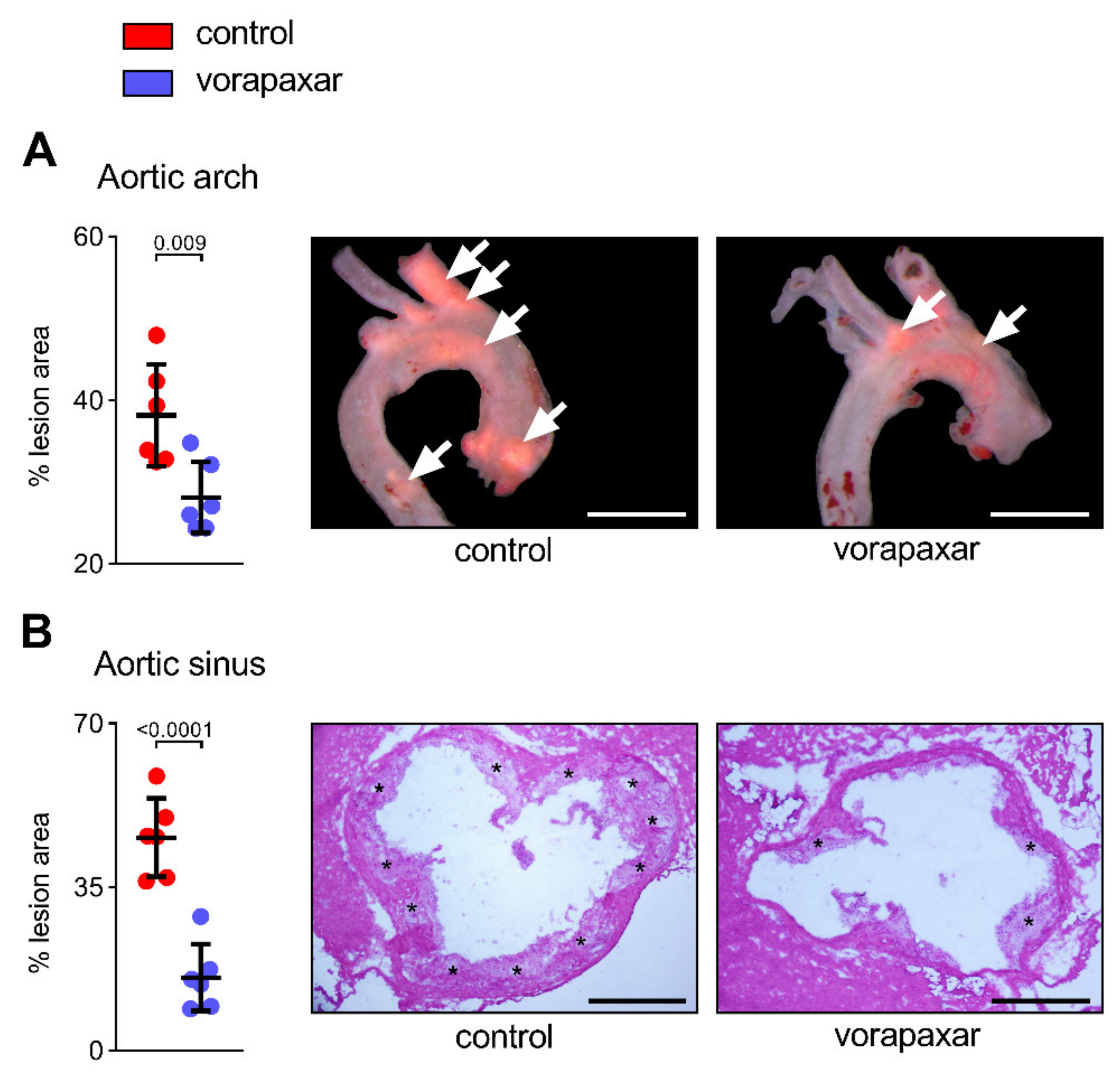
3.2.1. Vorapaxar Reduces de Novo Atherosclerosis in ApoEko Mice Fed an Atherogenic Diet
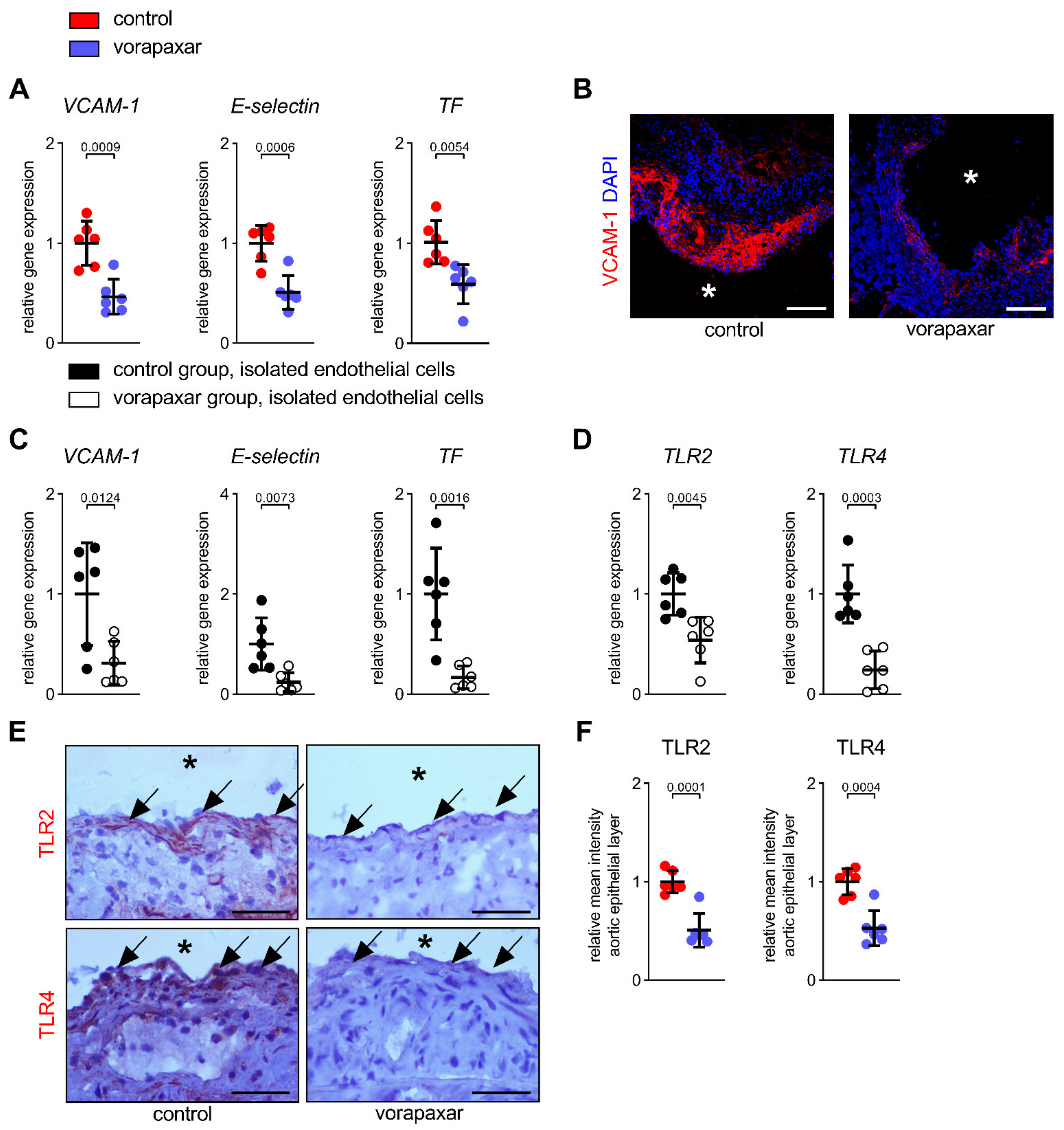
3.2.2. PAR1 Inhibition with Vorapaxar Attenuates Vascular Inflammation
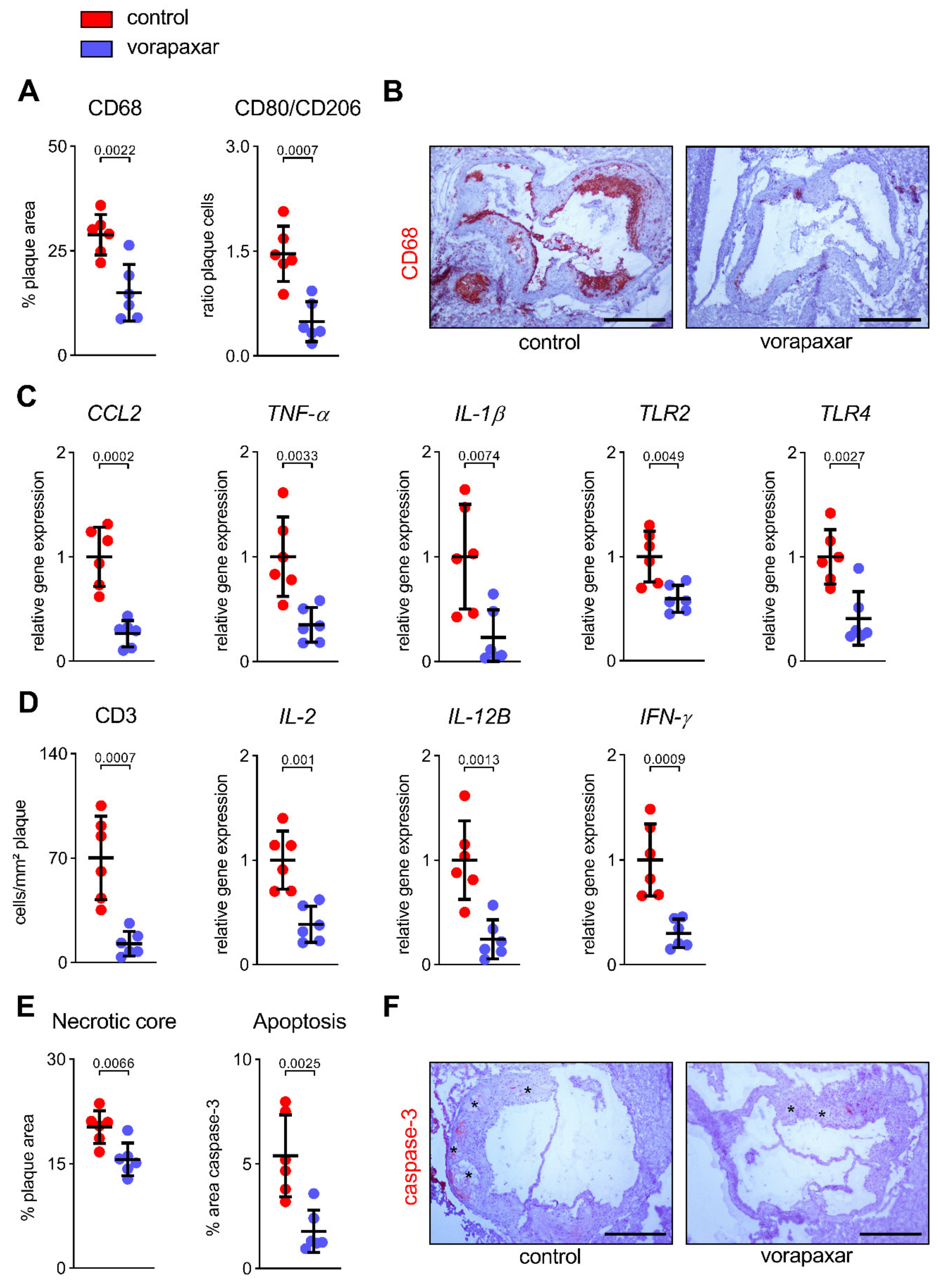
3.2.3. PAR1 Inhibition with Vorapaxar Impairs Foam Cell Formation
3.2.4. Vorapaxar Treatment Is Associated with an Anti-Inflammatory Cell and Cytokine Profile within Atherosclerotic Plaques
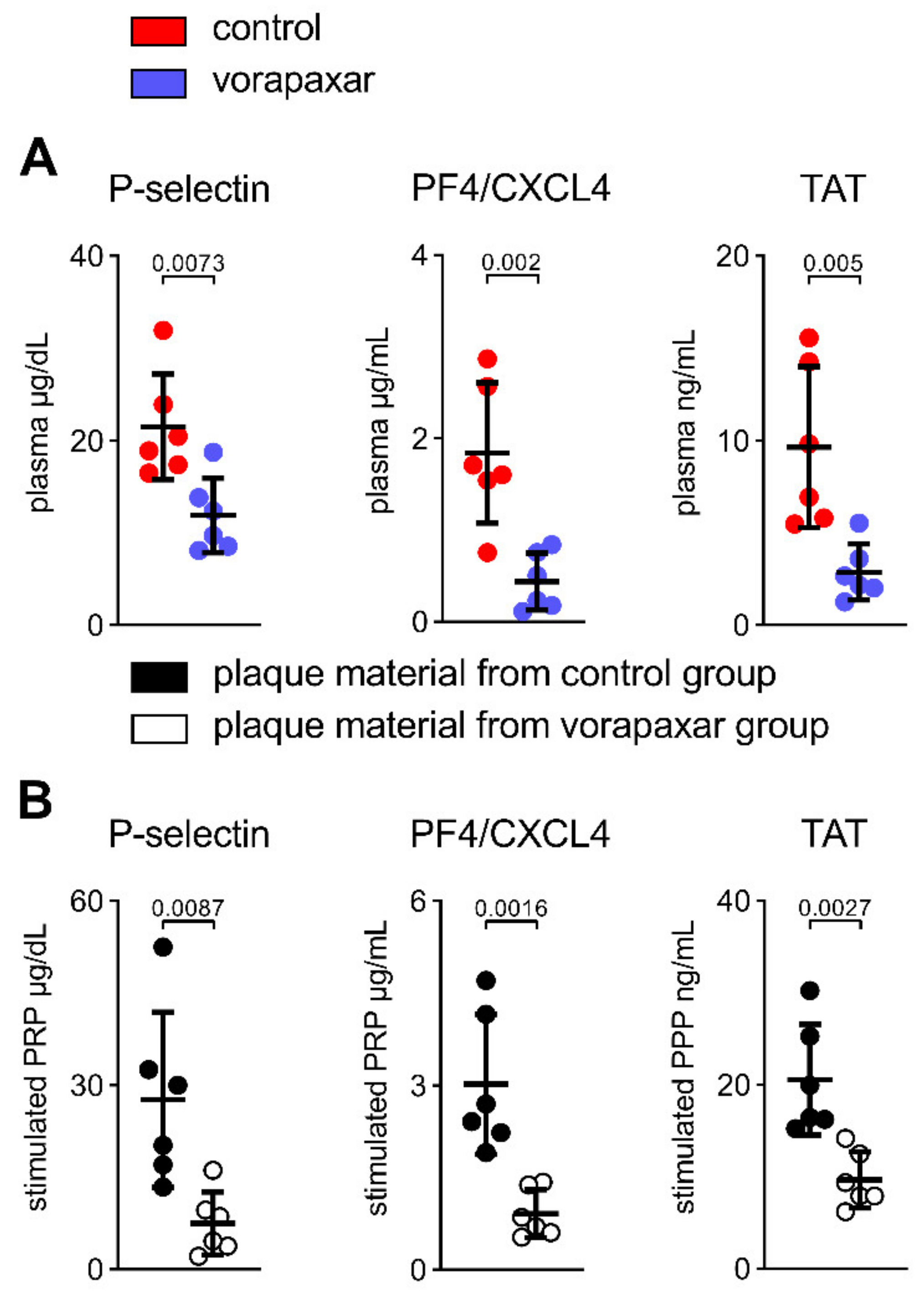
3.2.5. PAR1 Inhibition with Vorapaxar Reduced Thrombogenicity in Mice
4. Discussion
4.1. Target Identification of PAR1-Mediated Thrombo-Inflammation in Patients with Atherosclerotic Disease
4.2. The PAR1 Inhibitor Vorapaxar Attenuates de Novo Atherosclerosis
4.3. PAR1 Inhibition with Vorapaxar Attenuates Vascular Inflammation
4.4. PAR1 Inhibition with Vorapaxar Impairs Foam Cell Formation
4.5. Vorapaxar Treatment Is Associated with an Anti-Inflammatory Cell and Cytokine Profile within Atherosclerotic Plaques
4.6. Clinical Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- d’Alessandro, E.; Becker, C.; Bergmeier, W.; Bode, C.; Bourne, J.H.; Brown, H.; Buller, H.R.; Ten Cate-Hoek, A.J.; Ten Cate, V.; van Cauteren, Y.J.M.; et al. Thrombo-Inflammation in Cardiovascular Disease: An Expert Consensus Document from the Third Maastricht Consensus Conference on Thrombosis. Thromb. Haemost. 2020, 120, 538–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Cate, H.; Guzik, T.J.; Eikelboom, J.; Spronk, H.M.H. Pleiotropic actions of factor Xa inhibition in cardiovascular prevention: Mechanistic insights and implications for anti-thrombotic treatment. Cardiovasc. Res. 2021, 117, 2030–2044. [Google Scholar] [CrossRef] [PubMed]
- Kremers, B.M.M.; Ten Cate, H.; Spronk, H.M.H. Pleiotropic effects of the hemostatic system. J. Thromb. Haemost. 2018, 16, 1464–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüscher, T.F. Atherosclerosis, neoatherosclerosis, and vascular disease. Eur. Heart J. 2015, 36, 2121–2123. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, T.F. Understanding and preventing atherosclerosis: From bench to bedside. Eur. Heart J. 2019, 40, 323–327. [Google Scholar] [CrossRef] [Green Version]
- Aboyans, V.; Bauersachs, R.; Mazzolai, L.; Brodmann, M.; Palomares, J.F.R.; Debus, S.; Collet, J.P.; Drexel, H.; Espinola-Klein, C.; Lewis, B.S.; et al. Antithrombotic therapies in aortic and peripheral arterial diseases in 2021: A consensus document from the ESC working group on aorta and peripheral vascular diseases, the ESC working group on thrombosis, and the ESC working group on cardiovascular pharmacotherapy. Eur. Heart J. 2021, 42, 4013–4024. [Google Scholar] [CrossRef] [PubMed]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. J. Prev. Cardiol. 2021, 41, 3227–3337. [Google Scholar] [CrossRef]
