
Role of Kynurenine Pathway in Oxidative Stress during Neurodegenerative Disorders


1
Department of Pharmacodynamics, Medical University of Bialystok, Mickiewicza 2c, 15-222 Bialystok, Poland
2
Department of Monitored Pharmacotherapy, Medical University of Bialystok, Mickiewicza 2c, 15-222 Bialystok, Poland
3
Department of Internal Medicine and Metabolic, Medical University of Bialystok, M. Skłodowskiej-Curie 24a, 15-276 Bialystok, Poland
*
Author to whom correspondence should be addressed.
Cells 2021, 10(7), 1603; https://doi.org/10.3390/cells10071603
Submission received: 27 May 2021
/
Revised: 18 June 2021
/
Accepted: 24 June 2021
/
Published: 26 June 2021
(This article belongs to the Special Issue Cellular and Molecular Mechanisms of Neurodegenerative Diseases)
Abstract
:Neurodegenerative disorders are chronic and life-threatening conditions negatively affecting the quality of patients’ lives. They often have a genetic background, but oxidative stress and mitochondrial damage seem to be at least partly responsible for their development. Recent reports indicate that the activation of the kynurenine pathway (KP), caused by an activation of proinflammatory factors accompanying neurodegenerative processes, leads to the accumulation of its neuroactive and pro-oxidative metabolites. This leads to an increase in the oxidative stress level, which increases mitochondrial damage, and disrupts the cellular energy metabolism. This significantly reduces viability and impairs the proper functioning of central nervous system cells and may aggravate symptoms of many psychiatric and neurodegenerative disorders. This suggests that the modulation of KP activity could be effective in alleviating these symptoms. Numerous reports indicate that tryptophan supplementation, inhibition of KP enzymes, and administration or analogs of KP metabolites show promising results in the management of neurodegenerative disorders in animal models. This review gathers and systematizes the knowledge concerning the role of metabolites and enzymes of the KP in the development of oxidative damage within brain cells during neurodegenerative disorders and potential strategies that could reduce the severity of this process.
1. Introduction
Neurological disorders are chronic and life-threatening conditions, negatively affecting the quality of human life. Their treatment options are still limited. In recent years, due to population growth and aging, their incidence has been constantly increasing, and therefore they have become a leading cause of disability and the second cause of death in the world [1,2,3]. The global prevalence of these disorders remains at a level of 10.2% [3]. The rate of disability and mortality associated with neurological disorders is significantly higher compared with other human disorders [3,4,5]. During pathological neurodegeneration, causative factors and cellular disturbances occur earlier, are much more intensified than in the aging-related processes, and often attack specific structures of the central nervous system (CNS). The abovementioned mitochondrial energy failure and excessive ROS production accompany the development of such diseases as Alzheimer’s disease (AD), Huntington’s disease (HD), and Parkinson’s disease (PD) [6,7,8,9,10,11,12,13].
Neurological disorders often have a genetic background, but in many cases, their progression is triggered or intensified by various exogenous factors [14,15,16]. Oxidative stress and mitochondrial damage seem to be at least partly responsible for their development [6,17]. The primary events responsible for the initiation of neurodegenerative disorders are still unclear, but the mechanism of oxidative damage seems to be significant in the propagation of cellular injuries observed at their beginning. Abnormal accumulation of protein deposits, such as amyloid-β protein, tau protein, α-synuclein, transactive response DNA-binding protein-43, and myelin debris, in and around neurons, is a histopathological hallmark of neurodegenerative processes. Their accumulation is unquestionably related to the pathogenesis and progression of neurodegenerative disorders. It induces molecular alterations, which in conjunction with activation of microglia and astrocytes, cause the release of pro-inflammatory cytokines, leading to the increased production of reactive oxygen (ROS) and nitrogen species, which damage the surrounding tissues and lead to neuroinflammation, most probably the last common pathway leading to neurodegeneration. Neuroinflammation potentiates the accumulation of the aforementioned abnormal proteinaceous materials in the brain tissue and enhances the neuroinflammatory conditions, forming a cyclic potentiation. [18]. Furthermore, mitochondrial DNA damage, enhanced by neuroinflammation and protein deposit accumulation, causes a loss of mitochondrial polypeptide expression, leading to a subsequent decrease in electron transport, an increase in ROS generation, disturbances of mitochondrial membrane potential, and finally to the release of cell death induction signals, which further amplify neuronal damage [7,19,20].
Loss of mitochondrial function also causes ionic imbalance, calcium (Ca2+) overload, and adenosine triphosphate (ATP) depletion. The potent drop of the energy supply induces the process of necrotic death. In turn, even mild or gradual energy disturbances may induce the release of proapoptotic factors from the mitochondria, leading to the initiation of an apoptotic cascade [8,21]. Moreover, excessive Ca2+ accumulation reveals a detrimental effect on cell viability, inducing oxidative damage of proteins and lipids and opening mitochondrial permeability transition pores, which also triggers the apoptotic cascade. However, it should not be forgotten that non-excitable cells, such as astrocytes and microglia, are strongly dependent on the intracellular Ca2+ concentration, and Ca2+ signaling is necessary to maintain their proper function [8,9,22,23]. Furthermore, the cellular processes observed during physiological aging are similar to those involved in the pathomechanism of neurodegenerative processes.
The activation of the kynurenine pathway (KP) enzymes by interferon-γ, other proinflammatory cytokines, and cortisol links cell-mediated immunity, oxidative and nitrosative stress, and emotional stress with decreased serotonin levels and alterations in pain sensation and emotion regulation [11,12,13,24]. Some KP metabolites may enhance neuroinflammation and have detrimental effects on the mitochondria and energy metabolism. Therefore, they may generate oxidative stress, induce mitochondrial symptoms, and enhance neuronal dysfunction and apoptosis rate [11,12,13,24,25]. Measurement of redox status may reveal diagnostic, prognostic, or predictive value to diagnose and differentiate neurodegenerative disorders [26]. Therefore, the KP activity together with redox indicators seems to be a powerful battery of biomarkers in their diagnosis. Additionally targeting the components of the KP could be considered as a therapeutic solution, allowing efficient personalized treatment plans for this type of disorder to be built [27].
2. Role of Kynurenine Pathway Metabolites in Oxidative Stress-Associated Neurodegeneration
Tryptophan (TRP) is the essential amino acid involved in the process of protein biosynthesis. Considering its role in the development of neurodegenerative disorders, it cannot be forgotten that it undergoes extensive metabolism through several metabolic pathways, resulting in the production of many biologically-active compounds, exerting differential effects on numerous physiological and pathological processes [20,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46]. TRP is transformed into serotonin in approximately 1% of its total body amount, while about 95% of this amino acid is metabolized by the KP, leading to the synthesis of the oxidized form of nicotinamide adenine dinucleotide (NAD+), a coenzyme participating in many cellular processes (Figure 1).
As mentioned above, the KP is the main path responsible for its catabolism. Its metabolites demonstrate diverse, sometimes opposite, activities in numerous biological processes [28,29,30,31]. Their accumulation may induce oxidative cell damage, which triggers the inflammatory process. They also modulate the activity of numerous signaling pathways, leading to the disruption of homeostasis and the functioning of various organs and systems. Thus, they contribute to the development of many systemic disorders, including neurological ones [11,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46]. Within the KP, TRP is oxidized to N-formylkynurenine by tryptophan 2,3-dioxygenase (TDO) and two 2,3-indoleamine dioxygenase isoforms (IDO-1 and IDO-2) [47,48]. In physiological conditions, they catalyze the same reaction in parallel, but their tissue distribution is different [49,50,51,52,53]. N-formylkynurenine is catabolized into kynurenine (KYN), which is metabolized by the three branches of the KP, resulting in the formation of kynurenic acid (KYNA), anthranilic acid (AA), and 3-hydroxykynurenine (3-HKYN) [54,55,56,57]. The conversion of KYN to 3-HKYN occurs using kynurenine 3-monooxygenase (KMO), and to KYNA with the participation of kynurenine aminotransferase (KAT) [52,53]. 3-HKYN is metabolized into xanthurenic acid (XA) by KAT and then to 3-hydroxyanthranillic acid (3-HAA) by kynureninase (KYNU) [58,59,60]. The presence of enzymes that catalyze the transformation of both KYN to 3-HKYN and 3-HKYN to XA and 3-HAA was discovered in almost all tissues of the body [59,60,61]. 3-HAA is metabolized by 3-hydroxyanthranilic acid 3,4-dioxygenase (3-HAO) to aminocarboxymuconatesemialdehyde (ACMS), which is converted by ACMS decarboxylase to aminomuconicsemialdehyde. This compound undergoes non-enzymatic cyclization to picolinic acid (PA) or is non-enzymatically transformed to quinolinic acid (QA), which is converted by quinolinate phosphoribosyltransferase (QPRT) into NAD+ (Table 1) [61,62,63,64].
Among the abovementioned metabolites, KYN, 3-HKYN, KYNA, AA, and QA turned out to be neuroactive [35,58,65,66]. KYN, 3-HKYN, and AA penetrate well through the blood–brain barrier (BBB), in opposition to KYNA, QA, and 3-HAA [24,67,68,69]. Further research indicated that 3-HKYN, 3-HAA, and QA have neurotoxic properties [24,35,65]. The peripheral increase in the activity of the KP, described by the elevated serum KYN/TRP ratio, was found in the course of numerous neurological and psychiatric disorders [24,70]. It is mainly associated with increased activity of peripheral IDO isoforms. Over 60% of the central KYN amount is delivered to the brain from peripheral circulation [71]. Peripheral KYN is transported through the BBB by the large neutral amino acid transporter 1 (LAT-1) as well as through organic anion transporters 1 and 3 (Figure 2) [72,73,74,75]. Therefore, it may easily enter into the CNS. In the brain, it is uptaken by glial cells, where it is metabolized. Neuroactive KP metabolites can directly or indirectly affect neuronal functions [72,73]. Abnormalities in their concentrations have been linked to numerous psychiatric and neurodegenerative symptoms [24,76,77]. However, it is still unclear if alterations of KP activity occur only during the progression of these disorders or also contribute to their initiation. Evaluation of the impact of the disruptions in KP activity on the pathogenesis of various neurological and psychiatric diseases will allow for a better understanding of their pathophysiology and the development of novel, effective therapeutic solutions.
It is worth mentioning that KYN and KYNA are the main metabolites of the KP in the brain cells with a predominance of TDO activity, such as astrocytes and hippocampal neurons. In turn, in CNS cells with predominant IDO activity, such as microglia and macrophages, the authors observed further activation of the KP, leading to increased synthesis of 3-HKYN, recognized as a major inhibitor of neurogenesis (Figure 2) [72]. This method of KP activation depletes TRP, decreases serotonin and melatonin synthesis, and increases the production of active metabolites, which have deleterious effects on neuronal activity, survival, and the neurogenesis rate [78]. For this reason, numerous studies have focused on the possibility of KP activity modulation to reduce its detrimental impact on the CNS. The main strategies to restore and maintain a proper balance of the KP proposed by the authors include TRP supplementation, the use of inhibitors of KP enzymes, and the administration of analogs of KP metabolites, especially KYNA [61,79,80,81,82].
2.1. Tryptophan (TRP)
Several studies have already demonstrated that the administration of TRP may enhance oxidative stress levels and lead to an imbalance between the free radical content and the capacity of the cellular scavenging systems (Table 1) [83,84,85,86]. This effect is the most severe within the cerebral cortex. It can be induced both directly by an excess of TRP and by the accumulation of its active metabolites [83]. As mentioned above, changes in ROS levels may be associated with numerous alterations in brain cell functioning. Therefore, elevated TRP levels in the brain observed in the course of hypertryptophanemia and neurodegenerative disorders seem to be involved in the mechanisms leading to brain injury, observed in patients with these diseases. This detrimental effect could be diminished by antioxidant use. Although their use is a controversial issue, it seems to be reasonable that the administration of taurine or vitamins E and C might be useful as an adjuvant method in the treatment of patients with symptoms related to an excess of TRP [83,85,86]. Therefore, the impact of supplementation with vitamins E and C or taurine in these patients still needs further evaluation.
Despite the fact that excess TRP was linked with an increase in oxidative stress levels and severity of psychiatric and neurodegenerative symptoms, the first strategy was considered because, except for KYN, TRP is also metabolized to serotonin and melatonin [87,88,89,132]. Moreover, TRP supplementation has been widely used as an effective therapy to alleviate behavior problems in animals. A low-protein diet supplemented with TRP is a solution to manage excessive aggression in dogs [133]. The detailed mechanism associated with this effect of TRP is still unclear but seems to be at least partly associated with the impact of its neuroactive metabolites. In turn, Ciji et al. discovered that fish exposed to nitrites have increased cortisol serum levels and decreased testosterone and estradiol concentrations. This effect was prevented by the supplementation of vitamin E and TRP. The beneficial effect of vitamin E was associated with its antioxidant properties, but the exact mechanism responsible for the TRP action was also unknown. Its protective properties may result from both its direct redox properties as well as the impact of some of its antioxidant metabolites [134]. Additionally, TRP supplementation can also positively affect intracellular NAD+ concentrations, which improves cell viability and function, and protects cells against energy depletion [135]. However, a diet rich in TRP in healthy women increased the serum levels and urinary excretion of KYN, KYNA, 3-HKYN, 3-HAA, AA, and QA, the excesses of which are toxic. This indicates that redundant TRP supplementation could aggravate symptoms caused by oxidative stress due to the accumulation of its active metabolites [135,136].
2.2. Kynurenine (KYN)
KYN can scavenge hydrogen peroxide and superoxide in specific ROS producer systems. Increased KYN levels also lead to a decrease in ROS production by activated neutrophils [90]. Blanco-Ayala et al. showed that this compound, both in vitro and in vivo, reacts with hydroxyl radical and peroxynitrite, transforming into KYNA in these reactions [91,97]. In effect, it decreases the DNA and protein degradation process induced by these radicals. Additionally, KYN can attenuate the ROS production and lipid peroxidation induced by pro-oxidant compounds, such as iron(II) sulfate, peroxynitrite, and 3-nitropropionic acid, in rat brain homogenates [92]. Although KYN has been employed in many neurotoxic models as a neuroprotective agent, the observed effects were rather attributed to its metabolite, KYNA [11,92,93,97]. However, recent reports also suggest that they are at least partially induced by KYN’s own scavenging activity. This seems to be confirmed by the fact that KYN can donate electrons and protect macromolecules against oxidative modifications both in vitro and in vivo [94]. On the other hand, Song et al. claim that it cannot be ruled out that KYN, especially in excessive levels, may also contribute to apoptosis through ROS generation, because its co-incubation with antioxidants blocks cytochrome c release and activation of caspase 3, leading to an inhibition of apoptosis in a way associated with its activity (Table 1) [95].
2.3. Kynurenic Acid (KYNA)
Solvag et al. observed that plasma levels of KYNA are decreased in AD patients, suggesting the shift toward neurotoxic metabolites over neuroprotection in peripheral KP accompanying these diseases [137]. Interestingly, a similar trend was observed in PD and HD patients and children with autism spectrum disorders, while in vascular cognitive dementia, its serum concentration was increased [138]. This observation is extremely important because decreased levels of KYNA may provoke an inadequate anti-inflammatory response, resulting in enhanced tissue damage and exceeding cell proliferation during inflammatory in AD, PD, HD, and Lewy’s bodies dementia, contributing to the aggravation of their symptoms [98].
