
Therapeutic Benefit of Galectin-1: Beyond Membrane Repair, a Multifaceted Approach to LGMD2B

 , , , ,
, , , ,
Abstract
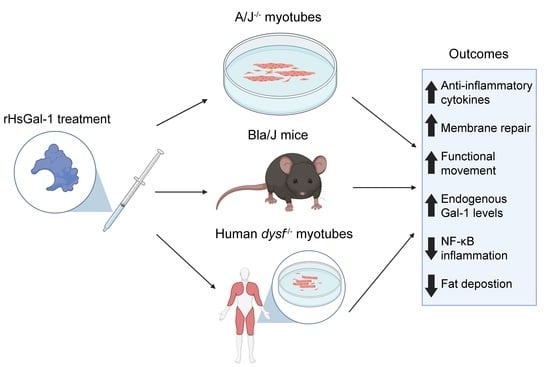
:
1. Introduction
2. Materials and Methods
2.1. Galectin-1 Construction, Production, and Purification
2.2. Animal Care
2.3. Cell Culture
2.4. Muscle Fiber Isolation
2.5. Laser Injury
2.6. RayBio Mouse Inflammation Array
2.7. Immunofluorescence
2.8. Western Blotting
2.9. Histology
2.10. Statistical Analyses
3. Results
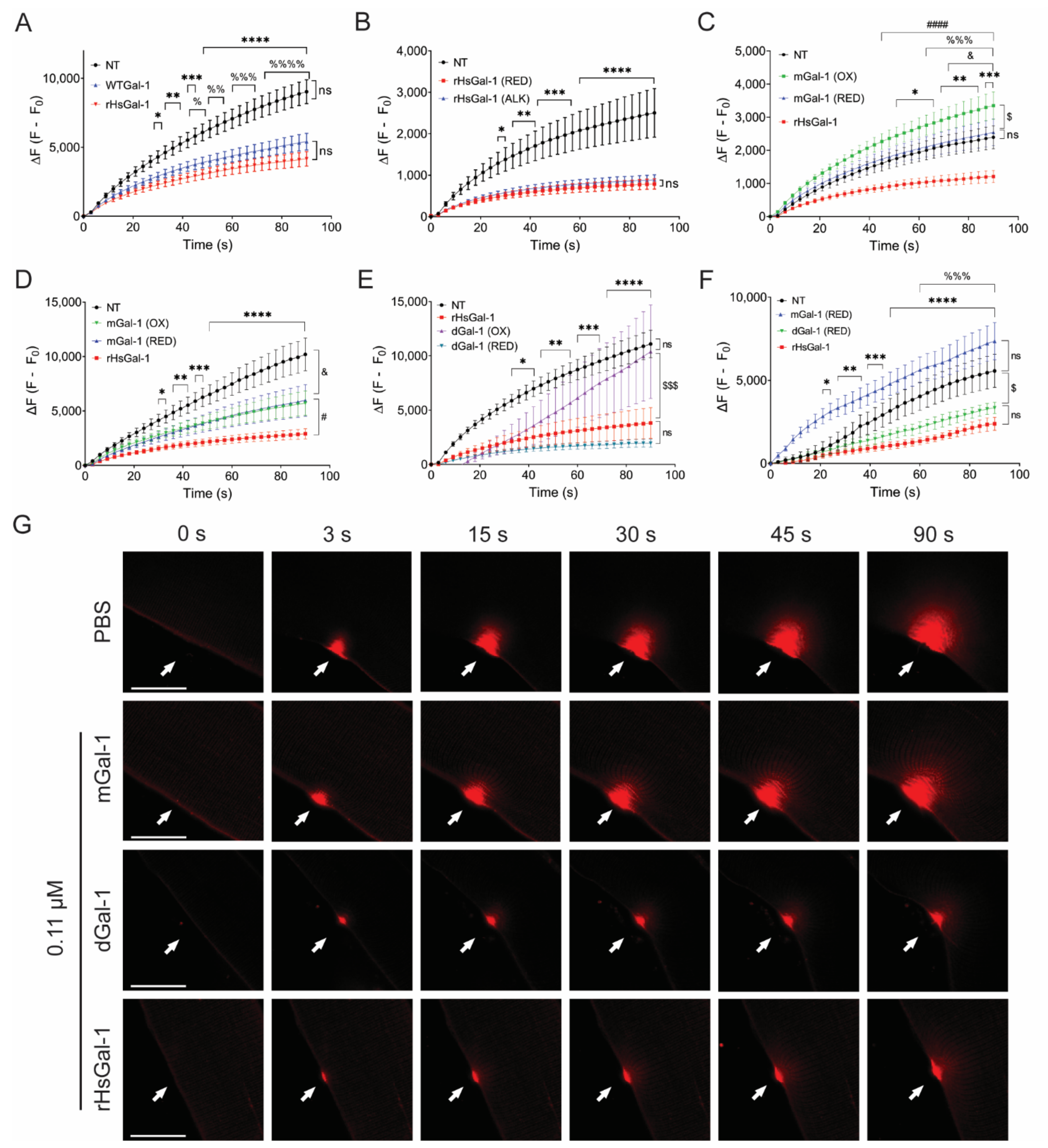
3.1. Reduced Dimeric Galectin-1 Is Responsible for Optimal Membrane Repair
3.2. rHsGal-1 Lowers Expression of Proteins in the NF-κB Pathway
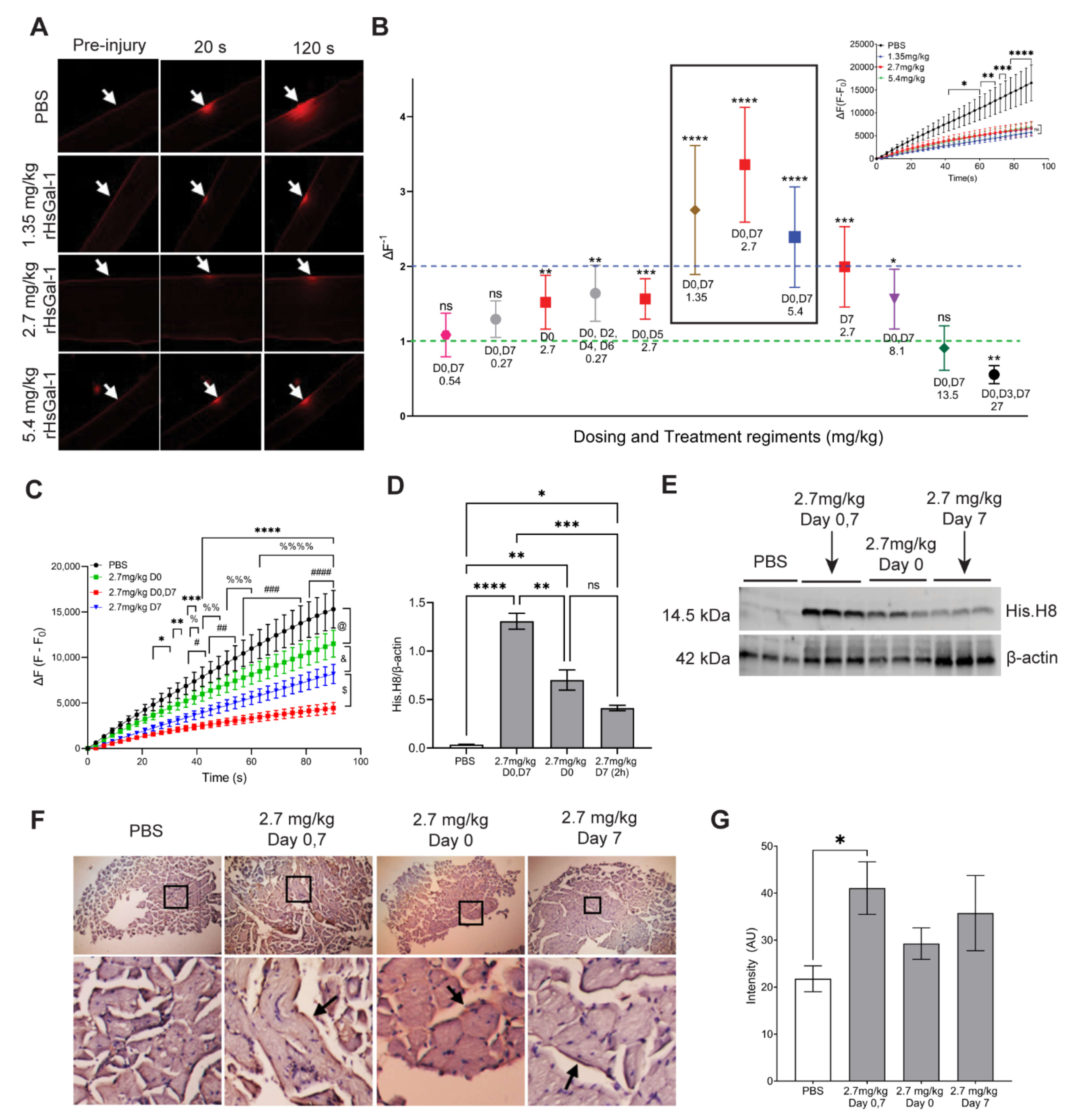
3.3. The Therapeutic Window for In Vivo rHsGal-1 Is from 1.35 to 8.1 mg/kg
3.4. One-Month rHsGal-1 Treatment Improves Translational and Biochemical Measures of LGMD2B
3.5. rHsGal-1 Treatment Upregulates Anti-Inflammatory Cytokine Secretion In Vitro and In Vivo
3.6. rHsGal-1 Improves Membrane Repair in Dysferlin Deficient, Patient-Derived Myotubes
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, R.; Campbell, K.P. Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 2007, 19, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef]
- Defour, A.; Van der Meulen, J.; Bhat, R.; Bigot, A.; Bashir, R.; Nagaraju, K.; Jaiswal, J. Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion. Cell Death Dis. 2014, 5, e1306. [Google Scholar] [CrossRef] [Green Version]
- Davenport, N.R.; Sonnemann, K.J.; Eliceiri, K.W.; Bement, W.M. Membrane dynamics during cellular wound repair. Mol. Biol. Cell 2016, 27, 2272–2285. [Google Scholar] [CrossRef]
- Bansal, D.; Campbell, K.P. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004, 14, 206–213. [Google Scholar] [CrossRef]
- Cárdenas, A.M.; González-Jamett, A.M.; Cea, L.A.; Bevilacqua, J.A.; Caviedes, P. Dysferlin function in skeletal muscle: Possible pathological mechanisms and therapeutical targets in dysferlinopathies. Exp. Neurol. 2016, 283, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M. Dysferlinopathy. In GeneReviews®; University of Washington: Seattle, WA, USA, 2015. [Google Scholar]