- Willis Fox, O.; Preston, R.J.S. Molecular basis of protease-activated receptor 1 signaling diversity. J. Thromb. Haemost. 2020, 18, 6–16. [Google Scholar] [CrossRef]
- Flaumenhaft, R.; De Ceunynck, K. Targeting PAR1: Now What? Trends Pharmacol. Sci. 2017, 38, 701–716. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Nieman, M.T.; Kerlin, B.A. Protease-activated receptors: An illustrated review. Res. Pract. Thromb. Haemost. 2021, 5, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Gieseler, F.; Ungefroren, H.; Settmacher, U.; Hollenberg, M.D.; Kaufmann, R. Proteinase-activated receptors (PARs)—Focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun. Signal. CCS 2013, 11, 86. [Google Scholar] [CrossRef] [Green Version]
- Posma, J.J.; Posthuma, J.J.; Spronk, H.M. Coagulation and non-coagulation effects of thrombin. J. Thromb. Haemost. 2016, 14, 1908–1916. [Google Scholar] [CrossRef] [Green Version]
- Posma, J.J.; Grover, S.P.; Hisada, Y.; Owens, A.P., 3rd; Antoniak, S.; Spronk, H.M.; Mackman, N. Roles of Coagulation Proteases and PARs (Protease-Activated Receptors) in Mouse Models of Inflammatory Diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Nelken, N.A.; Soifer, S.J.; O’Keefe, J.; Vu, T.K.; Charo, I.F.; Coughlin, S.R. Thrombin receptor expression in normal and atherosclerotic human arteries. J. Clin. Investig. 1992, 90, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, M.A.C.; Prange, K.H.M.; Slenders, L.; Örd, T.; Elbersen, D.; Boltjes, A.; de Jager, S.C.A.; Asselbergs, F.W.; de Borst, G.J.; Aavik, E.; et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res. 2020, 127, 1437–1455. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Insel, P.A. Proteinase-activated receptor 1: A target for repurposing in the treatment of COVID-19? Br. J. Pharmacol. 2020, 177, 4971–4974. [Google Scholar] [CrossRef]
- Raghavan, S.; Singh, N.K.; Mani, A.M.; Rao, G.N. Protease-activated receptor 1 inhibits cholesterol efflux and promotes atherogenesis via cullin 3-mediated degradation of the ABCA1 transporter. J. Biol. Chem. 2018, 293, 10574–10589. [Google Scholar] [CrossRef] [Green Version]
- Rana, R.; Huang, T.; Koukos, G.; Fletcher, E.K.; Turner, S.E.; Shearer, A.; Gurbel, P.A.; Rade, J.J.; Kimmelstiel, C.D.; Bliden, K.P.; et al. Noncanonical Matrix Metalloprotease 1-Protease-Activated Receptor 1 Signaling Drives Progression of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1368–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jirouskova, M.; Shet, A.S.; Johnson, G.J. A guide to murine platelet structure, function, assays, and genetic alterations. J. Thromb. Haemost. 2007, 5, 661–669. [Google Scholar] [CrossRef]