Brain levels of KYNA are elevated in brain tissue, mainly within the striatum and the hippocampus of AD patients [98,137,139]. In contrast, in HD patients, its levels are decreased in the precentral gyrus, frontal, and temporal cortex [98]. This compound acts mostly as a neuroprotectant and exerts a wide spectrum of endogenous antagonism on ionotropic excitatory amino acid receptors [12,13,97,140,141]. Its brain pool is mainly endogenous; however, a small part of brain KYNA is also derived from the peripheral circulation because it can cross the BBB through organic anion transporters 1 and 3 [142]. In micromolar concentrations, it acts as a competitive antagonist at the strychnine-insensitive glycine-binding site of the N-methyl-D-aspartate (NMDA) receptor and reveals a weak antagonistic effect on α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) and kainite receptors in higher concentrations [97,141,143,144,145]. Furthermore, KYNA is capable of facilitating AMPAR responses in low concentrations, but the concentration range seems to be controversial. At the micromolar level, KYNA shows neuroinhibitory effects, while in nanomolar concentrations it turned out to be a facilitator of neurotransmission [97,141,145,146]. In addition to its direct impact on NMDA receptors, KYNA in a non-competitive way inhibits the 7-nicotinic acetylcholine receptors, in which presynaptic activation is involved in the regulation of NMDA release [97,141,147,148]. It has also been reported that KYNA, through the activation of the G protein-coupled receptor 35 (GPR35), can induce inositol trisphosphate synthesis and Ca2+ mobilization, but the significance of this phenomenon regarding the CNS is questionable, because GPR35 in the CNS has a low expression level, while relatively high KYNA concentrations are necessary for its activation [97,141,149]. Furthermore, KYNA significantly inhibited the in vivo transformation of hippocampal microglia cells from changing into the activated forms and the blood IL-6 level and significantly inhibited the in vitro phagocytosis of fluorescent microbeads in microglia cells induced by LPS treatment [150]. Moreover, KYNA also exhibits strong antioxidant activity by scavenging ROS in a way independent of NMDA and nicotinic receptor-associated mechanisms. In vitro models shows that in concentrations above 100 µM, KYNA can potently abolish ROS synthesis and prevent tissue damage triggered by overshooting inflammation (Table 1) [96,97,98]. However, an effect as potent as that observed at this concentration is difficult to observe in vivo, because its serum concentration observed in AD, HD, PD, and Lewy’s bodies dementia patients usually do not reach 70 µM, while in brain tissue homogenates it rarely exceeds the level of 10 µmol/g of protein [98,137,139].
2.4. 3-Hydroxykynurenine (3-HKYN) and 3-Hydroxyanthranillic Acid (3-HAA)
3-HKYN, an intermediate metabolite of KP, has been hypothesized to be a neurotoxic molecule, contributing to the development of various neurodegenerative lesions in the experimental models and clinical conditions [58,99,100,151]. The brain cortex and striatum are most sensitive to the impact of 3-HKYN, which is transferred into neurons through the LAT-1. Differences in the susceptibility to 3-HKYN in various brain regions result mainly from differences in LAT-1 expression in their cells [58]. Only by interacting with cellular xanthine oxidase can 3-HKYN generate sufficient amounts of ROS, such as superoxide radicals, hydrogen peroxide, and hydroxyl radical, to induce inter-nucleosomal DNA damage, leading to apoptosis [100,151]. However, even relatively low levels of 3-HKYN and 3-HAA may cause neurotoxicity via the induction of oxidative stress (Figure 3). They can promote the apoptotic death of neurons and enhance the severity of QA-induced excitotoxicity (Table 1) [58,101]. In addition, their excess is also associated with depressive symptoms [58,80,102]. The authors suggest a link between ROS overproduction and increased monoamine oxidase activity, which lowers brain levels of catecholamines and serotonin [152,153]. Moreover, polyunsaturated fatty acids are also sensitive to oxidation. ROS overproduction may damage phospholipids and reduce cell membrane viscosity [154]. Alterations in membrane viscosity may negatively impact the density and function of serotonergic or catecholaminergic receptors [152]. All these changes may intensify the development of depressive symptoms [155,156,157]. Various agents with antioxidant activity may prevent the detrimental effects induced by 3-HKYN and 3-HAA in neurons. Furthermore, selective serotonin reuptake inhibitors also revealed antioxidant properties and the ability to reverse the overproduction of ROS. This effect seems to enhance their antidepressant effect [158].
The neurotoxicity of KYN metabolites has been well demonstrated in vitro, in vivo, and in patients [11,13,25,33,35,55,56,58,65,103,107,108,139,151,158,159,160,161,162,163,164,165,166,167]. Preclinical and clinical data demonstrated that their elevated levels occur in several degenerative diseases. Increased synthesis of 3-HKYN and 3-HAA was confirmed in the course of AD, HD, and PD [11,12,13,24,33,56,58,151,159,160,161,162,163,164,165,166,167]. In the pathogenesis of AD, oxidative stress induced by their excess significantly enhances neuronal damage and may potentiate the neurodegenerative processes associated with the accumulation of Aβ-amyloid, glial activation, and upregulation of proinflammatory cytokine expression [139,159,160,161,162]. Additionally, the pathomechanism of HD may be at least partly associated with alterations in TRP metabolism and mitochondrial dysfunction. Significantly elevated concentrations of 3-HKYN and 3-HAA have been reported in the brains of HD patients, even at early stages of the disease [55,70,162,163,164,165,166,167]. Additionally, in the course of PD, decreased KYNA and increased 3-HKYN concentrations were found in brain samples [110,168,169,170,171,172,173,174]. Increased production of 3-HKYN and 3-HAA may contribute to the neuronal damage responsible for the cognitive disorders observed during aging, malaria, ischemia, traumatic injury, birth hypoxia, and epilepsy [11,56,175,176,177]. Furthermore, their accumulation may be involved in the development of various psychiatric diseases, such as anxiety, depression, and schizophrenia. All this evidence demonstrates that excess 3-HKYN and 3-HAA is at least partly responsible for the development of neurodegenerative and psychiatric symptoms [56,178,179,180].
It is worth noting that the issue of the impact of 3-HKYN and 3-HAA on the development of detrimental lesions in the brain cells associated with oxidative stress is controversial. The literature indicates the dual role of 3-HKYN in the CNS [99,104,105]. In vitro studies show that it has both neurotoxic and antioxidant properties, whereas in vivo studies suggest its role rather as a weak neurotoxin. However, there are some discrepancies in the research results. Colín-González et al. showed that 3-HKYN in rat striatal slices revealed a concentration- and time-dependent impact on lipid peroxidation, inducing both pro-oxidative at low (5–20 nM) and antioxidative properties at higher (100 nM) concentrations [99]. In turn, Braidy et al. achieved different results. They demonstrated that both in primary human astrocytes and neurons, 3-HKYN promoted NAD+ synthesis at concentrations below 100 nM, while at concentrations above 100 nM it caused a decrease in intracellular NAD+ levels and an increase in extracellular lactate dehydrogenase (LDH) activity, leading to a decrease in the viability of human cerebral neurons (Table 1) [104].
This phenomenon is important due to the fact that NAD+ biosynthesis is essential for the energetic balance and maintenance of cell viability and functions. However, it seems to be rather a form of a compensatory mechanism, because animals receiving 3-HKYN in striatal injection in high concentrations showed no behavioral or morphological changes in their striata. Moreover, in the range of high concentrations, 3-HKYN was unable to induce mitochondrial dysfunction in brain slices and prevented the detrimental lesions caused by its downstream metabolite, QA, the mitochondrial toxin 3-nitropropionic acid, and iron(II) sulfate. These protective properties were associated with the induction of glutathione S-transferase (GST) and superoxide dismutase (SOD) activities (Table 1) [99].
Moreover, 3-HKYN stimulates the synthesis and activation of Nrf2, a transcription factor and antioxidant regulator, and the proteins associated with this molecule, which caused a partial increase in cell resistance against oxidative stress [99,106]. Accordingly, 3-HKYN exhibited reductive properties at high concentrations, although the striatal tissue infused with 3-HKYN exhibited a significant increase in lipid and protein peroxidation levels in a short time after infusion. After long-time exposition to 3-HKYN, it substantially decreased. This seems to be associated with an increase in Nrf2 expression, as well as glutathione (GS) reductase and GST activity [99].
Altogether, despite the divergences observed by the authors, these findings suggest that although 3-HKYN may exert pro-oxidative effects under certain conditions, it is more likely to modulate redox activity and reduce the risk of cellular oxidative damage. This suggests that in physiological conditions, 3-HKYN appears to be rather a redox modulator than a neurotoxin. Additionally, 3-HAA revealed both and pro-oxidant effects. It can reduce ROS concentrations by the formation of complexes with iron, inhibiting its autoxidation and physiologically acting rather as redox modulator and antioxidant [11,109].
2.5. Anthranilic Acid (AA) and Xanthurenic Acid (XA)
AA is almost exclusively known for its antioxidant activity [11]. It revealed the ability to inhibit the citric acid cycle and respiratory chain complexes I–III from interfering with mitochondrial function. Moreover, similarly to 3-HAA, it downregulates ROS formation due to its iron-complexing properties [180]. In addition, it has anti-inflammatory and weak neuroprotective properties associated with its ability to form complexes with copper ions and to inactivate hydroxyl radicals [110,111,112]. In turn, xanthurenic acid, similarly to its precursor, 3-HKYN, revealed both pro-oxidative as well as antioxidative properties, based on similar mechanisms (Table 1) [113,114].
2.6. Picolinic Acid (PA)
PA was found to be a non-selective metal ion chelating agent and neuroprotectant. It revealed the ability to prevent QA-induced neuronal loss after injection of QA into the rat nucleus basalis magnocellularis and to activate brain macrophages. It inhibits excitotoxic but not neuroexcitatory responses within the CNS. Its mechanism of anti-excitotoxic activity is unclear but seems to be related to its zinc chelation capacity. Its effectiveness against QA-induced neurotoxicity was lower than KYNA but higher than AA. It also can modulate kainate-induced glutamate release from the striatum (Table 1) [115].
2.7. Quinolinic Acid (QA)
QA is a moderate, specific competitive agonist of the NMDA receptors. The hippocampus and striatum are the most sensitive to its neurotoxicity. The vulnerability to this compound observed in human cerebral neurons is associated with the fact that they can uptake exogenous QA in large amounts, but catabolize only its small part because QPRT is rapidly saturated [11,115,116]. There are two mechanisms responsible for the neurotoxic effect of QA on the glutamatergic system. The first one is associated with the direct overstimulation of the NMDA receptors by QA, which induces increased Ca2+ influx into the target neurons, leading to their damage, and death. It is named excitotoxicity, and neurons also affect oligodendrocytes. The second mechanism is associated with the ability of QA to contribute to ROS formation. The influx of Ca2+ into neurons induced by the activation of NMDA receptors contributes to the increase in the oxidative stress level (Figure 3) [93,116,117,119].
Furthermore, QA can induce oxidative damage in a manner independent of the NMDA receptor-associated mechanism. It creates complexes with iron(II) ions (QA–Fe2+). They enhance the formation of hydroxyl radicals. Especially lipid peroxidation induced by QA is intensified after an interaction with Fe2+ from these complexes. In addition, QA inhibits iron autoxidation due to their formation [117,120,121,122]. Both these pro-oxidative mechanisms lead to increased lipid and protein peroxidation levels, superoxide anion generation, and in effect disturbances in neuronal functioning and viability, especially within the hippocampus [116,117,118,119,120,121,122,123]. The direct pro-oxidative properties of QA expand its neurotoxic activity. Moreover, QA and 3-HKYN act synergistically in the production of ROS [124,125]. Therefore, even low doses of QA significantly potentiate the neurotoxicity of 3-HKYN. The QA–Fe2+ complexes also proved to be responsible for the breakage of DNA chains and the intensification of the lipid peroxidation process mediated by hydroxyl radicals observed in vitro (Table 1, Figure 3) [116,125].
In turn, Tronel et al. showed that hemin, a heme oxygenase 1 inducer, increases the neurotoxic effect of QA in the animal model and leads to an increase in the loss of brain tissue and microglia activation. This effect is mainly linked to increased ROS generation and iron accumulation [117,126]. Moreover, QA can enhance ROS synthesis by inducing nitric oxide synthases and intracellular poly(adenosine diphosphate (ADP)-ribose) polymerase activity in astrocytes and neurons, as well as activating extracellular LDH. This is confirmed by the fact that striatal slices exposed to QA showed a significant increase in both lipid peroxidation and LDH activity levels and a decrease in mitochondrial function (Table 1). These changes appear to be also related to the activation of cellular proteases [77,117,127].
Furthermore, in vivo studies proved that QA can modify the expression and activity of certain endogenous antioxidants in the brain, such as reduced GS and SOD. It can generate toxic peroxide radicals and peroxynitrite, even after a short-time impact on the cell [128]. It is worth mentioning that intracerebral injection of QA in the rat brain led to a significant increase in SOD expression levels in a time-dependent manner. This is recognized as a neuroprotective response to limit the oxidative damage caused by QA [129]. In support of this, it was found that QA infusion, in addition to increasing the ROS level, induces a delayed and prolonged upregulation in the antioxidant capacity in mice hippocampus. It also appears to be a cellular adaptive mechanism that may contribute to reducing oxidative stress levels [130]. Additionally, in synaptosomal fractions exposed to QA and 3-nitropropionic acid, even at nontoxic concentrations, Pérez-De La Cruz et al. observed a synergic increase in the oxidative stress levels, which was partially reduced by the administration of dizocilpine, a non-competitive antagonist of the NMDA receptor. This suggests that the severity of the lipid and protein peroxidation process in neurons induced by 3-nitropropionic acid, and probably by other pro-oxidants, is strongly dependent on cellular Ca2+ levels (Table 1). In turn, QA may increase the influx of Ca2+ into glutamatergic neurons [131]. Therefore, inhibitors of the NMDA receptors could be effective protectants against neurotoxic mechanisms induced or enhanced by QA.
The main symptom of AD is memory impairment. It is associated with the atrophy of the temporal lobes containing the hippocampal formations responsible for memory formation. The pathological processes within the hippocampus involve an excessive stimulation of excitatory glutamatergic neurons, especially the NMDA type. Although the NMDA receptors play an important role in the memory formation process, their overstimulation may cause neuronal dysfunction, damage, or even death. The accumulation of the Aβ-amyloid in AD brains causes several toxic effects, involving mitochondrial arrest, increased ROS generation, proteasomal dysfunction, as well as IDO induction [93,139,159,160,161,162,181,182]. QA seems to play an important role in the neuropathogenesis of AD due to its associations with inflammatory, pro-oxidative, and excitotoxic mechanisms. It can induce, among others, interleukin-1β synthesis and excessive NMDA transmission and inhibit GS function [159,181,182]. Additionally, in AIDS-associated dementia, the level of QA is increased in the cerebrospinal fluid and correlates with the severity of cognitive dysfunctions [183,184]. These findings can be useful for the development of novel therapies for AD and other neuroinflammatory diseases with accompanying dementia. In addition to for the upregulation of IDO isoforms and enhanced production of QA, the authors also confirmed the presence of elevated KYNA levels in the area of the putamen and caudate nuclei in AD patients. It appears to be a compensatory mechanism developed for counteracting the QA effects [182]. However, it may also contribute to the deterioration of cognitive functions due to the inhibition of the NMDA receptors. Interestingly, intrastriatal injection of QA in rodents causes neuropathological changes similar to those observed in the course of HD [185]. As mentioned earlier, QA can amplify the inflammatory response, mainly by the upregulation of several proinflammatory cytokine and chemokine receptor expressions. In vitro studies discovered that the potency of QA to induce inflammation is comparable to tumor necrosis factor-α or interferon-γ [181]. This indicates that its excess may worsen the course of numerous neuroinflammatory diseases.