- Matsuda, C.; Kiyosue, K.; Nishino, I.; Goto, Y.; Hayashi, Y.K. Dysferlinopathy Fibroblasts Are Defective in Plasma Membrane Repair. PLoS Curr. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Haynes, V.R.; Keenan, S.N.; Bayliss, J.; Lloyd, E.M.; Meikle, P.J.; Grounds, M.D.; Watt, M.J. Dysferlin deficiency alters lipid metabolism and remodels the skeletal muscle lipidome in mice. J. Lipid Res. 2019, 60, 1350–1364. [Google Scholar] [CrossRef]
- Cohen, T.V.; Cohen, J.E.; Partridge, T.A. Myogenesis in dysferlin-deficient myoblasts is inhibited by an intrinsic inflammatory response. Neuromuscul. Disord. 2012, 22, 648–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraju, K.; Rawat, R.; Veszelovszky, E.; Thapliyal, R.; Kesari, A.; Sparks, S.; Raben, N.; Plotz, P.; Hoffman, E.P. Dysferlin deficiency enhances monocyte phagocytosis: A model for the inflammatory onset of limb-girdle muscular dystrophy 2B. Am. J. Pathol. 2008, 172, 774–785. [Google Scholar] [CrossRef] [Green Version]
- Rawat, R.; Cohen, T.V.; Ampong, B.; Francia, D.; Henriques-Pons, A.; Hoffman, E.P.; Nagaraju, K. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am. J. Pathol. 2010, 176, 2891–2900. [Google Scholar] [CrossRef]
- Cohen, T.V.; Many, G.M.; Fleming, B.D.; Gnocchi, V.F.; Ghimbovschi, S.; Mosser, D.M.; Hoffman, E.P.; Partridge, T.A. Upregulated IL-1β in dysferlin-deficient muscle attenuates regeneration by blunting the response to pro-inflammatory macrophages. Skelet Muscle 2015, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toegel, S.; Weinmann, D.; André, S.; Walzer, S.M.; Bilban, M.; Schmidt, S.; Chiari, C.; Windhager, R.; Krall, C.; Bennani-Baiti, I.M. Galectin-1 Couples Glycobiology to Inflammation in Osteoarthritis through the Activation of an NF-κB–Regulated Gene Network. J. Immunol. 2016, 196, 1910–1921. [Google Scholar] [CrossRef] [Green Version]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-κβ: A potential target in the management of vascular complications of diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, V.F.-S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnerz, T.; Hall, C.J. The diverse roles of phagocytes during bacterial and fungal infections and sterile inflammation: Lessons from zebrafish. Front. Immunol. 2020, 11, 1094. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hu, P. Skeletal muscle regeneration is modulated by inflammation. J. Orthop. Transl. 2018, 13, 25–32. [Google Scholar] [CrossRef]