- French, S.L.; Paramitha, A.C.; Moon, M.J.; Dickins, R.A.; Hamilton, J.R. Humanizing the Protease-Activated Receptor (PAR) Expression Profile in Mouse Platelets by Knocking PAR1 into the Par3 Locus Reveals PAR1 Expression Is Not Tolerated in Mouse Platelets. PLoS ONE 2016, 11, e0165565. [Google Scholar] [CrossRef] [PubMed]
- Friebel, J.; Weithauser, A.; Witkowski, M.; Rauch, B.H.; Savvatis, K.; Dörner, A.; Tabaraie, T.; Kasner, M.; Moos, V.; Bösel, D.; et al. Protease-activated receptor 2 deficiency mediates cardiac fibrosis and diastolic dysfunction. Eur. Heart J. 2019, 40, 3318–3332. [Google Scholar] [CrossRef]
- Gast, M.; Rauch, B.H.; Nakagawa, S.; Haghikia, A.; Jasina, A.; Haas, J.; Nath, N.; Jensen, L.; Stroux, A.; Böhm, A.; et al. Immune system-mediated atherosclerosis caused by deficiency of long non-coding RNA MALAT1 in ApoE-/-mice. Cardiovasc. Res. 2019, 115, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Vogt, K.; Mahajan-Thakur, S.; Wolf, R.; Bröderdorf, S.; Vogel, C.; Böhm, A.; Ritter, C.A.; Gräler, M.; Oswald, S.; Greinacher, A.; et al. Release of Platelet-Derived Sphingosine-1-Phosphate Involves Multidrug Resistance Protein 4 (MRP4/ABCC4) and Is Inhibited by Statins. Thromb. Haemost. 2018, 118, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Borissoff, J.I.; Heeneman, S.; Kilinç, E.; Kassák, P.; Van Oerle, R.; Winckers, K.; Govers-Riemslag, J.W.; Hamulyák, K.; Hackeng, T.M.; Daemen, M.J.; et al. Early atherosclerosis exhibits an enhanced procoagulant state. Circulation 2010, 122, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Jaén, R.I.; Val-Blasco, A.; Prieto, P.; Gil-Fernández, M.; Smani, T.; López-Sendón, J.L.; Delgado, C.; Boscá, L.; Fernández-Velasco, M. Innate Immune Receptors, Key Actors in Cardiovascular Diseases. JACC Basic Transl. Sci. 2020, 5, 735–749. [Google Scholar] [CrossRef]
- Carnevale, R.; Sciarretta, S.; Valenti, V.; di Nonno, F.; Calvieri, C.; Nocella, C.; Frati, G.; Forte, M.; d’Amati, G.; Pignataro, M.G.; et al. Low-grade endotoxaemia enhances artery thrombus growth via Toll-like receptor 4: Implication for myocardial infarction. Eur. Heart J. 2020, 41, 3156–3165. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Lüscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef]