In addition to the mechanisms described above, QA can exert early redox modifications also through alterations in the expression and activity of the Nrf2 transcription factor (Table 1). They are dependent on the time of exposure, the QA concentration, and the extent of oxidative damage occurring in the cells. Nrf2 can induce the upregulation of phase 2 enzymes, and therefore it seems to be a mechanism of compensatory response induced by QA-associated toxic processes in the cell. The loss of Nrf2 expression only moderately contributes to the severity of oxidative damage in cells, although striatal neurons from animals with this mutation showed higher sensitivity to the toxic effects of QA. In turn, the active role of the NMDA receptor in modulating oxidative stress and Nrf2-mediated response was observed only in the animals expressing this factor, but not in those with the loss of its function [186]. This indicates the need for further studies towards a better assessment of the impact of QA on Nrf2 activity and the potential role of this factor as a therapeutic tool for reducing neurodegenerative lesions associated with oxidative damage.
Numerous endo- and exogenous ROS scavengers, molecules with antioxidant properties, and activators of endogenous antioxidant enzymes revealed an efficiency against QA toxicity. The number of mechanisms and compounds that have evolved to counteract the adverse effects of this compound indicates the important role of the oxidative damage induced by QA in the development of neurodegenerative lesions [117]. Oxidative stress seems to be one of the most important mechanisms responsible for the QA-mediated neurotoxicity because free radicals can activate numerous signaling pathways, leading to extensive deleterious changes in the brain cells. Inflammation and oxidative stress are also involved in brain damage following a stroke. Increased KP activity contributes to an increase in oxidative stress levels by the impact of QA as well as other redox-active compounds, such as 3-HKYN and 3-HAA. The proinflammatory factors are detectable even in the early period following a stroke and proved to be associated with the rapid activation of the KP. Changes in the levels of 3-HKYN, 3-HAA, and QA appear to contribute to oxidative stress and brain damage after stroke. This indicates the need for the development of a new form of early anti-inflammatory interventions, effective in the modulation of the synthesis of such factors as KP metabolites, to reduce the inflammatory response, oxidative stress level, excitotoxicity, and in effect the development of delayed neurodegeneration [187]. Inhibitors of KP enzymes efficiently reduce ischemic damage in experimental models of stroke and brain inflammation in cerebral malaria [187,188,189,190]. Therefore, they could be useful for such interventions. In turn, the antioxidant properties of non-steroidal anti-inflammatory drugs, tolmetin and sulindac, can also protect hippocampal neurons against the oxidative damage induced by the accumulation of KP metabolites. Moreover, these agents prevent the depletion of reduced GS, decrease protein oxidation levels within the rat hippocampus, and improve spatial memory adversely affected by the accumulation of QA [191]. This finding supports the assumption that non-steroidal anti-inflammatory drugs may play a beneficial role in the management of neurodegenerative disorders.
3. Enzymes of the Kynurenine Pathway and the Possibility of Pharmacological Modulation of Their Activity
3.1. 2,3-Indoleamine Dioxygenase and 2,3-Tryptophan Dioxygenase
IDO-1 and IDO-2 participated in the oxidation of TRP to N-formylkynurenine, the first step of KYN synthesis. IDO-1 occurs in almost all body tissues. Its high activity was detected in the small intestine, spleen, lungs, epididymis, kidneys, blood vessel endothelium, and the brain. In physiological conditions, IDO-1 expression remains low, while interferon-γ, amyloid peptides, lipopolysaccharide, and other pro-inflammatory factors potently upregulate its expression and activation. IDO-2 plays a similar role and tissue distribution to IDO-1, but its physiological significance is still the subject of research [38,47,151,160,192].
The activation of IDO isoforms is mainly related to antioxidant capacities [11]. However, the downstream metabolites of KYN, which is a product of IDO, have both antioxidant and pro-oxidative properties. For example, as described above, 3-HKYN, 3-HAA, and QA play important roles in the development of various pathophysiological systemic disorders, including neurodegenerative ones, by the generation of oxidative and nitrosative damage, including lipid and protein peroxidation and excitotoxicity. Their excess enhances neuronal dysfunction and the apoptosis rate [193].
In the CNS, astrocytes play an important role in the maintenance of neuronal function and viability [192]. Inflammation within the CNS increases the concentration of pro-oxidative metabolites and the potential for NAD+ depletion through an increase in the poly (ADP-ribose) polymerase activity. On the other hand, the activity of IDO, the enzyme responsible for de novo NAD+ synthesis, is also significantly enhanced in astrocytes during inflammation [194,195]. Pretreatment of astroglial cells with interferon-γ increases IDO activity and intracellular NAD+ levels in vitro, leading to a decrease in their death ratio after exposure to hydrogen peroxide. These findings suggest that the induction of IDO and the subsequent de novo NAD+ synthesis may contribute to the maintenance of the proper intracellular energy supply and in effect cell viability and function in conditions of increased oxidative stress levels [194,196]. In turn, the inhibition of KP enzymes may in this case significantly decrease cellular functions and viability, unless alternate precursors for NAD+ synthesis are available (Table 2) [197]. This is confirmed by the fact that in vitro inhibition of IDO activity by pharmacological inhibitors, such as 1-methyl-L-tryptophan and QPRT using phthalic acid, resulted in a decreased intracellular NAD+ concentration and in effect cell viability both in astrocytes and neurons. Furthermore, this effect was more significant in neurons than astrocytes [11,198]. This suggests that changes in KP activity have a greater impact on the neurons than glial cells. It is also worth mentioning that IDO inhibition intensifies the progression of multiple sclerosis in a mouse model [196]. This seems to be associated with an increased CNS cell death rate caused by the decrease in NAD+ synthesis following IDO inhibition.
However, some studies indicate that inhibition of KYN synthesis does not always induce deleterious effects. Jovanovic et al. indicated that an administration of 1-methyl-tryptophan seems to be a useful approach in migraine and neuropathic pain treatment. In turn, Kanai et al. demonstrated that TDO modulates anxiety-related behavior in mice, while Campesan et al. showed that inhibition of TDO activity, by silencing its gene, and protects against the severity of HD symptoms in the transgenic Drosophila melanogaster model [163,200,209]. In turn, Sorgdrager et al. proved that long-term administration of 680C91, the oral inhibitor of TDO, significantly reversed recognition memory deficits in the mouse model of AD, however without affecting spatial learning and memory or anxiety-related behavior [199]. Moreover, preclinical studies also found that its inhibition is effective in alleviating depressive symptoms, which are also, to some extent, associated with an increase in oxidative stress levels caused by inflammation and KP overactivity in neurons (Table 2) [68,151,200].
In turn, Bonda et al. showed that the levels of 3-HKYN and IDO activity were elevated in the brain samples taken from AD patients in comparison with healthy individuals. Importantly, the authors confirmed associations between IDO activation and senile plaque accumulation. Its activity was also related to the number of neurofibrillary tangles in neurons. Both factors are the pathological lesions typical for AD; therefore, the KP seems to be related to the development of destructive neurodegenerative changes observed in the course of this disease [160].
3.2. Kynurenine 3-Monooxygenase
When cellular energy flow is disturbed, the activity of the KMO branch of the KP is enhanced [55,104]. This seems to be a compensatory mechanism, necessary to increase the NAD+ production required for ATP synthesis and proper functioning of the electron transport system in the mitochondria. While acute KMO activation may enable cells to temporarily meet higher energy demands, its chronic overactivity leads to oxidative damage and increases the apoptosis rate. Accordingly, prolonged KMO induction decreases the mitochondrial spare-respiratory capacity and increases ROS production. This indicates that KMO activity may have the opposite effects for neurons. Its moderate activation may promote favorable bioenergetics processes, but excessive activation would deplete energy stores and induce cell damage, leading to its dysfunction [104,201,202,203]. This mechanism seems to be important in terms of neurodegenerative diseases because neurons are highly sensitive to oxidative stress, energy depletion, and mitochondrial impairment (Table 2) [201,202,203]. Therefore, pharmacological inhibition of KMO activity seems to be a promising therapeutic tool in the management of various neurodegenerative disorders [104,163,201]. As mentioned above, the inhibition of IDO and QPRT activity may reduce the viability of astrocytes and neurons and exacerbate the clinical and histologic disease parameters during experimental autoimmune encephalomyelitis. In turn, preclinical studies indicate that targeting KMO in neurological disorders is a more efficient and safe solution [11,198].
In support of the above, Campesan et al. proved that KMO inhibition alleviates the severity of HD symptoms in the Drosophila melanogaster model (Table 2) [163]. Additionally, in the mouse model of HD, Sathyasaikumar et al. demonstrated a decrease in KYNU activity, the enzyme responsible for 3-HKYN degradation [162]. Treatment with the Ro 61-8048 and other KMO inhibitors, such as UPF648 and CHDI-340246, was effective in relieving neurological symptoms in the animal models of AD, HD, PD, and depression [55,162,163,209,210,211,212,213,214,215,216,217]. In turn, recent studies have shown that the administration of JM6, a new oral prodrug of Ro 61-8048, protects against behavioral deficits and synaptic loss, and, similarly to UPF648, increases the brain KYNA levels in the murine AD model as well as significantly inhibits microglial activation and prolongs life span in the mouse model of HD [218,219]. Therefore, KMO inhibitors seem to be a valuable therapeutic solution in the management of patients with AD, HD, and PD to ameliorate symptoms associated with 3-HKYN, 3-HAA, and QA accumulation in the brain.
In addition, treatment with KMO inhibitors could be beneficial in other neurological disorders. It has been proven that Ro 61-8048 administration prevents ataxia and reduces the mortality of mice infected with the plasmodium parasite [189]. All these reports indicate the need for further research concerning the use of KMO inhibitors in the therapy of neurological disorders with accompanying accumulation of KP metabolites. Importantly, their clinical utility in these diseases is still limited because most of them do not cross the BBB [55,217]. Therefore, the development of novel compounds with this ability is crucial.
An increase in brain KYNA levels can be achieved by modulating KP enzyme activity. Nicotinylalanine, an inhibitor of KYNU and KMO, administered together with KYN and probenecid, which is an inhibitor of organic anion transporters 1 and 3, significantly increases brain KYNA levels and reduces the neurotoxic symptoms induced by QA [172,204,220,221,222,223]. Giorgini et al. found that the administration of Ro 61-8048, a high-affinity KMO inhibitor, significantly reduced 3-HKYN production in vitro. However, its effectiveness seems to be limited because it did not inhibit QA synthesis nor alleviate ROS generation [212]. What is interesting, Amori et al. showed that the selective inhibition of KMO in vivo, using UPF648, leads to a decrease in the brain levels of both 3-HKYN and QA and a moderate increase in KYNA synthesis. This effect was observed even in animals pretreated with intrastriatal QA injection [55]. Moreover, in vitro research also proved that a loss of function mutation of KMO significantly reduced the toxicity of the mutant huntingtin fragment in a yeast HD transgenic model due to silencing the mechanism associated with ROS generation [212]. All of this indicates that the inhibition of 3-HKYN synthesis allows the severity of pathological changes during the neurodegenerative process to be reduced; therefore, the targeting of KMO could be an effective way to treat such diseases.
3.3. Kynurenine Aminotransferase and 3-Hydroxyanthranilic Acid 3,4-Dioxygenase
Although KAT isoform activity and brain KYNA levels were decreased in patients with neurodegenerative disorders, and this compound is widely recognized as a neuroprotectant [91,203,224,225], preclinical studies have demonstrated that excessive levels of both neurotoxic QA as well as neuroinhibitory KYNA can have detrimental effects on CNS cell function. KAT II inhibition by BFF816, although it decreases brain KYNA levels, may enhance cognitive abilities (Table 2) [73,204,205,206,207]. Yates et al. observed that the administration of 4-chloro-3-hydroxyanthranilate, a synthetic inhibitor of 3-HAO, leads to a decrease in QA synthesis and alleviates functional deficits in the experimental model of spinal cord injury in guinea pigs (Table 2) [208]. In turn, Vallerini et al. discovered that 2-aminonicotinic acid 1-oxide derivatives are stable, low molecular weight, and BBB-permeable inhibitors of 3-HAO. The most active of them can effectively shift the flux of the KP from QA toward KYNA in neurodegenerative conditions [226]. The downstream enzymes of KP appear to be a novel tool for better understanding the impact of the KP in the development of neurodegenerative disorders, such as AD, HD, epilepsy, and amyotrophic lateral sclerosis. Recent in vivo studies suggest that targeting them may open up new treatment options for the aforementioned diseases.
Excess 3-HKYN and its metabolites, 3-HAA and QA, can impair ATP production, and reduce the respiratory control index and the ADP/oxygen ratio in mitochondria. This fact justifies KP enzyme inhibitor use to counteract these effects [163,227]. KMO inhibition appears to be the most promising solution in preventing the accumulation of 3-HKYN, 3-HAA, and QA, and consequently interleukin-1β-dependent reduction of neurogenesis. In turn, the impact of the proinflammatory cytokines on KP metabolite concentration and their relationship with the neurogenesis rate indicates an important role of the KP in an inflammation-induced decrease in neurogenesis [70,224,228,229,230,231].
4. Analogs of Kynurenine Pathway Metabolites
Over the past years, several types of analogs of KP metabolites have been developed. Most of them belong to synthetic derivatives of KYNA. Their halogenated analogs, such as 7-Cl-KYNA, showed potent neuroprotective properties. However, their therapeutic significance is still limited, because most of them have poor BBB permeability. To date, only a few compounds, such as SZR72, SZR81, SZR104, and a prodrug 4-Cl-KYN, revealed the ability to cross this barrier. Therefore, this compound has the greatest potential to be used in the management of neurodegenerative disorders such as AD [13,232,233].
Synthetic analogs of KYNA are an important and numerous part of the several types of analogs of KP metabolites that have been developed over the last years [13,232]. A novel one, a KYNA amide derivative, 2-(2-N,N-dimethylaminoethylamine-1-carbonyl)-1H-quinolin-4-one hydrochloride, exhibits similar neuroprotective and neuroinhibitory properties to KYNA [13,81]. It showed beneficial effects in numerous areas. In the micromolar range, it significantly lowers the amplitude of the excitatory postsynaptic potentials within the hippocampus CA1 region. In turn, at the nanomolar level, it exerts a facilitatory effect on neurotransmission [81]. Similar to KYNA, it is capable of facilitating AMPAR responses. In the transgenic mouse model of HD, its administration prolongs survival time and prevents loss of striatal neurons [234]. Assessment of its anti-inflammatory properties proved that it has a greater ability to inhibit tumor necrosis factor-α synthesis than endogenous KYNA, which suggests its potent immunoregulatory properties [235]. It also effectively reduces oxidative stress levels associated with inflammation development and revealed a neuroprotective effect against ischemia-induced neuronal loss. Although its exact mechanism is unknown, the lack of cognitive adverse effects makes this compound a promising candidate for further investigations [236,237]. It could be useful in the management of neurodegenerative disorders such as AD and HD.
Halogenated analogs of KYNA, such as 7-Cl-KYNA, demonstrated high effectiveness as neuroprotectants because they are a selective antagonist of the glycine site of NMDA receptors and significantly reduce kainite-induced seizure activity [82,233,238]. Another synthetic KYNA analog, 5,7-ichlorokynurenic, despite being a potent antagonist of NMDA, does not decrease the level of hyperphosphorylation induced by protein phosphatase 2 inhibition, detrimental to neuron viability [239]. The clinical significance of most of them is questionable because their bioavailability within the CNS is poor [232]. Therefore, researchers focused on the development of agents with this property. It led them to the discovery of 4-Cl-KYN, a prodrug that enters the brain cells, where it is transformed into 7-Cl-KYNA by endogenous KAT isoforms. Systemic administration of 4-Cl-KYN leads to a significant increase in 7-Cl-KYNA levels in hippocampal neurons. Finally, it effectively alleviates the neurotoxic effects of QA in the rat hippocampus and striatum. This compound has completed a phase I clinical trial. It passed the evaluation of safety, tolerability, and pharmacokinetic profile. Therefore, it has a good chance to enter clinical use in the treatment of neurodegenerative diseases [81].