- Costamagna, D.; Costelli, P.; Sampaolesi, M.; Penna, F. Role of Inflammation in Muscle Homeostasis and Myogenesis. Mediat. Inflamm. 2015, 2015, 805172. [Google Scholar] [CrossRef] [Green Version]
- Londhe, P.; Guttridge, D.C. Inflammation induced loss of skeletal muscle. Bone 2015, 80, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlini, L.; Cicognani, A.; Malaspina, E.; Gennari, M.; Gnudi, S.; Talim, B.; Franzoni, E. Early prednisone treatment in Duchenne muscular dystrophy. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 2003, 27, 222–227. [Google Scholar] [CrossRef]
- Sreetama, S.C.; Chandra, G.; Van der Meulen, J.H.; Ahmad, M.M.; Suzuki, P.; Bhuvanendran, S.; Nagaraju, K.; Hoffman, E.P.; Jaiswal, J.K. Membrane Stabilization by Modified Steroid Offers a Potential Therapy for Muscular Dystrophy Due to Dysferlin Deficit. Mol. Ther. 2018, 26, 2231–2242. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.C.; Reilich, P.; Thiele, S.; Schessl, J.; Schreiber, H.; Reiners, K.; Kress, W.; Müller-Reible, C.; Vorgerd, M.; Urban, P. Treatment of dysferlinopathy with deflazacort: A double-blind, placebo-controlled clinical trial. Orphanet J. Rare Dis. 2013, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Camby, I.; Le Mercier, M.; Lefranc, F.; Kiss, R. Galectin-1: A small protein with major functions. Glycobiology 2006, 16, 137R–157R. [Google Scholar] [CrossRef]
- Yu, X.; Scott, S.A.; Pritchard, R.; Houston, T.A.; Ralph, S.J.; Blanchard, H. Redox state influence on human galectin-1 function. Biochimie 2015, 116, 8–16. [Google Scholar] [CrossRef]
- Arthur, C.M.; Baruffi, M.D.; Cummings, R.D.; Stowell, S.R. Evolving Mechanistic Insights into Galectin Functions; Springer: New York, NY, USA, 2015; pp. 1–35. [Google Scholar]
- Guardia, C.M.; Caramelo, J.J.; Trujillo, M.; Méndez-Huergo, S.P.; Radi, R.; Estrin, D.A.; Rabinovich, G.A. Structural basis of redox-dependent modulation of galectin-1 dynamics and function. Glycobiology 2014, 24, 428–441. [Google Scholar] [CrossRef] [Green Version]
- Stowell, S.R.; Arthur, C.M.; Cummings, R.D.; Feasley, C.L. Alkylation of Galectin-1 with Iodoacetamide and Mass Spectrometric Mapping of the Sites of Incorporation; Springer: New York, NY, USA, 2015; pp. 51–62. [Google Scholar]