- Witkowski, M.; Weithauser, A.; Tabaraie, T.; Steffens, D.; Kränkel, N.; Witkowski, M.; Stratmann, B.; Tschoepe, D.; Landmesser, U.; Rauch-Kroehnert, U. Micro-RNA-126 Reduces the Blood Thrombogenicity in Diabetes Mellitus via Targeting of Tissue Factor. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1263–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkowski, M.; Witkowski, M.; Saffarzadeh, M.; Friebel, J.; Tabaraie, T.; Ta Bao, L.; Chakraborty, A.; Dörner, A.; Stratmann, B.; Tschoepe, D.; et al. Vascular miR-181b controls tissue factor-dependent thrombogenicity and inflammation in type 2 diabetes. Cardiovasc. Diabetol. 2020, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Antoniak, S.; Mackman, N. New Cellular Source of TF (Tissue Factor)-Positive Extracellular Vesicles in the Circulation. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef] [Green Version]
- Grover, S.P.; Mackman, N. Tissue factor in atherosclerosis and atherothrombosis. Atherosclerosis 2020, 307, 80–86. [Google Scholar] [CrossRef]
- Bogdanov, V.Y.; Versteeg, H.H. “Soluble Tissue Factor” in the 21st Century: Definitions, Biochemistry, and Pathophysiological Role in Thrombus Formation. Semin. Thromb. Hemost. 2015, 41, 700–707. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, R.; Ozhegov, E.; van den Berg, Y.W.; Aronow, B.J.; Franco, R.S.; Palascak, M.B.; Fallon, J.T.; Ruf, W.; Versteeg, H.H.; Bogdanov, V.Y. Splice variants of tissue factor promote monocyte-endothelial interactions by triggering the expression of cell adhesion molecules via integrin-mediated signaling. J. Thromb. Haemost. 2011, 9, 2087–2096. [Google Scholar] [CrossRef]
- Witkowski, M.; Witkowski, M.; Friebel, J.; Buffa, J.A.; Li, X.S.; Wang, Z.; Sangwan, N.; Li, L.; DiDonato, J.A.; Tizian, C.; et al. Vascular endothelial Tissue Factor contributes to trimethylamine N-oxide-enhanced arterial thrombosis. Cardiovasc. Res. 2021, in press. [Google Scholar] [CrossRef]
- Jones, S.M.; Mann, A.; Conrad, K.; Saum, K.; Hall, D.E.; McKinney, L.M.; Robbins, N.; Thompson, J.; Peairs, A.D.; Camerer, E.; et al. PAR2 (Protease-Activated Receptor 2) Deficiency Attenuates Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1271–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruf, W. Proteases, Protease-Activated Receptors, and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1252–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versteeg, H.H.; Ruf, W. Emerging insights in tissue factor-dependent signaling events. Semin. Thromb. Hemost. 2006, 32, 24–32. [Google Scholar] [CrossRef]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Liu, W.; Hua, X.; Chen, X.; Chang, Y.; Hu, Y.; Xu, Z.; Song, J. Single-Cell Transcriptomic Atlas of Different Human Cardiac Arteries Identifies Cell Types Associated with Vascular Physiology. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1408–1427. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, S.; Sidiropoulou, S.; Moschonas, I.C.; Tselepis, A.D. Factor Xa and thrombin induce endothelial progenitor cell activation. The effect of direct oral anticoagulants. Platelets 2020, 32, 807–814. [Google Scholar] [CrossRef]