The development of KYNA analogs exerting complex anti-excitotoxic properties and capable of crossing the BBB was the main goal of the research on compounds belonging to the SZR series [240]. SZR72, SZR81, and SZR104 seem to be the most promising agents from this group. They demonstrated the BBB permeability and significantly inhibited tumor necrosis factor-α synthesis in vitro [240,241,242]. Furthermore, administration of SZR72 downregulated protein kinase-like endoplasmic reticulum kinase and IL-1β activation in the trigeminal ganglions in Sprague–Dawley rats, while SZR81 reduced immobility time and increased swimming time in Charles Dawley mice [150,241,242,243]. It suggests that these compounds have potential in the treatment of migraines and depression [241,242]. In turn, SZR104, the newest representative of this family, significantly decreased the number of microglial cells in the epileptic hippocampus and prevented microglia proliferation and decreased microglial phagocytosis in vitro. However, it did not prevent behavioral convulsions and did not decrease the mortality in the epilepsy mice model. This suggests that it may not cross the BBB in vivo [240].
5. Aryl Hydrocarbon Receptor and Oxidative Stress
The toxic effects of polycyclic aromatic hydrocarbons, dioxins, and other compounds with a similar chemical structure are mainly mediated by the activation of an aryl hydrocarbon receptor (AhR). Research indicates that it operates beyond xenobiotic metabolism and exerts numerous pleiotropic functions. Physiologically it plays an important role in the regulation of the numerous cellular signaling pathways and is responsible for the maintenance of immune homeostasis. Its activation upregulates the IL-6 and signal transducer and activator of transcription 3 expressions, and in effect it induces the general control nonderepressible-2 kinase and mammalian target of rapamycin kinase pathways. In this manner, increased synthesis of KYN, mainly by IDO, suppresses the immune system response. It causes the inactivation and apoptosis of TH1 and effector T cells, as well as activation of immunosuppressive T regulatory cells, preventing an excessive immune response [43,44,46,244,245]. The excessive activation of AhR, caused by elevated concentrations of KYN and its metabolites, increases oxidative stress levels, proinflammatory cytokine release, and as a result enhances cell aging and death rate [32,42,43,46,245]. In the transgenic AhR-null mouse model, Garcia-Lara et al. observed elevated KYNA levels in specific brain areas. This suggests that AhR activity inhibition leads to an increase in the brain amount of neuroprotective KYNA and contributes to its effectiveness against excitotoxicity and oxidative stress within the CNS. Furthermore, AhR is a physiological receptor for KYN and KYNA, while KYNA has a higher affinity to AhR than KYN. Thus far, these compounds have been considered neuroprotectants and antioxidants. Furthermore, AhR also links them to a cascade of toxic processes leading to brain damage, observed in, among others, HD [245]. These data suggest that the pharmacological inhibition of AhR activity could be another interesting option for reducing the deleterious effects of excess KYN and its metabolites on neurons and has potential in the treatment of neurological disorders, including HD.
6. Summary and Therapeutic Perspective
Oxidative damage causes harmful alterations in the functioning of neurons. It can be prevented through antioxidant use [6,7,8,9,10,11,12,13]. In the course of hypertryptophanemia or neurodegenerative disorders, the oxidative stress associated with excess TRP and its metabolites may contribute to the mechanisms leading to brain injury. Although the use of antioxidant supplementation is a controversial issue, it appears to be reasonable that the administration of taurine or vitamin C and E supplementation in patients with symptoms associated with TRP accumulation could be useful as an adjuvant procedure during their therapy, due to their antioxidant properties [83,84,85,86]. It is worth mentioning that despite evidence of the pro-oxidative effects of this amino acid, Ciji et al. demonstrated that its administration could reduce the oxidative stress induced by exposition to nitrites. However, its exact mechanism in this case is unclear. Its protective properties might be the result of its own redox properties as well as the impact of its antioxidant metabolites [134].
Various compounds with antioxidant capacity prevent cell death induced by 3-HKYN and its metabolites [117,187]. The antioxidant properties of non-steroidal anti-inflammatory drugs, tolmetin and sulindac, can protect hippocampal neurons against 3-HKYN-, 3-HAA-, and QA-induced oxidative damage. This finding supports the hypothesis that the use of non-steroidal anti-inflammatory drugs could be effective in the treatment of various neurodegenerative disorders, such as AD and HD [191]. In turn, the inhibition of IDO and QPRT activities by 1-methyl-tryptophan and phthalic acid, respectively, leads to a decrease in intracellular NAD+ levels and cell viability, both in astrocytes and neurons. Therefore, their use in the treatment of neurodegenerative diseases is questionable [11,196,198]. However, it is worth mentioning that inhibition of IDO activity has some potential in neurological disorder therapy. Recent reports indicate that 1-methyl-tryptophan appears to be a promising agent for migraine and neuropathic pain therapies [209,246].
Inhibition of KMO by Ro 61-8048 significantly reduces 3-HKYN synthesis induced by the mutant huntingtin in the transgenic yeast model [212]. Moreover, its administration leads to an increase in brain KYNA levels in the PD monkey model [211], while in combination with levodopa, it reduces the severity of dyskinesias, both acute and after long-time administration. In turn, the combination of the KMO inhibitor with KYNA supplementation reveals a neuroprotective effect in the mouse model of PD [247]. Much preclinical research has proved that the administration of Ro 61-8048, UPF648, and CHDI-340246 was effective in the management of neurodegenerative and psychiatric disorders such as AD, HD, PD, and depression [55,162,163,209,210,211,212,213,214,215,216,217]. In turn, the oral prodrug JM6 ameliorated anxiety-related behavior, memory deficits, and synaptic loss, and increased the brain KYNA levels in the murine model of AD. It also efficiently decreased microglial activation and extended life span in transgenic mice with HD [218,219]. In addition to decreasing 3-HKYN and QA, treatment with UPF648 moderately elevated KYNA synthesis in rat brains [248]. In turn, in the mouse model of malaria, Ro 61–8048 averted ataxia and decreased the mortality rate [178]. The clinical utility of most of the currently available KMO inhibitors in brain disorders is limited because their BBB permeability is very weak [217,249]. Meanwhile, the novel competitive KMO inhibitors belonging to derivatives of aryl pyrimidine carboxylic acid can penetrate the BBB. Thus, they could be effective in the treatment of neurodegenerative diseases accompanied by alterations in KMO activity [55,217,249].
What is interesting is that the activation of KAT II in neurons appears to have an impact on such processes as programmed cell death. N-acetylcysteine, a brain-penetrant drug with antioxidant properties and procognitive effects in humans, increases the brain levels of reduced GS and indirectly inhibits KAT II. This mechanism seems to enhance the beneficial neuropharmacological effects of this drug and could be potentially useful in alleviating cognitive deficits in schizophrenia and other major brain diseases, such as AD [250].
Inhibition of 3-HAO activity by 4-chloro-3-hydroxyanthranilate and 2-aminonicotinic acid 1-oxide derivatives leads to a significant decrease in QA synthesis and alleviates the deleterious effects associated with its accumulation. This indicates that the inhibitors of downstream KP enzymes could be an effective pharmacological tool in the treatment of such neurodegenerative diseases as AD, HD, epilepsy, and amyotrophic lateral sclerosis [226,227]. Moreover, pharmacological inhibition of AhR could be a valuable option for a reduction of the harmful impact of excess KP metabolites on brain cells and has potential in the treatment of HD [245]. In turn, combined administration of KYN and probenecid increased the cortical levels of KYNA. This combination attenuated morphological lesions and improved spatial memory in the mouse model of AD. These compounds can also be co-administered with nicotinylalanine, an inhibitor of KYNU and KMO. In this setting, they relieve QA-induced changes in animal behavior [172,220,221,222,223]. Moreover, mitigation of the toxic effects caused by excess QA in the brain could be achieved using synthetic analogs of KP metabolites, such as 4-Cl-KYN, a BBB-permeable prodrug of 7Cl-KYNA [81,233].
7. Conclusions
Much research has proved the accumulation of neuroactive metabolites of the KP within the brain during numerous neurodegenerative and psychiatric disorders. Some of them are neurotoxic and stimulate the development of detrimental processes within the CNS. They can induce excitotoxicity, oxidative damage, and inflammation, leading to neuronal dysfunction and death. Preclinical studies indicate that strategies including targeting KP activity using TRP supplementation, KP enzyme inhibitors, or analogs of KP metabolites appear to be noteworthy therapeutic options against the excitotoxicity and oxidative stress accompanying neurological disorders as well as a neuroprotective solution. The greatest number of preclinical reports indicate that the use of KP enzyme inhibitors brings the most promising results in the treatment of these diseases. However, there are still many unknowns, and clinical solutions are far from being developed. The role of KP metabolites, oxidative stress, and excitotoxicity in the development of CNS pathologies still require better understanding to develop novel therapies effective in the management of neurodegenerative disorders. This indicates the need for further research on this topic, especially in terms of clinical application.
Author Contributions
Conceptualization, A.M. and A.T.-K.; writing—original draft preparation, A.M.; writing—review and editing, A.T.-K., A.K. and D.P.; figures preparation, A.T.-K.; supervision, D.P.; funding acquisition, A.T.-K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Medical University of Bialystok, Poland (grant number SUB/1/DN/20/001/2228).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 3-Hydroxyanthranilic acid | 3-HAA |
| 3-Hydroxyanthranilic acid 3,4-dioxygenase | 3-HAO |
| 3-Hydroxykynurenine | 3-HKYN |
| α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor | AMPAR |
| Adenosine diphosphonate | ADP |
| Adenosine triphosphate | ATP |
| Alzheimer’s disease | AD |
| Aminocarboxymuconatesemialdehyde | ACMS |
| Anthranilic acid | AA |
| Aryl hydrocarbon receptor | AhR |
| Blood–brain barrier | BBB |
| Calcium | Ca2+ |
| Central nervous system | CNS |
| Iron(II)G protein-coupled receptor 35 | Fe2+GPR35 |
| Glutathione | GS |
| Glutathione S-transferase | GST |
| Huntington’s disease | HD |
| Indoleamine 2,3-dioxygenase | IDO |
| Kynurenic acid | KYNA |
| Kynureninase | KYNU |
| Kynurenine | KYN |
| Kynurenine 3-monooxygenase | KMO |
| Kynurenine aminotransferase | KAT |
| Kynurenine pathway | KP |
| Lactate dehydrogenase | LDH |
| Large neutral amino acid transporter 1 | LAT-1 |
| Nicotinamide adenine dinucleotide | NAD |
| N-methyl-D-aspartate | NMDA |
| Parkinson’s disease | PD |
| Picolinic acid | PA |
| Quinolinate phosphoribosylotransferase | QPRT |
| Quinolinic acid | QA |
| Reactive oxygen species | ROS |
| Superoxide dismutase | SOD |
| Tryptophan | TRP |
| Tryptophan 2,3-dioxygenase | TDO |
| Xanthurenic acid | XA |
References
- Ghorbani, R.; Ghousi, R. Predictive Data Mining Approaches in Medical Diagnosis: A Review of Some Diseases Prediction. Int. J. Data Netw. Sci. 2019, 3, 47–70. [Google Scholar] [CrossRef]
- Kennedy, D.P.; Adolphs, R. The Social Brain in Psychiatric and Neurological Disorders. Trends Cogn. Sci. 2012, 16, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, R.; Sharma, M. Prevalence and Diagnosis of Neurological Disorders Using Different Deep Learning Techniques: A Meta-Analysis. J. Med. Syst. 2020, 44, 49. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Abajobir, A.A.; Abate, K.H.; Abd-Allah, F.; Abdulle, A.M.; Abera, S.F.; Abyu, G.Y.; Ahmed, M.B.; Aichour, A.N.; Aichour, I.; et al. Global, Regional, and National Burden of Neurological Disorders during 1990–2015: A Systematic Analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef] [Green Version]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, Regional, and National Burden of Neurological Disorders, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca2+ and Apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Perez-Gracia, E.; Blanco, R.; Carmona, M.; Carro, E.; Ferrer, I. Oxidative Stress Damage and Oxidative Stress Responses in the Choroid Plexus in Alzheimer’s Disease. Acta Neuropathol. 2009, 118, 497–504. [Google Scholar] [CrossRef]