- Sundblad, V.; Morosi, L.G.; Geffner, J.R.; Rabinovich, G.A. Galectin-1: A Jack-of-All-Trades in the Resolution of Acute and Chronic Inflammation. J. Immunol. 2017, 199, 3721–3730. [Google Scholar] [CrossRef] [Green Version]
- Van Ry, P.M.; Wuebbles, R.D.; Key, M.; Burkin, D.J. Galectin-1 protein therapy prevents pathology and improves muscle function in the mdx mouse model of Duchenne muscular dystrophy. Mol. Ther. 2015, 23, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- Vallecillo-Zúniga, M.L.; Rathgeber, M.F.; Poulson, P.D.; Hayes, S.; Luddington, J.S.; Gill, H.N.; Teynor, M.; Kartchner, B.C.; Valdoz, J.; Stowell, C. Treatment with galectin-1 improves myogenic potential and membrane repair in dysferlin-deficient models. PLoS ONE 2020, 15, e0238441. [Google Scholar] [CrossRef] [PubMed]
- Stowell, S.R. Human galectin-1 recognition of poly-N-acetyllactosamine and chimeric polysaccharides. Glycobiology 2003, 14, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, M.; Cummings, R.D. Characterization of monomeric forms of galectin-1 generated by site-directed mutagenesis. Biochemistry 1996, 35, 13081–13088. [Google Scholar] [CrossRef] [PubMed]
- Philippi, S.; Bigot, A.; Marg, A.; Mouly, V.; Spuler, S.; Zacharias, U. Dysferlin-deficient immortalized human myoblasts and myotubes as a useful tool to study dysferlinopathy. PLoS Curr. 2012, 4, RRN1298. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; McNally, E.M. DNA Electroporation, Isolation and Imaging of Myofibers. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Earl, L.A.; Bi, S.; Baum, L.G. Galectin multimerization and lattice formation are regulated by linker region structure. Glycobiology 2010, 21, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.; Tymoczko, J.; Stryer, L. The amino acid sequence of a protein determines its three-dimensional structure. In Biochemistry, 2nd ed.; W.H. Freeman and Company: New York, NY, USA, 2002. [Google Scholar]
- Miura, T.; Takahashi, M.; Horie, H.; Kurushima, H.; Tsuchimoto, D.; Sakumi, K.; Nakabeppu, Y. Galectin-1β, a natural monomeric form of galectin-1 lacking its six amino-terminal residues promotes axonal regeneration but not cell death. Cell Death Differ. 