- Van den Eshof, B.L.; Hoogendijk, A.J.; Simpson, P.J.; van Alphen, F.P.J.; Zanivan, S.; Mertens, K.; Meijer, A.B.; van den Biggelaar, M. Paradigm of Biased PAR1 (Protease-Activated Receptor-1) Activation and Inhibition in Endothelial Cells Dissected by Phosphoproteomics. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1891–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinozawa, E.; Nakayama, M.; Imura, Y. TAK-442, a Direct Factor Xa Inhibitor, Inhibits Monocyte Chemoattractant Protein 1 Production in Endothelial Cells via Involvement of Protease-Activated Receptor 1. Front. Pharmacol. 2018, 9, 1431. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Hu, P.; Wang, J.; Jiang, J.; Lai, J. Vorapaxar stabilizes permeability of the endothelial barrier under cholesterol stimulation via the AKT/JNK and NF-κB signaling pathways. Mol. Med. Rep. 2019, 19, 5291–5300. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.K.; Yang, X.V.; Fernández, J.A.; Xu, X.; Mosnier, L.O.; Griffin, J.H. Apolipoprotein E Receptor 2 Mediates Activated Protein C-Induced Endothelial Akt Activation and Endothelial Barrier Stabilization. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 518–524. [Google Scholar] [CrossRef] [Green Version]
- Stannard, A.K.; Riddell, D.R.; Sacre, S.M.; Tagalakis, A.D.; Langer, C.; von Eckardstein, A.; Cullen, P.; Athanasopoulos, T.; Dickson, G.; Owen, J.S. Cell-derived apolipoprotein E (ApoE) particles inhibit vascular cell adhesion molecule-1 (VCAM-1) expression in human endothelial cells. J. Biol. Chem. 2001, 276, 46011–46016. [Google Scholar] [CrossRef] [Green Version]
- Soh, U.J.; Trejo, J. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through β-arrestin and dishevelled-2 scaffolds. Proc. Natl. Acad. Sci. USA 2011, 108, E1372–E1380. [Google Scholar] [CrossRef] [Green Version]
- Niessen, F.; Furlan-Freguia, C.; Fernández, J.A.; Mosnier, L.O.; Castellino, F.J.; Weiler, H.; Rosen, H.; Griffin, J.H.; Ruf, W. Endogenous EPCR/aPC-PAR1 signaling prevents inflammation-induced vascular leakage and lethality. Blood 2009, 113, 2859–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riewald, M.; Petrovan, R.J.; Donner, A.; Mueller, B.M.; Ruf, W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 2002, 296, 1880–1882. [Google Scholar] [CrossRef]