- Reyes Ocampo, J.; Lugo Huitrón, R.; González-Esquivel, D.; Ugalde-Muñiz, P.; Jiménez-Anguiano, A.; Pineda, B.; Pedraza-Chaverri, J.; Ríos, C.; Pérez de la Cruz, V. Kynurenines with Neuroactive and Redox Properties: Relevance to Aging and Brain Diseases. Oxid. Med. Cell. Longev. 2014, 2014, 646909. [Google Scholar] [CrossRef]
- Wigner, P.; Czarny, P.; Galecki, P.; Sliwinski, T. Oxidative and Nitrosative Stress as Well as the Tryptophan Catabolites Pathway in Depressive Disorders. Psychiatr. Danub. 2017, 29, 394–400. [Google Scholar] [CrossRef] [Green Version]
- Bohár, Z.; Toldi, J.; Fülöp, F.; Vécsei, L. Changing the Face of Kynurenines and Neurotoxicity: Therapeutic Considerations. Int. J. Mol. Sci. 2015, 16, 9772–9793. [Google Scholar] [CrossRef] [Green Version]
- Bertram, L.; Tanzi, R.E. The Genetic Epidemiology of Neurodegenerative Disease. J. Clin. Investig. 2005, 115, 1449–1457. [Google Scholar] [CrossRef] [Green Version]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The Genetic Architecture of Parkinson’s Disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- García, J.-C.; Bustos, R.-H. The Genetic Diagnosis of Neurodegenerative Diseases and Therapeutic Perspectives. Brain Sci. 2018, 8, 222. [Google Scholar] [CrossRef] [Green Version]
- Trushina, E.; McMurray, C.T. Oxidative Stress and Mitochondrial Dysfunction in Neurodegenerative Diseases. Neuroscience 2007, 145, 1233–1248. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Douarre, C.; Sourbier, C.; Dalla Rosa, I.; Brata Das, B.; Redon, C.E.; Zhang, H.; Neckers, L.; Pommier, Y. Mitochondrial Topoisomerase I Is Critical for Mitochondrial Integrity and Cellular Energy Metabolism. PLoS ONE 2012, 7, e41094. [Google Scholar] [CrossRef] [Green Version]
- Hollensworth, S.B.; Shen, C.; Sim, J.E.; Spitz, D.R.; Wilson, G.L.; LeDoux, S.P. Glial Cell Type-Specific Responses to Menadione-Induced Oxidative Stress. Free Radic. Biol. Med. 2000, 28, 1161–1174. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Toescu, E.C. Normal Brain Ageing: Models and Mechanisms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2347–2354. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and Endoplasmic Reticulum Calcium Homeostasis and Cell Death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the Mammalian Brain: When Physiology Meets Pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Garrison, A.M.; Parrott, J.M.; Tuñon, A.; Delgado, J.; Redus, L.; O’Connor, J.C. Kynurenine Pathway Metabolic Balance Influences Microglia Activity: Targeting Kynurenine Monooxygenase to Dampen Neuroinflammation. Psychoneuroendocrinology 2018, 94, 1–10. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Monitoring the Redox Status in Multiple Sclerosis. Biomedicines 2020, 8, 406. [Google Scholar] [CrossRef]
- Török, N.; Tanaka, M.; Vécsei, L. Searching for Peripheral Biomarkers in Neurodegenerative Diseases: The Tryptophan-Kynurenine Metabolic Pathway. Int. J. Mol. Sci. 2020, 21, 9338. [Google Scholar] [CrossRef] [PubMed]
- Bender, D.A. Biochemistry of Tryptophan in Health and Disease. Mol. Asp. Med. 1983, 6, 101–197. [Google Scholar] [CrossRef]
- Badawy, A.A. Tryptophan Metabolism in Alcoholism. Adv. Exp. Med. Biol. 1999, 467, 265–274. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Tryptophan Metabolism, Disposition and Utilization in Pregnancy. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Pellagra and Alcoholism: A Biochemical Perspective. Alcohol Alcohol. 2014, 49, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, D.; Song, P.; Zou, M.-H. Deregulated Tryptophan-Kynurenine Pathway Is Linked to Inflammation, Oxidative Stress, and Immune Activation Pathway in Cardiovascular Diseases. Front. Biosci. Landmark Ed. 2015, 20, 1116–1143. [Google Scholar]
- Heyes, M.P.; Saito, K.; Crowley, J.S.; Davis, L.E.; Demitrack, M.A.; Der, M.; Dilling, L.A.; Elia, J.; Kruesi, M.J.; Lackner, A. Quinolinic Acid and Kynurenine Pathway Metabolism in Inflammatory and Non-Inflammatory Neurological Disease. Brain J. Neurol. 1992, 115 (Pt 5), 1249–1273. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T Cell Apoptosis by Tryptophan Catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Topczewska-Bruns, J.; Pawlak, D.; Chabielska, E.; Tankiewicz, A.; Buczko, W. Increased Levels of 3-Hydroxykynurenine in Different Brain Regions of Rats with Chronic Renal Insufficiency. Brain Res. Bull. 2002, 58, 423–428. [Google Scholar] [CrossRef]
- Forrest, C.M.; Mackay, G.M.; Oxford, L.; Stoy, N.; Stone, T.W.; Darlington, L.G. Kynurenine Pathway Metabolism in Patients with Osteoporosis after 2 Years of Drug Treatment. Clin. Exp. Pharm. Physiol. 2006, 33, 1078–1087. [Google Scholar] [CrossRef]
- Dayer, M.R.; Safari, I.; Dayer, M.S. New Evidence on Hypoglycemic Effect of Quinolinic Acid in Diabetic Rats. Pak. J. Biol. Sci. 2009, 12, 1025–1030. [Google Scholar] [CrossRef] [Green Version]
- Munipally, P.K.; Agraharm, S.G.; Valavala, V.K.; Gundae, S.; Turlapati, N.R. Evaluation of Indoleamine 2,3-Dioxygenase Expression and Kynurenine Pathway Metabolites Levels in Serum Samples of Diabetic Retinopathy Patients. Arch. Physiol. Biochem. 2011, 117, 254–258. [Google Scholar] [CrossRef]
- Prendergast, G.C. Cancer: Why Tumours Eat Tryptophan. Nature 2011, 478, 192–194. [Google Scholar] [CrossRef]
- Apalset, E.M.; Gjesdal, C.G.; Ueland, P.M.; Midttun, Ø.; Ulvik, A.; Eide, G.E.; Meyer, K.; Tell, G.S. Interferon (IFN)-γ-Mediated Inflammation and the Kynurenine Pathway in Relation to Bone Mineral Density: The Hordaland Health Study. Clin. Exp. Immunol. 2014, 176, 452–460. [Google Scholar] [CrossRef] [Green Version]
- González Esquivel, D.; Ramírez-Ortega, D.; Pineda, B.; Castro, N.; Ríos, C.; Pérez de la Cruz, V. Kynurenine Pathway Metabolites and Enzymes Involved in Redox Reactions. Neuropharmacology 2017, 112, 331–345. [Google Scholar] [CrossRef]
- Kawajiri, K.; Fujii-Kuriyama, Y. The Aryl Hydrocarbon Receptor: A Multifunctional Chemical Sensor for Host Defense and Homeostatic Maintenance. Exp. Anim. 2017, 66, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.C.; Macchiarulo, A.; Vacca, C.; et al. Aryl Hydrocarbon Receptor Control of a Disease Tolerance Defence Pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M.; et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. 2018, 29, 1063–1072. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Nakahama, T.; Le, D.H.; Van Son, L.; Chu, H.H.; Kishimoto, T. Aryl Hydrocarbon Receptor and Kynurenine: Recent Advances in Autoimmune Disease Research. Front. Immunol. 2014, 5, 551. [Google Scholar] [CrossRef] [Green Version]
- King, N.J.C.; Thomas, S.R. Molecules in Focus: Indoleamine 2,3-Dioxygenase. Int. J. Biochem. Cell Biol. 2007, 39, 2167–2172. [Google Scholar] [CrossRef]
- Capece, L.; Lewis-Ballester, A.; Marti, M.A.; Estrin, D.A.; Yeh, S.-R. Molecular Basis for the Substrate Stereoselectivity in Tryptophan Dioxygenase. Biochemistry 2011, 50, 10910–10918. [Google Scholar] [CrossRef] [Green Version]
- Knox, W.E.; Greengard, O. The Regulation of Some Enzymes of Nitrogen Metabolism—An Introduction to Enzyme Physiology. Adv. Enzym. Regul. 1965, 3, 247–313. [Google Scholar] [CrossRef]
- Feigelson, M.; Feigelson, P. Metabolic Effects of Glucocorticoids as Related to Enzyme Induction. Adv. Enzym. Regul. 1965, 3, 11–27. [Google Scholar] [CrossRef]
- Schimke, R.T.; Sweeney, E.W.; Berlin, C.M. The roles of synthesis and degradation in the control of rat liver tryptophan pyrrolase. J. Biol. Chem. 1965, 240, 322–331. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawy, A.A.-B.; Bano, S. Tryptophan Metabolism in Rat Liver After Administration of Tryptophan, Kynurenine Metabolites, and Kynureninase Inhibitors. Int. J. Tryptophan Res. 2016, 9, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouns, R.; Verkerk, R.; Aerts, T.; De Surgeloose, D.; Wauters, A.; Scharpé, S.; De Deyn, P.P. The Role of Tryptophan Catabolism along the Kynurenine Pathway in Acute Ischemic Stroke. Neurochem. Res. 2010, 35, 1315–1322. [Google Scholar] [CrossRef]
- Amori, L.; Guidetti, P.; Pellicciari, R.; Kajii, Y.; Schwarcz, R. On the Relationship between the Two Branches of the Kynurenine Pathway in the Rat Brain in Vivo. J. Neurochem. 2009, 109, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Barone, P. The “Yin” and the “Yang” of the Kynurenine Pathway: Excitotoxicity and Neuroprotection Imbalance in Stress-Induced Disorders. Behav. Pharm. 2019, 30, 163–186. [Google Scholar] [CrossRef]
- Walsh, H.A.; Botting, N.P. Purification and Biochemical Characterization of Some of the Properties of Recombinant Human Kynureninase. Eur. J. Biochem. 2002, 269, 2069–2074. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an Endogenous Oxidative Stress Generator, Causes Neuronal Cell Death with Apoptotic Features and Region Selectivity. J. Neurochem. 1998, 70, 299–307. [Google Scholar] [CrossRef]
- Stone, T.W. Neuropharmacology of Quinolinic and Kynurenic Acids. Pharm. Rev. 1993, 45, 309–379. [Google Scholar]
- Schwarcz, R.; Pellicciari, R. Manipulation of Brain Kynurenines: Glial Targets, Neuronal Effects, and Clinical Opportunities. J. Pharm. Exp. Ther. 2002, 303, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Nishizuka, Y.; Hayaishi, O. Studies on the biosynthesis of nicotinamide adenine dinucleotide: I. Enzymic synthesis of niacin ribonucleotides from 3-hydroxyanthranilic acid in mammalian tissues. J. Biol. Chem. 1963, 238, 3369–3377. [Google Scholar] [CrossRef]
- Bender, D.A. Inhibition in Vitro of the Enzymes of the Oxidative Pathway of Tryptophan Metabolism and of Nicotinamide Nucleotide Synthesis by Benserazide, Carbidopa and Isoniazid. Biochem. Pharm. Ther. 1980, 29, 707–712. [Google Scholar] [CrossRef]
- Pawlak, D.; Pawlak, K.; Malyszko, J.; Mysliwiec, M.; Buczko, W. Accumulation of Toxic Products Degradation of Kynurenine in Hemodialyzed Patients. Int. Urol. Nephrol. 2001, 33, 399–404. [Google Scholar] [CrossRef]
- Debnath, S.; Velagapudi, C.; Redus, L.; Thameem, F.; Kasinath, B.; Hura, C.E.; Lorenzo, C.; Abboud, H.E.; O’Connor, J.C. Tryptophan Metabolism in Patients with Chronic Kidney Disease Secondary to Type 2 Diabetes: Relationship to Inflammatory Markers. Int. J. Tryptophan Res. 2017, 10, 1178646917694600. [Google Scholar] [CrossRef]
- Topczewska-Bruns, J.; Tankiewicz, A.; Pawlak, D.; Buczko, W. Behavioral Changes in the Course of Chronic Renal Insufficiency in Rats. Pol. J. Pharm. Ther. 2001, 53, 263–269. [Google Scholar]
- Lovelace, M.D.; Varney, B.; Sundaram, G.; Lennon, M.J.; Lim, C.K.; Jacobs, K.; Guillemin, G.J.; Brew, B.J. Recent Evidence for an Expanded Role of the Kynurenine Pathway of Tryptophan Metabolism in Neurological Diseases. Neuropharmacology 2017, 112, 373–388. [Google Scholar] [CrossRef]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood-Brain Barrier Transport of Kynurenines: Implications for Brain Synthesis and Metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef]
- Agudelo, L.Z.; Femenía, T.; Orhan, F.; Porsmyr-Palmertz, M.; Goiny, M.; Martinez-Redondo, V.; Correia, J.C.; Izadi, M.; Bhat, M.; Schuppe-Koistinen, I.; et al. Skeletal Muscle PGC-1α1 Modulates Kynurenine Metabolism and Mediates Resilience to Stress-Induced Depression. Cell 2014, 159, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Gobaille, S.; Kemmel, V.; Brumaru, D.; Dugave, C.; Aunis, D.; Maitre, M. Xanthurenic Acid Distribution, Transport, Accumulation and Release in the Rat Brain. J. Neurochem. 2008, 105, 982–993. [Google Scholar] [CrossRef] [Green Version]
- Zunszain, P.A.; Anacker, C.; Cattaneo, A.; Choudhury, S.; Musaelyan, K.; Myint, A.M.; Thuret, S.; Price, J.; Pariante, C.M. Interleukin-1β: A New Regulator of the Kynurenine Pathway Affecting Human Hippocampal Neurogenesis. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2012, 37, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Maes, M.; Rief, W. Diagnostic Classifications in Depression and Somatization Should Include Biomarkers, Such as Disorders in the Tryptophan Catabolite (TRYCAT) Pathway. Psychiatry Res. 2012, 196, 243–249. [Google Scholar] [CrossRef]
- Walker, A.K.; Wing, E.E.; Banks, W.A.; Dantzer, R. Leucine Competes with Kynurenine for Blood-to-Brain Transport and Prevents Lipopolysaccharide-Induced Depression-like Behavior in Mice. Mol. Psychiatry 2019, 24, 1523–1532. [Google Scholar] [CrossRef]
- Sekine, A.; Kuroki, Y.; Urata, T.; Mori, N.; Fukuwatari, T. Inhibition of Large Neutral Amino Acid Transporters Suppresses Kynurenic Acid Production Via Inhibition of Kynurenine Uptake in Rodent Brain. Neurochem. Res. 2016, 41, 2256–2266. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bush, K.T.; Martovetsky, G.; Ahn, S.-Y.; Liu, H.C.; Richard, E.; Bhatnagar, V.; Wu, W. The Organic Anion Transporter (OAT) Family: A Systems Biology Perspective. Physiol. Rev. 2015, 95, 83–123. [Google Scholar] [CrossRef]
- Wu, W.; Bush, K.T.; Nigam, S.K. Key Role for the Organic Anion Transporters, OAT1 and OAT3, in the in Vivo Handling of Uremic Toxins and Solutes. Sci. Rep. 2017, 7, 4939. [Google Scholar] [CrossRef] [Green Version]
- Biernacki, T.; Sandi, D.; Bencsik, K.; Vécsei, L. Kynurenines in the Pathogenesis of Multiple Sclerosis: Therapeutic Perspectives. Cells 2020, 9, 1564. [Google Scholar] [CrossRef]