2004, 11, 1076–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuebbles, R.D.; Cruz, V.; Van Ry, P.; Barraza-Flores, P.; Brewer, P.D.; Jones, P.; Burkin, D.J. Human Galectin-1 Improves Sarcolemma Stability and Muscle Vascularization in the mdx Mouse Model of Duchenne Muscular Dystrophy. Mol. Ther. Methods Clin. Dev. 2019, 13, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, N.; Nonneman, R.J.; Llanga, T.; Dial, C.F.; Riddick, N.V.; Hampton, T.; Moy, S.S.; Lehtimäki, K.K.; Ahtoniemi, T.; Puoliväli, J.; et al. Hip region muscular dystrophy and emergence of motor deficits in dysferlin-deficient Bla/J mice. Physiol. Rep. 2017, 5, e13173. [Google Scholar] [CrossRef] [Green Version]
- Collao, N.; Farup, J.; De Lisio, M. Role of metabolic stress and exercise in regulating fibro/adipogenic progenitors. Front. Cell Dev. Biol. 2020, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Sawano, S.; Mashima, D.; Ichitsubo, R.; Nakamura, M.; Tatsumi, R.; Ikeuchi, Y.; Mizunoya, W. Mouse soleus (slow) muscle shows greater intramyocellular lipid droplet accumulation than EDL (fast) muscle: Fiber type-specific analysis. J. Muscle Res. Cell Motil. 2017, 38, 163–173. [Google Scholar] [CrossRef]
- Grounds, M.D.; Terrill, J.R.; Radley-Crabb, H.G.; Robertson, T.; Papadimitriou, J.; Spuler, S.; Shavlakadze, T. Lipid accumulation in dysferlin-deficient muscles. Am. J. Pathol. 2014, 184, 1668–1676. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Celik, M.Ö.; Labuz, D.; Keye, J.; Glauben, R.; Machelska, H. IL-4 induces M2 macrophages to produce sustained analgesia via opioids. JCI Insight 2020, 5, e133093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casella, G.; Garzetti, L.; Gatta, A.T.; Finardi, A.; Maiorino, C.; Ruffini, F.; Martino, G.; Muzio, L.; Furlan, R. IL4 induces IL6-producing M2 macrophages associated to inhibition of neuroinflammation in vitro and in vivo. J. Neuroinflamm. 2016, 13, 139. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, E.; Rojas–García, R.; de Luna, N.; Pou, A.; Brown, R.H.; Illa, I. Inflammation in dysferlin myopathy: Immunohistochemical characterization of 13 patients. Neurology 2001, 57, 2136. [Google Scholar] [CrossRef]
- Gayathri, N.; Alefia, R.; Nalini, A.; Yasha, T.; Anita, M.; Santosh, V.; Shankar, S. Dysferlinopathy: Spectrum of pathological changes in skeletal muscle tissue. Indian J. Pathol. Microbiol. 2011, 54, 350–354. [Google Scholar] [CrossRef]