- Roshan, M.H.; Tambo, A.; Pace, N.P. The Role of TLR2, TLR4, and TLR9 in the Pathogenesis of Atherosclerosis. Int. J. Inflamm. 2016, 2016, 1532832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edfeldt, K.; Swedenborg, J.; Hansson, G.K.; Yan, Z.Q. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation 2002, 105, 1158–1161. [Google Scholar] [CrossRef] [Green Version]
- Weithauser, A.; Witkowski, M.; Rauch, U. The Role of Protease-Activated Receptors for the Development of Myocarditis: Possible Therapeutic Implications. Curr. Pharm. Des. 2016, 22, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Weithauser, A.; Rauch, U. Role of protease-activated receptors for the innate immune response of the heart. Trends Cardiovasc. Med. 2014, 24, 249–255. [Google Scholar] [CrossRef]
- Antoniak, S.; Owens, A.P., 3rd; Baunacke, M.; Williams, J.C.; Lee, R.D.; Weithauser, A.; Sheridan, P.A.; Malz, R.; Luyendyk, J.P.; Esserman, D.A.; et al. PAR-1 contributes to the innate immune response during viral infection. J. Clin. Investig. 2013, 123, 1310–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weithauser, A.; Bobbert, P.; Antoniak, S.; Bohm, A.; Rauch, B.H.; Klingel, K.; Savvatis, K.; Kroemer, H.K.; Tschope, C.; Stroux, A.; et al. Protease-activated receptor-2 regulates the innate immune response to viral infection in a coxsackievirus B3-induced myocarditis. J. Am. Coll. Cardiol. 2013, 62, 1737–1745. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, S.; Singh, N.K.; Gali, S.; Mani, A.M.; Rao, G.N. Protein Kinase Cθ Via Activating Transcription Factor 2-Mediated CD36 Expression and Foam Cell Formation of Ly6C(hi) Cells Contributes to Atherosclerosis. Circulation 2018, 138, 2395–2412. [Google Scholar] [CrossRef]
- Boro, M.; Govatati, S.; Kumar, R.; Singh, N.K.; Pichavaram, P.; Traylor, J.G., Jr.; Orr, A.W.; Rao, G.N. Thrombin-Par1 signaling axis disrupts COP9 signalosome subunit 3-mediated ABCA1 stabilization in inducing foam cell formation and atherogenesis. Cell Death Differ. 2021, 28, 780–798. [Google Scholar] [CrossRef]
- Izem, L.; Bialkowska, K.; Pluskota, E.; Das, M.; Das, R.; Nieman, M.T.; Plow, E.F. Plasminogen-induced foam cell formation by macrophages occurs through a histone 2B (H2B)-PAR1 pathway and requires integrity of clathrin-coated pits. J. Thromb. Haemost. 2021, 19, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Hurley, A.; Smith, M.; Karpova, T.; Hasley, R.B.; Belkina, N.; Shaw, S.; Balenga, N.; Druey, K.M.; Nickel, E.; Packard, B.; et al. Enhanced effector function of CD8(+) T cells from healthy controls and HIV-infected patients occurs through thrombin activation of protease-activated receptor 1. J. Infect. Dis. 2013, 207, 638–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Zambrano, M.; Rodriguez-Montesinos, J.; Crespo-Avilan, G.E.; Muñoz-Vega, M.; Preissner, K.T. Thrombin Promotes Macrophage Polarization into M1-Like Phenotype to Induce Inflammatory Responses. Thromb. Haemost. 2020, 120, 658–670. [Google Scholar] [CrossRef] [PubMed]