- Braidy, N.; Grant, R.; Adams, S.; Guillemin, G.J. Neuroprotective Effects of Naturally Occurring Polyphenols on Quinolinic Acid-Induced Excitotoxicity in Human Neurons. FEBS J. 2010, 277, 368–382. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Oxidative/Nitrosative Stress and Immuno-Inflammatory Pathways in Depression: Treatment Implications. Curr. Pharm. Des. 2014, 20, 3812–3847. [Google Scholar] [CrossRef]
- Beadle, G.W.; Mitchell, H.K.; Nyc, J.F. Kynurenine as an Intermediate in the Formation of Nicotinic Acid from Tryptophane by Neurospora. Proc. Natl. Acad. Sci. USA 1947, 33, 155–158. [Google Scholar] [CrossRef] [Green Version]
- Moroni, F.; Russi, P.; Carlá, V.; Lombardi, G. Kynurenic Acid Is Present in the Rat Brain and Its Content Increases during Development and Aging Processes. Neurosci. Lett. 1988, 94, 145–150. [Google Scholar] [CrossRef]
- Marosi, M.; Nagy, D.; Farkas, T.; Kis, Z.; Rózsa, E.; Robotka, H.; Fülöp, F.; Vécsei, L.; Toldi, J. A Novel Kynurenic Acid Analogue: A Comparison with Kynurenic Acid. An in Vitro Electrophysiological Study. J. Neural Transm. 2010, 117, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Domenici, M.R.; Longo, R.; Sagratella, S. 7-Chlorokynurenic Acid Prevents in Vitro Epileptiform and Neurotoxic Effects Due to Kainic Acid. Gen. Pharm. Ther. 1996, 27, 113–116. [Google Scholar] [CrossRef]
- Feksa, L.R.; Latini, A.; Rech, V.C.; Feksa, P.B.; Koch, G.D.W.; Amaral, M.F.A.; Leipnitz, G.; Dutra-Filho, C.S.; Wajner, M.; Wannmacher, C.M.D. Tryptophan Administration Induces Oxidative Stress in Brain Cortex of Rats. Metab. Brain Dis. 2008, 23, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxygen Radicals as Key Mediators in Neurological Disease: Fact or Fiction? Ann. Neurol. 1992, 32 (Suppl. S10–S15). [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative Stress and Neurodegeneration: Where Are We Now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive Species and Antioxidants. Redox Biology Is a Fundamental Theme of Aerobic Life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Adams, G.E.; Aldrich, J.E.; Bisby, R.H.; Cundall, R.B.; Redpath, J.L.; Willson, R.L. Selective Free Radical Reactions with Proteins and Enzymes: Reactions of Inorganic Radical Anions with Amino Acids. Radiat. Res. 1972, 49, 278–289. [Google Scholar] [CrossRef]
- Land, E.J.; Prütz, W.A. Fast One-Electron Oxidation of Tryptophan by Azide Radicals. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1977, 32, 203–207. [Google Scholar] [CrossRef] [Green Version]
- Gracanin, M.; Hawkins, C.L.; Pattison, D.I.; Davies, M.J. Singlet-Oxygen-Mediated Amino Acid and Protein Oxidation: Formation of Tryptophan Peroxides and Decomposition Products. Free Radic. Biol. Med. 2009, 47, 92–102. [Google Scholar] [CrossRef]
- Genestet, C.; Le Gouellec, A.; Chaker, H.; Polack, B.; Guery, B.; Toussaint, B.; Stasia, M.J. Scavenging of Reactive Oxygen Species by Tryptophan Metabolites Helps Pseudomonas Aeruginosa Escape Neutrophil Killing. Free Radic. Biol. Med. 2014, 73, 400–410. [Google Scholar] [CrossRef]
- Blanco Ayala, T.; Lugo Huitrón, R.; Carmona Aparicio, L.; Ramírez Ortega, D.; González Esquivel, D.; Pedraza Chaverrí, J.; Pérez de la Cruz, G.; Ríos, C.; Schwarcz, R.; Pérez de la Cruz, V. Alternative Kynurenic Acid Synthesis Routes Studied in the Rat Cerebellum. Front. Cell. Neurosci. 2015, 9, 178. [Google Scholar] [CrossRef] [Green Version]
- Giles, G.I.; Collins, C.A.; Stone, T.W.; Jacob, C. Electrochemical and in Vitro Evaluation of the Redox-Properties of Kynurenine Species. Biochem. Biophys. Res. Commun. 2003, 300, 719–724. [Google Scholar] [CrossRef]
- Rios, C.; Santamaria, A. Quinolinic Acid Is a Potent Lipid Peroxidant in Rat Brain Homogenates. Neurochem. Res. 1991, 16, 1139–1143. [Google Scholar] [CrossRef]
- Reszka, K.J.; Matuszak, Z.; Chignell, C.F. Lactoperoxidase-Catalyzed Oxidation of the Anticancer Agent Mitoxantrone by Nitrogen Dioxide (NO2.) Radicals. Chem. Res. Toxicol. 1997, 10, 1325–1330. [Google Scholar] [CrossRef]
- Song, H.; Park, H.; Kim, Y.-S.; Kim, K.D.; Lee, H.-K.; Cho, D.-H.; Yang, J.-W.; Hur, D.Y. L-Kynurenine-Induced Apoptosis in Human NK Cells Is Mediated by Reactive Oxygen Species. Int. Immunopharmacol. 2011, 11, 932–938. [Google Scholar] [CrossRef]
- Lugo-Huitrón, R.; Blanco-Ayala, T.; Ugalde-Muñiz, P.; Carrillo-Mora, P.; Pedraza-Chaverrí, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzón, E.; Ortiz-Islas, E.; et al. On the Antioxidant Properties of Kynurenic Acid: Free Radical Scavenging Activity and Inhibition of Oxidative Stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef]
- Ramos-Chávez, L.A.; Lugo Huitrón, R.; González Esquivel, D.; Pineda, B.; Ríos, C.; Silva-Adaya, D.; Sánchez-Chapul, L.; Roldán-Roldán, G.; Pérez de la Cruz, V. Relevance of Alternative Routes of Kynurenic Acid Production in the Brain. Oxid. Med. Cell. Longev. 2018, 2018, 5272741. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Bohár, Z.; Vécsei, L. Are Kynurenines Accomplices or Principal Villains in Dementia? Maintenance of Kynurenine Metabolism. Molecules 2020, 25, 564. [Google Scholar] [CrossRef] [Green Version]
- Colín-González, A.L.; Maya-López, M.; Pedraza-Chaverrí, J.; Ali, S.F.; Chavarría, A.; Santamaría, A. The Janus Faces of 3-Hydroxykynurenine: Dual Redox Modulatory Activity and Lack of Neurotoxicity in the Rat Striatum. Brain Res. 2014, 1589, 1–14. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. Hydrogen Peroxide-Mediated Neuronal Cell Death Induced by an Endogenous Neurotoxin, 3-Hydroxykynurenine. Proc. Natl. Acad. Sci. USA 1996, 93, 12553–12558. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Ortega, D.; Ramiro-Salazar, A.; González-Esquivel, D.; Ríos, C.; Pineda, B.; Pérez de la Cruz, V. 3-Hydroxykynurenine and 3-Hydroxyanthranilic Acid Enhance the Toxicity Induced by Copper in Rat Astrocyte Culture. Oxid. Med. Cell. Longev. 2017, 2017, 2371895. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, K. Etiological Classification of Depression Based on the Enzymes of Tryptophan Metabolism. BMC Psychiatry 2014, 14, 372. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Ocampo, J.; Ramírez-Ortega, D.; Cervantes, G.I.V.; Pineda, B.; Balderas, P.M.d.O.; González-Esquivel, D.; Sánchez-Chapul, L.; Lugo-Huitrón, R.; Silva-Adaya, D.; Ríos, C.; et al. Mitochondrial Dysfunction Related to Cell Damage Induced by 3-Hydroxykynurenine and 3-Hydroxyanthranilic Acid: Non-Dependent-Effect of Early Reactive Oxygen Species Production. Neurotoxicology 2015, 50, 81–91. [Google Scholar] [CrossRef]
- Braidy, N.; Grant, R.; Brew, B.J.; Adams, S.; Jayasena, T.; Guillemin, G.J. Effects of Kynurenine Pathway Metabolites on Intracellular NAD Synthesis and Cell Death in Human Primary Astrocytes and Neurons. Int. J. Tryptophan Res. 2009, 2, 61–69. [Google Scholar] [CrossRef]
- Leipnitz, G.; Schumacher, C.; Dalcin, K.B.; Scussiato, K.; Solano, A.; Funchal, C.; Dutra-Filho, C.S.; Wyse, A.T.S.; Wannmacher, C.M.D.; Latini, A.; et al. In Vitro Evidence for an Antioxidant Role of 3-Hydroxykynurenine and 3-Hydroxyanthranilic Acid in the Brain. Neurochem. Int. 2007, 50, 83–94. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharm. Ther. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Dykens, J.A.; Sullivan, S.G.; Stern, A. Oxidative Reactivity of the Tryptophan Metabolites 3-Hydroxyanthranilate, Cinnabarinate, Quinolinate and Picolinate. Biochem. Pharm. Ther. 1987, 36, 211–217. [Google Scholar] [CrossRef]
- Quagliariello, E.; Papa, S.; Saccone, C.; Alifano, A. Effect of 3-Hydroxyanthranilic Acid on the Mitochondrial Respiratory System. Biochem. J. 1964, 91, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Chobot, V.; Hadacek, F.; Weckwerth, W.; Kubicova, L. Iron Chelation and Redox Chemistry of Anthranilic Acid and 3-Hydroxyanthranilic Acid: A Comparison of Two Structurally Related Kynurenine Pathway Metabolites to Obtain Improved Insights into Their Potential Role in Neurological Disease Development. J. Organomet. Chem. 2015, 782, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Schuck, P.F.; Tonin, A.; da Costa Ferreira, G.; Viegas, C.M.; Latini, A.; Duval Wannmacher, C.M.; de Souza Wyse, A.T.; Dutra-Filho, C.S.; Wajner, M. Kynurenines Impair Energy Metabolism in Rat Cerebral Cortex. Cell. Mol. Neurobiol. 2007, 27, 147–160. [Google Scholar] [CrossRef]
- Gaubert, S.; Bouchaut, M.; Brumas, V.; Berthon, G. Copper--Ligand Interactions and the Physiological Free Radical Processes. Part 3. Influence of Histidine, Salicylic Acid and Anthranilic Acid on Copper-Driven Fenton Chemistry in Vitro. Free Radic. Res. 2000, 32, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Miche, H.; Brumas, V.; Berthon, G. Copper(II) Interactions with Nonsteroidal Antiinflammatory Agents. II. Anthranilic Acid as a Potential. OH-Inactivating Ligand. J. Inorg. Biochem. 1997, 68, 27–38. [Google Scholar] [CrossRef]
- Christen, S.; Peterhans, E.; Stocker, R. Antioxidant Activities of Some Tryptophan Metabolites: Possible Implication for Inflammatory Diseases. Proc. Natl. Acad. Sci. USA 1990, 87, 2506–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, K.; Haneda, M.; Qiao, S.; Naruse, M.; Yoshino, M. Prooxidant Action of Rosmarinic Acid: Transition Metal-Dependent Generation of Reactive Oxygen Species. Toxicol. Vitr. Int. J. Publ. Assoc. Bibra 2007, 21, 613–617. [Google Scholar] [CrossRef]
- Aggett, P.J.; Fenwick, P.K.; Kirk, H. An in Vitro Study of the Effect of Picolinic Acid on Metal Translocation across Lipid Bilayers. J. Nutr. 1989, 119, 1432–1437. [Google Scholar] [CrossRef] [Green Version]
- Goda, K.; Kishimoto, R.; Shimizu, S.; Hamane, Y.; Ueda, M. Quinolinic Acid and Active Oxygens. Possible Contribution of Active Oxygens during Cell Death in the Brain. Adv. Exp. Med. Biol. 1996, 398, 247–254. [Google Scholar]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic Acid: An Endogenous Neurotoxin with Multiple Targets. Oxid. Med. Cell. Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.C.; Tse, H.W.; Skifter, D.A.; Christie, J.M.; Andaloro, V.J.; Kemp, M.C.; Watkins, J.C.; Jane, D.E.; Monaghan, D.T. [3H]Homoquinolinate Binds to a Subpopulation of NMDA Receptors and to a Novel Binding Site. J. Neurochem. 1998, 71, 1464–1470. [Google Scholar] [CrossRef]
- De Carvalho, L.P.; Bochet, P.; Rossier, J. The Endogenous Agonist Quinolinic Acid and the Non Endogenous Homoquinolinic Acid Discriminate between NMDAR2 Receptor Subunits. Neurochem. Int. 1996, 28, 445–452. [Google Scholar] [CrossRef]
- Schwarcz, R.; Whetsell, W.O.; Mangano, R.M. Quinolinic Acid: An Endogenous Metabolite That Produces Axon-Sparing Lesions in Rat Brain. Science 1983, 219, 316–318. [Google Scholar] [CrossRef]
- Rodríguez-Martínez, E.; Camacho, A.; Maldonado, P.D.; Pedraza-Chaverrí, J.; Santamaría, D.; Galván-Arzate, S.; Santamaría, A. Effect of Quinolinic Acid on Endogenous Antioxidants in Rat Corpus Striatum. Brain Res. 2000, 858, 436–439. [Google Scholar] [CrossRef]
- Iwahashi, H.; Kawamori, H.; Fukushima, K. Quinolinic Acid, Alpha-Picolinic Acid, Fusaric Acid, and 2,6-Pyridinedicarboxylic Acid Enhance the Fenton Reaction in Phosphate Buffer. Chem. Biol. Interact. 1999, 118, 201–215. [Google Scholar] [CrossRef]
- Sahm, F.; Oezen, I.; Opitz, C.A.; Radlwimmer, B.; von Deimling, A.; Ahrendt, T.; Adams, S.; Bode, H.B.; Guillemin, G.J.; Wick, W.; et al. The Endogenous Tryptophan Metabolite and NAD+ Precursor Quinolinic Acid Confers Resistance of Gliomas to Oxidative Stress. Cancer Res. 2013, 73, 3225–3234. [Google Scholar] [CrossRef] [Green Version]
- Santamaría, A.; Galván-Arzate, S.; Lisý, V.; Ali, S.F.; Duhart, H.M.; Osorio-Rico, L.; Ríos, C.; St’astný, F. Quinolinic Acid Induces Oxidative Stress in Rat Brain Synaptosomes. Neuroreport 2001, 12, 871–874. [Google Scholar] [CrossRef]
- Nakagami, Y.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine Toxicity on the Rat Striatum in Vivo. Jpn. J. Pharm. Ther. 1996, 71, 183–186. [Google Scholar] [CrossRef] [Green Version]
- Tronel, C.; Rochefort, G.Y.; Arlicot, N.; Bodard, S.; Chalon, S.; Antier, D. Oxidative Stress Is Related to the Deleterious Effects of Heme Oxygenase-1 in an in Vivo Neuroinflammatory Rat Model. Oxid. Med. Cell. Longev. 2013, 2013, 264935. [Google Scholar] [CrossRef] [Green Version]
- Pérez-De La Cruz, V.; Elinos-Calderón, D.; Carrillo-Mora, P.; Silva-Adaya, D.; Konigsberg, M.; Morán, J.; Ali, S.F.; Chánez-Cárdenas, M.E.; Pérez-De La Cruz, G.; Santamaría, A. Time-Course Correlation of Early Toxic Events in Three Models of Striatal Damage: Modulation by Proteases Inhibition. Neurochem. Int. 2010, 56, 834–842. [Google Scholar] [CrossRef]
- Santamaría, A.; Jiménez-Capdeville, M.E.; Camacho, A.; Rodríguez-Martínez, E.; Flores, A.; Galván-Arzate, S. In Vivo Hydroxyl Radical Formation after Quinolinic Acid Infusion into Rat Corpus Striatum. Neuroreport 2001, 12, 2693–2696. [Google Scholar] [CrossRef]
- Noack, H.; Lindenau, J.; Rothe, F.; Asayama, K.; Wolf, G. Differential Expression of Superoxide Dismutase Isoforms in Neuronal and Glial Compartments in the Course of Excitotoxically Mediated Neurodegeneration: Relation to Oxidative and Nitrergic Stress. Glia 1998, 23, 285–297. [Google Scholar] [CrossRef]