- Han, R. Muscle membrane repair and inflammatory attack in dysferlinopathy. Skelet. Muscle 2011, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- Maguire, O.; O’Loughlin, K.; Minderman, H. Simultaneous assessment of NF-κB/p65 phosphorylation and nuclear localization using imaging flow cytometry. J. Immunol. Methods 2015, 423, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guha, T.K.; Pichavant, C.; Calos, M.P. Plasmid-Mediated Gene Therapy in Mouse Models of Limb Girdle Muscular Dystrophy. Molecular Therapy-Methods & Clinical Development 2019, 15, 294–304. [Google Scholar]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Therap. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.-C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallabhapurapu, S.; Karin, M. Regulation and Function of NF-κB Transcription Factors in the Immune System. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, T.M.; Mooney, D.J. Anti-inflammatory nanoparticles significantly improve muscle function in a murine model of advanced muscular dystrophy. Sci. Adv. 2021, 7, eabh3693. [Google Scholar] [CrossRef]
- Bosurgi, L.; Cao, Y.G.; Cabeza-Cabrerizo, M.; Tucci, A.; Hughes, L.D.; Kong, Y.; Weinstein, J.S.; Licona-Limon, P.; Schmid, E.T.; Pelorosso, F.; et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017, 356, 1072–1076. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Zhou, Y.; Liang, Q.; Ge, C.; Yang, J.; Shan, B.; Liu, Y.; Zhou, X.; Yin, L. Inflammation-Instructed Hierarchical Delivery of IL-4/miR-21 Orchestrates Osteoimmune Microenvironment toward the Treatment of Rheumatoid Arthritis. Adv. Funct. Mater. 2021, 2101033. [Google Scholar] [CrossRef]
- Lin, T.; Pajarinen, J.; Nabeshima, A.; Lu, L.; Nathan, K.; Yao, Z.; Goodman, S.B. Establishment of NF-κB sensing and interleukin-4 secreting mesenchymal stromal cells as an “on-demand” drug delivery system to modulate inflammation. Cytotherapy 2017, 19, 1025–1034. [Google Scholar] [CrossRef]
- Baek, J.-H.; Many, G.M.; Evesson, F.J.; Kelley, V.R. Dysferlinopathy promotes an intramuscle expansion of macrophages with a cyto-destructive phenotype. Am. J. Pathol. 2017, 187, 1245–1257. [Google Scholar] [CrossRef] [Green Version]
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Murphy, G.; Stanton, H.; Cowell, S.; Butler, G.; Knäuper, V.; Atkinson, S.; Gavrilovic, J. Mechanisms for pro matrix metalloproteinase activation. APMIS 1999, 107, 38–44. [Google Scholar] [CrossRef]