- López, M.L.; Bruges, G.; Crespo, G.; Salazar, V.; Deglesne, P.A.; Schneider, H.; Cabrera-Fuentes, H.; Schmitz, M.L.; Preissner, K.T. Thrombin selectively induces transcription of genes in human monocytes involved in inflammation and wound healing. Thromb. Haemost. 2014, 112, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Baron, M.; Bouhlel, M.A.; Vanhoutte, J.; Copin, C.; Sebti, Y.; Derudas, B.; Mayi, T.; Bories, G.; Tailleux, A.; et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARγ and LXRα pathways. Circ. Res. 2011, 108, 985–995. [Google Scholar] [CrossRef]
- Moon, J.Y.; Franchi, F.; Rollini, F.; Angiolillo, D.J. Role for Thrombin Receptor Antagonism with Vorapaxar in Secondary Prevention of Atherothrombotic Events: From Bench to Bedside. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 23–37. [Google Scholar] [CrossRef]
- Weitz, J.I.; Angiolillo, D.J.; Geisler, T.; Heitmeier, S. Dual Pathway Inhibition for Vascular Protection in Patients with Atherosclerotic Disease: Rationale and Review of the Evidence. Thromb. Haemost. 2020, 120, 1147–1158. [Google Scholar] [CrossRef]
- Hofmann, C.; Völkers, M.; Katus, H.A. Targeting coagulation in heart failure with preserved ejection fraction and cardiac fibrosis. Eur. Heart J. 2019, 40, 3333–3335. [Google Scholar] [CrossRef]
- Friebel, J.; Witkowski, M.; Rauch, U. Treating the unstable atherosclerotic plaque by targeting activated factor X—Anticoagulation and beyond. Circ. J. Off. J. Jpn. Circ. Soc. 2015, 79, 2329–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoergenhofer, C.; Schwameis, M.; Gelbenegger, G.; Buchtele, N.; Thaler, B.; Mussbacher, M.; Schabbauer, G.; Wojta, J.; Jilma-Stohlawetz, P.; Jilma, B. Inhibition of Protease-Activated Receptor (PAR1) Reduces Activation of the Endothelium, Coagulation, Fibrinolysis and Inflammation during Human Endotoxemia. Thromb. Haemost. 2018, 118, 1176–1184. [Google Scholar] [CrossRef]
- Vergallo, R.; Crea, F. Atherosclerotic Plaque Healing. N. Engl. J. Med. 2020, 383, 846–857. [Google Scholar] [CrossRef]
- Leistner, D.M.; Kränkel, N.; Meteva, D.; Abdelwahed, Y.S.; Seppelt, C.; Stähli, B.E.; Rai, H.; Skurk, C.; Lauten, A.; Mochmann, H.C.; et al. Differential immunological signature at the culprit site distinguishes acute coronary syndrome with intact from acute coronary syndrome with ruptured fibrous cap: Results from the prospective translational OPTICO-ACS study. Eur. Heart J. 2020, 41, 3549–3560. [Google Scholar] [CrossRef]