- Ganzella, M.; Jardim, F.M.; Boeck, C.R.; Vendite, D. Time Course of Oxidative Events in the Hippocampus Following Intracerebroventricular Infusion of Quinolinic Acid in Mice. Neurosci. Res. 2006, 55, 397–402. [Google Scholar] [CrossRef]
- Pérez-De La Cruz, V.; Konigsberg, M.; Pedraza-Chaverri, J.; Herrera-Mundo, N.; Díaz-Muñoz, M.; Morán, J.; Fortoul-van der Goes, T.; Rondán-Zárate, A.; Maldonado, P.D.; Ali, S.F.; et al. Cytoplasmic Calcium Mediates Oxidative Damage in an Excitotoxic /Energetic Deficit Synergic Model in Rats. Eur. J. Neurosci. 2008, 27, 1075–1085. [Google Scholar] [CrossRef]
- Mahal, H.S.; Sharma, H.S.; Mukherjee, T. Antioxidant Properties of Melatonin: A Pulse Radiolysis Study. Free Radic. Biol. Med. 1999, 26, 557–565. [Google Scholar] [CrossRef]
- DeNapoli, J.S.; Dodman, N.H.; Shuster, L.; Rand, W.M.; Gross, K.L. Effect of Dietary Protein Content and Tryptophan Supplementation on Dominance Aggression, Territorial Aggression, and Hyperactivity in Dogs. J. Am. Vet. Med. Assoc. 2000, 217, 504–508. [Google Scholar] [CrossRef]
- Ciji, A.; Sahu, N.P.; Pal, A.K.; Akhtar, M.S. Nitrite-Induced Alterations in Sex Steroids and Thyroid Hormones of Labeo Rohita Juveniles: Effects of Dietary Vitamin E and L-Tryptophan. Fish Physiol. Biochem. 2013, 39, 1297–1307. [Google Scholar] [CrossRef]
- Penberthy, W.T. Pharmacological Targeting of IDO-Mediated Tolerance for Treating Autoimmune Disease. Curr. Drug Metab. 2007, 8, 245–266. [Google Scholar] [CrossRef]
- Hiratsuka, C.; Fukuwatari, T.; Sano, M.; Saito, K.; Sasaki, S.; Shibata, K. Supplementing Healthy Women with up to 5.0 g/d of L-Tryptophan Has No Adverse Effects. J. Nutr. 2013, 143, 859–866. [Google Scholar] [CrossRef] [Green Version]
- Hafstad Solvang, S.-E.; Nordrehaug, J.E.; Aarsland, D.; Lange, J.; Ueland, P.M.; McCann, A.; Midttun, Ø.; Tell, G.S.; Giil, L.M. Kynurenines, Neuropsychiatric Symptoms, and Cognitive Prognosis in Patients with Mild Dementia. Int. J. Tryptophan Res. 2019, 12, 1178646919877883. [Google Scholar] [CrossRef] [Green Version]
- Boros, F.A.; Vécsei, L. Immunomodulatory Effects of Genetic Alterations Affecting the Kynurenine Pathway. Front. Immunol. 2019, 10, 2570. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Jellinger, K.; Deecke, L. Kynurenine Metabolism in Alzheimer’s Disease. J. Neural Transm. 1999, 106, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Perkins, M.N.; Stone, T.W. An Iontophoretic Investigation of the Actions of Convulsant Kynurenines and Their Interaction with the Endogenous Excitant Quinolinic Acid. Brain Res. 1982, 247, 184–187. [Google Scholar] [CrossRef]
- Zádori, D.; Klivényi, P.; Plangár, I.; Toldi, J.; Vécsei, L. Endogenous Neuroprotection in Chronic Neurodegenerative Disorders: With Particular Regard to the Kynurenines. J. Cell. Mol. Med. 2011, 15, 701–717. [Google Scholar] [CrossRef]
- Uwai, Y.; Honjo, H.; Iwamoto, K. Interaction and Transport of Kynurenic Acid via Human Organic Anion Transporters HOAT1 and HOAT3. Pharm. Ther. Res. 2012, 65, 254–260. [Google Scholar] [CrossRef]
- Kessler, M.; Terramani, T.; Lynch, G.; Baudry, M. A Glycine Site Associated with N-Methyl-D-Aspartic Acid Receptors: Characterization and Identification of a New Class of Antagonists. J. Neurochem. 1989, 52, 1319–1328. [Google Scholar] [CrossRef]
- Birch, P.J.; Grossman, C.J.; Hayes, A.G. Kynurenate and FG9041 Have Both Competitive and Non-Competitive Antagonist Actions at Excitatory Amino Acid Receptors. Eur. J. Pharm. Ther. 1988, 151, 313–315. [Google Scholar] [CrossRef]
- Prescott, C.; Weeks, A.M.; Staley, K.J.; Partin, K.M. Kynurenic Acid Has a Dual Action on AMPA Receptor Responses. Neurosci. Lett. 2006, 402, 108–112. [Google Scholar] [CrossRef]
- Rózsa, E.; Robotka, H.; Vécsei, L.; Toldi, J. The Janus-Face Kynurenic Acid. J. Neural Transm. 2008, 115, 1087–1091. [Google Scholar] [CrossRef]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The Brain Metabolite Kynurenic Acid Inhibits Alpha7 Nicotinic Receptor Activity and Increases Non-Alpha7 Nicotinic Receptor Expression: Physiopathological Implications. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef]
- Marchi, M.; Risso, F.; Viola, C.; Cavazzani, P.; Raiteri, M. Direct Evidence That Release-Stimulating Alpha7* Nicotinic Cholinergic Receptors Are Localized on Human and Rat Brain Glutamatergic Axon Terminals. J. Neurochem. 2002, 80, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic Acid as a Ligand for Orphan G Protein-Coupled Receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [Green Version]
- Lajkó, N.; Kata, D.; Szabó, M.; Mátyás, A.; Dulka, K.; Földesi, I.; Fülöp, F.; Gulya, K.; Vécsei, L.; Mihály, A. Sensitivity of Rodent Microglia to Kynurenines in Models of Epilepsy and Inflammation In Vivo and In Vitro: Microglia Activation Is Inhibited by Kynurenic Acid and the Synthetic Analogue SZR104. Int. J. Mol. Sci. 2020, 21, 9333. [Google Scholar] [CrossRef]
- Wichers, M.C.; Maes, M. The Role of Indoleamine 2,3-Dioxygenase (IDO) in the Pathophysiology of Interferon-Alpha-Induced Depression. J. Psychiatry Neurosci. Jpn. 2004, 29, 11–17. [Google Scholar]
- Van der Vliet, A.; Bast, A. Effect of Oxidative Stress on Receptors and Signal Transmission. Chem. Biol. Interact. 1992, 85, 95–116. [Google Scholar] [CrossRef]
- Gutteridge, J.M. Lipid Peroxidation and Antioxidants as Biomarkers of Tissue Damage. Clin. Chem. 1995, 41, 1819–1828. [Google Scholar] [CrossRef]
- McIntyre, N. Familial LCAT Deficiency and Fish-Eye Disease. J. Inherit. Metab. Dis. 1988, 11 (Suppl. 1), 45–56. [Google Scholar] [CrossRef]
- Brigitta, B. Pathophysiology of Depression and Mechanisms of Treatment. Dialogues Clin. Neurosci. 2002, 4, 7–20. [Google Scholar]
- Maes, M.; Smith, R.; Christophe, A.; Cosyns, P.; Desnyder, R.; Meltzer, H. Fatty Acid Composition in Major Depression: Decreased Omega 3 Fractions in Cholesteryl Esters and Increased C20: 4 Omega 6/C20:5 Omega 3 Ratio in Cholesteryl Esters and Phospholipids. J. Affect. Disord. 1996, 38, 35–46. [Google Scholar] [CrossRef]
- Maes, M.; Christophe, A.; Delanghe, J.; Altamura, C.; Neels, H.; Meltzer, H.Y. Lowered Omega3 Polyunsaturated Fatty Acids in Serum Phospholipids and Cholesteryl Esters of Depressed Patients. Psychiatry Res. 1999, 85, 275–291. [Google Scholar] [CrossRef]
- Bilici, M.; Efe, H.; Köroğlu, M.A.; Uydu, H.A.; Bekaroğlu, M.; Değer, O. Antioxidative Enzyme Activities and Lipid Peroxidation in Major Depression: Alterations by Antidepressant Treatments. J. Affect. Disord. 2001, 64, 43–51. [Google Scholar] [CrossRef]
- Kincses, Z.T.; Toldi, J.; Vécsei, L. Kynurenines, Neurodegeneration and Alzheimer’s Disease. J. Cell. Mol. Med. 2010, 14, 2045–2054. [Google Scholar] [CrossRef] [Green Version]
- Bonda, D.J.; Mailankot, M.; Stone, J.G.; Garrett, M.R.; Staniszewska, M.; Castellani, R.J.; Siedlak, S.L.; Zhu, X.; Lee, H.; Perry, G.; et al. Indoleamine 2,3-Dioxygenase and 3-Hydroxykynurenine Modifications Are Found in the Neuropathology of Alzheimer’s Disease. Redox Rep. Commun. Free Radic. Res. 2010, 15, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, Metabolic Disturbances, Oxidative Stress and the Kynurenine System, with Focus on Neurodegenerative Disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef] [PubMed]
- Sathyasaikumar, K.V.; Stachowski, E.K.; Amori, L.; Guidetti, P.; Muchowski, P.J.; Schwarcz, R. Dysfunctional Kynurenine Pathway Metabolism in the R6/2 Mouse Model of Huntington’s Disease. J. Neurochem. 2010, 113, 1416–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campesan, S.; Green, E.W.; Breda, C.; Sathyasaikumar, K.V.; Muchowski, P.J.; Schwarcz, R.; Kyriacou, C.P.; Giorgini, F. The Kynurenine Pathway Modulates Neurodegeneration in a Drosophila Model of Huntington’s Disease. Curr. Biol. Cb 2011, 21, 961–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidetti, P.; Reddy, P.H.; Tagle, D.A.; Schwarcz, R. Early Kynurenergic Impairment in Huntington’s Disease and in a Transgenic Animal Model. Neurosci. Lett. 2000, 283, 233–235. [Google Scholar] [CrossRef]
- Guidetti, P.; Bates, G.P.; Graham, R.K.; Hayden, M.R.; Leavitt, B.R.; MacDonald, M.E.; Slow, E.J.; Wheeler, V.C.; Woodman, B.; Schwarcz, R. Elevated Brain 3-Hydroxykynurenine and Quinolinate Levels in Huntington Disease Mice. Neurobiol. Dis. 2006, 23, 190–197. [Google Scholar] [CrossRef]
- Sas, K.; Szabó, E.; Vécsei, L. Mitochondria, Oxidative Stress and the Kynurenine System, with a Focus on Ageing and Neuroprotection. Molecules 2018, 23, 191. [Google Scholar] [CrossRef] [Green Version]
- Pearson, S.J.; Reynolds, G.P. Increased Brain Concentrations of a Neurotoxin, 3-Hydroxykynurenine, in Huntington’s Disease. Neurosci. Lett. 1992, 144, 199–201. [Google Scholar] [CrossRef]
- Ménard, C.; Hodes, G.E.; Russo, S.J. Pathogenesis of Depression: Insights from Human and Rodent Studies. Neuroscience 2016, 321, 138–162. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.; Maes, M.; Berk, M. Biological Underpinnings of the Commonalities in Depression, Somatization, and Chronic Fatigue Syndrome. Med. Hypotheses 2012, 78, 752–756. [Google Scholar] [CrossRef]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vecsei, L. Kynurenine Metabolism in Plasma and in Red Blood Cells in Parkinson’s Disease. J. Neurol. Sci. 2005, 239, 31–35. [Google Scholar] [CrossRef]
- Knyihár-Csillik, E.; Chadaide, Z.; Mihály, A.; Krisztin-Péva, B.; Fenyo, R.; Vécsei, L. Effect of 6-Hydroxydopamine Treatment on Kynurenine Aminotransferase-I (KAT-I) Immunoreactivity of Neurons and Glial Cells in the Rat Substantia Nigra. Acta Neuropathol. 2006, 112, 127–137. [Google Scholar] [CrossRef]
- Silva-Adaya, D.; Pérez-De La Cruz, V.; Villeda-Hernández, J.; Carrillo-Mora, P.; González-Herrera, I.G.; García, E.; Colín-Barenque, L.; Pedraza-Chaverrí, J.; Santamaría, A. Protective Effect of L-Kynurenine and Probenecid on 6-Hydroxydopamine-Induced Striatal Toxicity in Rats: Implications of Modulating Kynurenate as a Protective Strategy. Neurotoxicol. Teratol. 2011, 33, 303–312. [Google Scholar] [CrossRef]
- Zádori, D.; Klivényi, P.; Toldi, J.; Fülöp, F.; Vécsei, L. Kynurenines in Parkinson’s Disease: Therapeutic Perspectives. J. Neural Transm. 2012, 119, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.; Maes, M. Neurodegeneration in Parkinson’s Disease: Interactions of Oxidative Stress, Tryptophan Catabolites and Depression with Mitochondria and Sirtuins. Mol. Neurobiol. 2014, 49, 771–783. [Google Scholar] [CrossRef]
- Sanni, L.A.; Thomas, S.R.; Tattam, B.N.; Moore, D.E.; Chaudhri, G.; Stocker, R.; Hunt, N.H. Dramatic Changes in Oxidative Tryptophan Metabolism along the Kynurenine Pathway in Experimental Cerebral and Noncerebral Malaria. Am. J. Pathol. 1998, 152, 611–619. [Google Scholar]
- Saito, K.; Nowak, T.S.; Markey, S.P.; Heyes, M.P. Mechanism of Delayed Increases in Kynurenine Pathway Metabolism in Damaged Brain Regions Following Transient Cerebral Ischemia. J. Neurochem. 1993, 60, 180–192. [Google Scholar] [CrossRef]
- Kazda, H.; Taylor, N.; Healy, D.; Walker, D. Maternal, Umbilical, and Amniotic Fluid Concentrations of Tryptophan and Kynurenine after Labor or Cesarean Section. Pediatr. Res. 1998, 44, 368–373. [Google Scholar] [CrossRef] [Green Version]
- Orlikov, A.B.; Prakhye, I.B.; Ryzov, I.V. Kynurenine in Blood Plasma and DST in Patients with Endogenous Anxiety and Endogenous Depression. Biol. Psychiatry 1994, 36, 97–102. [Google Scholar] [CrossRef]
- Mangoni, A. The “Kynurenine Shunt” and Depression. Adv. Biochem. Psychopharmacol. 1974, 11, 293–298. [Google Scholar]
- Issa, F.; Kirch, D.G.; Gerhardt, G.A.; Bartko, J.J.; Suddath, R.L.; Freedman, R.; Wyatt, R.J. A Multidimensional Approach to Analysis of Cerebrospinal Fluid Biogenic Amines in Schizophrenia: II. Correlations with Psychopathology. Psychiatry Res. 1994, 52, 251–258. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Croitoru-Lamoury, J.; Dormont, D.; Armati, P.J.; Brew, B.J. Quinolinic Acid Upregulates Chemokine Production and Chemokine Receptor Expression in Astrocytes. Glia 2003, 41, 371–381. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Williams, K.R.; Smith, D.G.; Smythe, G.A.; Croitoru-Lamoury, J.; Brew, B.J. Quinolinic Acid in the Pathogenesis of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2003, 527, 167–176. [Google Scholar] [CrossRef]
- Heyes, M.P.; Saito, K.; Lackner, A.; Wiley, C.A.; Achim, C.L.; Markey, S.P. Sources of the Neurotoxin Quinolinic Acid in the Brain of HIV-1-Infected Patients and Retrovirus-Infected Macaques. FASEB J. 1998, 12, 881–896. [Google Scholar] [CrossRef]
- Chao, C.C.; Hu, S.; Gekker, G.; Lokensgard, J.R.; Heyes, M.P.; Peterson, P.K. U50,488 Protection against HIV-1-Related Neurotoxicity: Involvement of Quinolinic Acid Suppression. Neuropharmacology 2000, 39, 150–160. [Google Scholar] [CrossRef]
- Beal, M.F.; Kowall, N.W.; Ellison, D.W.; Mazurek, M.F.; Swartz, K.J.; Martin, J.B. Replication of the Neurochemical Characteristics of Huntington’s Disease by Quinolinic Acid. Nature 1986, 321, 168–171. [Google Scholar] [CrossRef]
- Colín-González, A.L.; Luna-López, A.; Königsberg, M.; Ali, S.F.; Pedraza-Chaverrí, J.; Santamaría, A. Early Modulation of the Transcription Factor Nrf2 in Rodent Striatal Slices by Quinolinic Acid, a Toxic Metabolite of the Kynurenine Pathway. Neuroscience 2014, 260, 130–139. [Google Scholar] [CrossRef]