- Toscano, M.A.; Campagna, L.; Molinero, L.L.; Cerliani, J.P.; Croci, D.O.; Ilarregui, J.M.; Fuertes, M.B.; Nojek, I.M.; Fededa, J.P.; Zwirner, N.W.; et al. Nuclear factor (NF)-kappaB controls expression of the immunoregulatory glycan-binding protein galectin-1. Mol. Immunol. 2011, 48, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- De Freitas Zanon, C.; Sonehara, N.M.; Girol, A.P.; Gil, C.D.; Oliani, S.M. Protective effects of the galectin-1 protein on in vivo and in vitro models of ocular inflammation. Mol. Vis. 2015, 21, 1036. [Google Scholar]
- Hogarth, M.W.; Defour, A.; Lazarski, C.; Gallardo, E.; Manera, J.D.; Partridge, T.A.; Nagaraju, K.; Jaiswal, J.K. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| rHsGal-1 Dose | Treatment Schedule | Fold Improvement Over PBS |
|---|---|---|
| 0.27 mg/kg | Day 0, 7 | 1.29 (ns) |
| 0.27 mg/kg | Day 0, 2, 4, 6 | 1.64 (**) |
| 0.54 mg/kg | Day 0, 7 | 1.08 (ns) |
| 1.35 mg/kg | Day 0, 7 | 2.75 (****) |
| 2.7 mg/kg | Day 0, 7 | 3.35 (****) |
| 2.7 mg/kg | Day 0, 5 | 1.56 (***) |
| 2.7 mg/kg | Day 0 | 1.52 (**) |
| 2.7 mg/kg | Day 7 | 1.99 (***) |
| 5.4 mg/kg | Day 0, 7 | 2.39 (****) |
| 8.1 mg/kg | Day 0, 7 | 1.56 (*) |
| 13.5 mg/kg | Day 0, 7 | 0.98 (ns) |
| 27 mg/kg | Day 0, 3, 7 | 0.55 (**) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallecillo-Zúniga, M.L.; Poulson, P.D.; Luddington, J.S.; Arnold, C.J.; Rathgeber, M.; Kartchner, B.C.; Hayes, S.; Gill, H.; Valdoz, J.C.; Spallino, J.L.; et al. Therapeutic Benefit of Galectin-1: Beyond Membrane Repair, a Multifaceted Approach to LGMD2B. Cells 2021, 10, 3210. https://doi.org/10.3390/cells10113210
Vallecillo-Zúniga ML, Poulson PD, Luddington JS, Arnold CJ, Rathgeber M, Kartchner BC, Hayes S, Gill H, Valdoz JC, Spallino JL, et al. Therapeutic Benefit of Galectin-1: Beyond Membrane Repair, a Multifaceted Approach to LGMD2B. Cells. 2021; 10(11):3210. https://doi.org/10.3390/cells10113210
Chicago/Turabian StyleVallecillo-Zúniga, Mary L., Peter Daniel Poulson, Jacob S. Luddington, Christian J. Arnold, Matthew Rathgeber, Braden C. Kartchner, Spencer Hayes, Hailie Gill, Jonard C. Valdoz, Jonathan L. Spallino, and et al. 2021. "Therapeutic Benefit of Galectin-1: Beyond Membrane Repair, a Multifaceted Approach to LGMD2B" Cells 10, no. 11: 3210. https://doi.org/10.3390/cells10113210