- Tomaniak, M.; Katagiri, Y.; Modolo, R.; de Silva, R.; Khamis, R.Y.; Bourantas, C.V.; Torii, R.; Wentzel, J.J.; Gijsen, F.J.H.; van Soest, G.; et al. Vulnerable plaques and patients: State-of-the-art. Eur. Heart J. 2020, 41, 2997–3004. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation during the life cycle of the atherosclerotic plaque. Cardiovasc. Res. 2021, 117, 2525–2535. [Google Scholar] [CrossRef]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.K. Reassessing the Mechanisms of Acute Coronary Syndromes. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Schumski, A.; Ortega-Gómez, A.; Wichapong, K.; Winter, C.; Lemnitzer, P.; Viola, J.R.; Pinilla-Vera, M.; Folco, E.; Solis-Mezarino, V.; Völker-Albert, M.; et al. Endotoxinemia Accelerates Atherosclerosis Through Electrostatic Charge-Mediated Monocyte Adhesion. Circulation 2021, 143, 254–266. [Google Scholar] [CrossRef]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker Cohort | EMB Cohort | |
|---|---|---|
| (n = 190) | (n = 12) | |
| Age, yrs | 70.3 ± 11.1 | 62.1 ± 10.5 |
| Male | 104/190 | 9/12 |
| Coronary artery disease | 190/190 | 12/12 |
| History of MI | 19/190 | 1/12 |
| Polyvascular Disease | ||
| Peripheral artery disease | 12/190 | 2/12 |
| Carotid artery disease | 51/190 | 0/12 |
| History of TIA/Stroke | 14/190 | 1/12 |
| Hypertension | 123/190 | 5/12 |
| Diabetes | 35/190 | 3/12 |
| BMI, kg/m² | 27.8 ± 5.8 | 26.6 ± 4.9 |
| CRP, mg/L | 7.4 ± 6.7 | 4.2 ± 4.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friebel, J.; Moritz, E.; Witkowski, M.; Jakobs, K.; Strässler, E.; Dörner, A.; Steffens, D.; Puccini, M.; Lammel, S.; Glauben, R.; et al. Pleiotropic Effects of the Protease-Activated Receptor 1 (PAR1) Inhibitor, Vorapaxar, on Atherosclerosis and Vascular Inflammation. Cells 2021, 10, 3517. https://doi.org/10.3390/cells10123517
Friebel J, Moritz E, Witkowski M, Jakobs K, Strässler E, Dörner A, Steffens D, Puccini M, Lammel S, Glauben R, et al. Pleiotropic Effects of the Protease-Activated Receptor 1 (PAR1) Inhibitor, Vorapaxar, on Atherosclerosis and Vascular Inflammation. Cells. 2021; 10(12):3517. https://doi.org/10.3390/cells10123517
Chicago/Turabian StyleFriebel, Julian, Eileen Moritz, Marco Witkowski, Kai Jakobs, Elisabeth Strässler, Andrea Dörner, Daniel Steffens, Marianna Puccini, Stella Lammel, Rainer Glauben, and et al. 2021. "Pleiotropic Effects of the Protease-Activated Receptor 1 (PAR1) Inhibitor, Vorapaxar, on Atherosclerosis and Vascular Inflammation" Cells 10, no. 12: 3517. https://doi.org/10.3390/cells10123517