- Darlington, L.G.; Mackay, G.M.; Forrest, C.M.; Stoy, N.; George, C.; Stone, T.W. Altered Kynurenine Metabolism Correlates with Infarct Volume in Stroke. Eur. J. Neurosci. 2007, 26, 2211–2221. [Google Scholar] [CrossRef]
- Cozzi, A.; Carpenedo, R.; Moroni, F. Kynurenine Hydroxylase Inhibitors Reduce Ischemic Brain Damage: Studies with (m-Nitrobenzoyl)-Alanine (MNBA) and 3,4-Dimethoxy-[-N-4-(Nitrophenyl)Thiazol-2yl]-Benzenesulfonamide (Ro 61-8048) in Models of Focal or Global Brain Ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 1999, 19, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Clark, C.J.; Mackay, G.M.; Smythe, G.A.; Bustamante, S.; Stone, T.W.; Phillips, R.S. Prolonged Survival of a Murine Model of Cerebral Malaria by Kynurenine Pathway Inhibition. Infect. Immun. 2005, 73, 5249–5251. [Google Scholar] [CrossRef] [Green Version]
- Moroni, F.; Cozzi, A.; Peruginelli, F.; Carpenedo, R.; Pellegrini-Giampietro, D.E. Neuroprotective Effects of Kynurenine-3-Hydroxylase Inhibitors in Models of Brain Ischemia. Adv. Exp. Med. Biol. 1999, 467, 199–206. [Google Scholar] [CrossRef]
- Dairam, A.; Müller, A.C.; Daya, S. Non-Steroidal Anti-Inflammatory Agents, Tolmetin and Sulindac Attenuate Quinolinic Acid (QA)-Induced Oxidative Stress in Primary Hippocampal Neurons and Reduce QA-Induced Spatial Reference Memory Deficits in Male Wistar Rats. Life Sci. 2007, 80, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.S.; Naif, H.; Espinosa, M.; Kapoor, V. IDO Induction in IFN-Gamma Activated Astroglia: A Role in Improving Cell Viability during Oxidative Stress. Redox Rep. Commun. Free Radic. Res. 2000, 5, 101–104. [Google Scholar] [CrossRef]
- Maes, M.; Leonard, B.E.; Myint, A.M.; Kubera, M.; Verkerk, R. The New “5-HT” Hypothesis of Depression: Cell-Mediated Immune Activation Induces Indoleamine 2,3-Dioxygenase, Which Leads to Lower Plasma Tryptophan and an Increased Synthesis of Detrimental Tryptophan Catabolites (TRYCATs), Both of Which Contribute to the Onset of Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 702–721. [Google Scholar] [CrossRef] [PubMed]
- Szabó, C.; Dawson, V.L. Role of Poly(ADP-Ribose) Synthetase in Inflammation and Ischaemia-Reperfusion. Trends Pharm. Ther. Sci. 1998, 19, 287–298. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Hyslop, P.A.; Hinshaw, D.B.; Spragg, R.G.; Sklar, L.A.; Cochrane, C.G. Hydrogen Peroxide-Induced Injury of Cells and Its Prevention by Inhibitors of Poly(ADP-Ribose) Polymerase. Proc. Natl. Acad. Sci. USA 1986, 83, 4908–4912. [Google Scholar] [CrossRef] [Green Version]
- Perera, L.P.; Waldmann, T.A. Activation of Human Monocytes Induces Differential Resistance to Apoptosis with Rapid down Regulation of Caspase-8/FLICE. Proc. Natl. Acad. Sci. USA 1998, 95, 14308–14313. [Google Scholar] [CrossRef] [Green Version]
- Grant, R.; Kapoor, V. Inhibition of Indoleamine 2,3-Dioxygenase Activity in IFN-Gamma Stimulated Astroglioma Cells Decreases Intracellular NAD Levels. Biochem. Pharm. Ther. 2003, 66, 1033–1036. [Google Scholar] [CrossRef]
- Sakurai, K.; Zou, J.-P.; Tschetter, J.R.; Ward, J.M.; Shearer, G.M. Effect of Indoleamine 2,3-Dioxygenase on Induction of Experimental Autoimmune Encephalomyelitis. J. Neuroimmunol. 2002, 129, 186–196. [Google Scholar] [CrossRef]
- Sorgdrager, F.; van Der Ley, C.P.; van Faassen, M.; Calus, E.; Nollen, E.A.; Kema, I.P.; van Dam, D.; De Deyn, P.P. The Effect of Tryptophan 2,3-Dioxygenase Inhibition on Kynurenine Metabolism and Cognitive Function in the APP23 Mouse Model of Alzheimer’s Disease. Int. J. Tryptophan Res. 2020, 13, 1178646920972657. [Google Scholar] [CrossRef]
- Kanai, M.; Funakoshi, H.; Takahashi, H.; Hayakawa, T.; Mizuno, S.; Matsumoto, K.; Nakamura, T. Tryptophan 2,3-Dioxygenase Is a Key Modulator of Physiological Neurogenesis and Anxiety-Related Behavior in Mice. Mol. Brain 2009, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Maddison, D.C.; Alfonso-Núñez, M.; Swaih, A.M.; Breda, C.; Campesan, S.; Allcock, N.; Straatman-Iwanowska, A.; Kyriacou, C.P.; Giorgini, F. A Novel Role for Kynurenine 3-Monooxygenase in Mitochondrial Dynamics. PLoS Genet. 2020, 16, e1009129. [Google Scholar] [CrossRef]
- Ivatt, R.M.; Sanchez-Martinez, A.; Godena, V.K.; Brown, S.; Ziviani, E.; Whitworth, A.J. Genome-Wide RNAi Screen Identifies the Parkinson Disease GWAS Risk Locus SREBF1 as a Regulator of Mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 8494–8499. [Google Scholar] [CrossRef] [Green Version]
- Parrott, J.M.; O’Connor, J.C. Kynurenine 3-Monooxygenase: An Influential Mediator of Neuropathology. Front. Psychiatry 2015, 6, 116. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Kopcho, L.; Ghosh, K.; Witmer, M.; Parker, M.; Gupta, S.; Paul, M.; Krishnamurthy, P.; Laksmaiah, B.; Xie, D.; et al. Development of a RapidFire Mass Spectrometry Assay and a Fluorescence Assay for the Discovery of Kynurenine Aminotransferase II Inhibitors to Treat Central Nervous System Disorders. Anal. Biochem. 2016, 501, 56–65. [Google Scholar] [CrossRef]
- Koshy Cherian, A.; Gritton, H.; Johnson, D.E.; Young, D.; Kozak, R.; Sarter, M. A Systemically-Available Kynurenine Aminotransferase II (KAT II) Inhibitor Restores Nicotine-Evoked Glutamatergic Activity in the Cortex of Rats. Neuropharmacology 2014, 82, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Pocivavsek, A.; Wu, H.-Q.; Potter, M.C.; Elmer, G.I.; Pellicciari, R.; Schwarcz, R. Fluctuations in Endogenous Kynurenic Acid Control Hippocampal Glutamate and Memory. Neuropsychopharmacology 2011, 36, 2357–2367. [Google Scholar] [CrossRef]
- Bortz, D.M.; Wu, H.-Q.; Schwarcz, R.; Bruno, J.P. Oral Administration of a Specific Kynurenic Acid Synthesis (KAT II) Inhibitor Attenuates Evoked Glutamate Release in Rat Prefrontal Cortex. Neuropharmacology 2017, 121, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Yates, J.R.; Heyes, M.P.; Blight, A.R. 4-Chloro-3-Hydroxyanthranilate Reduces Local Quinolinic Acid Synthesis, Improves Functional Recovery, and Preserves White Matter after Spinal Cord Injury. J. Neurotrauma 2006, 23, 866–881. [Google Scholar] [CrossRef]
- Jovanovic, F.; Candido, K.D.; Knezevic, N.N. The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents. Int. J. Mol. Sci. 2020, 21, 6045. [Google Scholar] [CrossRef]
- Röver, S.; Cesura, A.M.; Huguenin, P.; Kettler, R.; Szente, A. Synthesis and Biochemical Evaluation of N-(4-Phenylthiazol-2-Yl)Benzenesulfonamides as High-Affinity Inhibitors of Kynurenine 3-Hydroxylase. J. Med. Chem. 1997, 40, 4378–4385. [Google Scholar] [CrossRef]
- Samadi, P.; Grégoire, L.; Rassoulpour, A.; Guidetti, P.; Izzo, E.; Schwarcz, R.; Bédard, P.J. Effect of Kynurenine 3-Hydroxylase Inhibition on the Dyskinetic and Antiparkinsonian Responses to Levodopa in Parkinsonian Monkeys. Mov. Disord. Off. J. Mov. Disord. Soc. 2005, 20, 792–802. [Google Scholar] [CrossRef]
- Giorgini, F.; Guidetti, P.; Nguyen, Q.; Bennett, S.C.; Muchowski, P.J. A Genomic Screen in Yeast Implicates Kynurenine 3-Monooxygenase as a Therapeutic Target for Huntington Disease. Nat. Genet. 2005, 37, 526–531. [Google Scholar] [CrossRef] [Green Version]
- Richter, A.; Hamann, M. The Kynurenine 3-Hydroxylase Inhibitor Ro 61-8048 Improves Dystonia in a Genetic Model of Paroxysmal Dyskinesia. Eur. J. Pharm. Ther. 2003, 478, 47–52. [Google Scholar] [CrossRef]
- Beaumont, V.; Mrzljak, L.; Dijkman, U.; Freije, R.; Heins, M.; Rassoulpour, A.; Tombaugh, G.; Gelman, S.; Bradaia, A.; Steidl, E.; et al. The Novel KMO Inhibitor CHDI-340246 Leads to a Restoration of Electrophysiological Alterations in Mouse Models of Huntington’s Disease. Exp. Neurol. 2016, 282, 99–118. [Google Scholar] [CrossRef]
- Dounay, A.B.; Tuttle, J.B.; Verhoest, P.R. Challenges and Opportunities in the Discovery of New Therapeutics Targeting the Kynurenine Pathway. J. Med. Chem. 2015, 58, 8762–8782. [Google Scholar] [CrossRef]
- Pellicciari, R.; Amori, L.; Costantino, G.; Giordani, A.; Macchiarulo, A.; Mattoli, L.; Pevarello, P.; Speciale, C.; Varasi, M. Modulation of the Kynurine Pathway of Tryptophan Metabolism in Search for Neuroprotective Agents. Focus on Kynurenine-3-Hydroxylase. Adv. Exp. Med. Biol. 2003, 527, 621–628. [Google Scholar] [CrossRef]
- Toledo-Sherman, L.M.; Prime, M.E.; Mrzljak, L.; Beconi, M.G.; Beresford, A.; Brookfield, F.A.; Brown, C.J.; Cardaun, I.; Courtney, S.M.; Dijkman, U.; et al. Development of a Series of Aryl Pyrimidine Kynurenine Monooxygenase Inhibitors as Potential Therapeutic Agents for the Treatment of Huntington’s Disease. J. Med. Chem. 2015, 58, 1159–1183. [Google Scholar] [CrossRef]
- Zwilling, D.; Huang, S.-Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.-Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-Monooxygenase Inhibition in Blood Ameliorates Neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Reinhart, P.H.; Kelly, J.W. Treating the Periphery to Ameliorate Neurodegenerative Diseases. Cell 2011, 145, 813–814. [Google Scholar] [CrossRef] [Green Version]
- Connick, J.H.; Heywood, G.C.; Sills, G.J.; Thompson, G.G.; Brodie, M.J.; Stone, T.W. Nicotinylalanine Increases Cerebral Kynurenic Acid Content and Has Anticonvulsant Activity. Gen. Pharm. Ther. 1992, 23, 235–239. [Google Scholar] [CrossRef]
- Russi, P.; Alesiani, M.; Lombardi, G.; Davolio, P.; Pellicciari, R.; Moroni, F. Nicotinylalanine Increases the Formation of Kynurenic Acid in the Brain and Antagonizes Convulsions. J. Neurochem. 1992, 59, 2076–2080. [Google Scholar] [CrossRef] [PubMed]
- Moroni, F.; Russi, P.; Gallo-Mezo, M.A.; Moneti, G.; Pellicciari, R. Modulation of Quinolinic and Kynurenic Acid Content in the Rat Brain: Effects of Endotoxins and Nicotinylalanine. J. Neurochem. 1991, 57, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Mora, P.; Méndez-Cuesta, L.A.; Pérez-De La Cruz, V.; Fortoul-van Der Goes, T.I.; Santamaría, A. Protective Effect of Systemic L-Kynurenine and Probenecid Administration on Behavioural and Morphological Alterations Induced by Toxic Soluble Amyloid Beta (25-35) in Rat Hippocampus. Behav. Brain Res. 2010, 210, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Matson, W.R.; Storey, E.; Milbury, P.; Ryan, E.A.; Ogawa, T.; Bird, E.D. Kynurenic Acid Concentrations Are Reduced in Huntington’s Disease Cerebral Cortex. J. Neurol. Sci. 1992, 108, 80–87. [Google Scholar] [CrossRef]
- Jauch, D.; Urbańska, E.M.; Guidetti, P.; Bird, E.D.; Vonsattel, J.P.; Whetsell, W.O.; Schwarcz, R. Dysfunction of Brain Kynurenic Acid Metabolism in Huntington’s Disease: Focus on Kynurenine Aminotransferases. J. Neurol. Sci. 1995, 130, 39–47. [Google Scholar] [CrossRef]
- Vallerini, G.P.; Amori, L.; Beato, C.; Tararina, M.; Wang, X.-D.; Schwarcz, R.; Costantino, G. 2-Aminonicotinic Acid 1-Oxides Are Chemically Stable Inhibitors of Quinolinic Acid Synthesis in the Mammalian Brain: A Step toward New Antiexcitotoxic Agents. J. Med. Chem. 2013, 56, 9482–9495. [Google Scholar] [CrossRef]
- Baran, H.; Staniek, K.; Kepplinger, B.; Stur, J.; Draxler, M.; Nohl, H. Kynurenines and the Respiratory Parameters on Rat Heart Mitochondria. Life Sci. 2003, 72, 1103–1115. [Google Scholar] [CrossRef]
- Ting, K.K.; Brew, B.J.; Guillemin, G.J. Effect of Quinolinic Acid on Gene Expression in Human Astrocytes: Implications for Alzheimer’s Disease. Int. Congr. Ser. 2007, 1304, 384–388. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Kerr, S.J.; Brew, B.J. Involvement of Quinolinic Acid in AIDS Dementia Complex. Neurotox. Res. 2005, 7, 103–123. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Meininger, V.; Brew, B.J. Implications for the Kynurenine Pathway and Quinolinic Acid in Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2005, 2, 166–176. [Google Scholar] [CrossRef]
- Castellano-Gonzalez, G.; Jacobs, K.R.; Don, E.; Cole, N.J.; Adams, S.; Lim, C.K.; Lovejoy, D.B.; Guillemin, G.J. Kynurenine 3-Monooxygenase Activity in Human Primary Neurons and Effect on Cellular Bioenergetics Identifies New Neurotoxic Mechanisms. Neurotox. Res. 2019, 35, 530–541. [Google Scholar] [CrossRef]
- Fülöp, F.; Szatmári, I.; Vámos, E.; Zádori, D.; Toldi, J.; Vécsei, L. Syntheses, Transformations and Pharmaceutical Applications of Kynurenic Acid Derivatives. Curr. Med. Chem. 2009, 16, 4828–4842. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.Q.; Lee, S.C.; Schwarcz, R. Systemic Administration of 4-Chlorokynurenine Prevents Quinolinate Neurotoxicity in the Rat Hippocampus. Eur. J. Pharm. Ther. 2000, 390, 267–274. [Google Scholar] [CrossRef]
- Zádori, D.; Nyiri, G.; Szonyi, A.; Szatmári, I.; Fülöp, F.; Toldi, J.; Freund, T.F.; Vécsei, L.; Klivényi, P. Neuroprotective Effects of a Novel Kynurenic Acid Analogue in a Transgenic Mouse Model of Huntington’s Disease. J. Neural Transm. 2011, 118, 865–875. [Google Scholar] [CrossRef]
- Tiszlavicz, Z.; Németh, B.; Fülöp, F.; Vécsei, L.; Tápai, K.; Ocsovszky, I.; Mándi, Y. Different Inhibitory Effects of Kynurenic Acid and a Novel Kynurenic Acid Analogue on Tumour Necrosis Factor-α (TNF-α) Production by Mononuclear Cells, HMGB1 Production by Monocytes and HNP1-3 Secretion by Neutrophils. Naunyn. Schmiedebergs Arch. Pharm. Ther. 2011, 383, 447–455. [Google Scholar] [CrossRef]
- Gellért, L.; Fuzik, J.; Göblös, A.; Sárközi, K.; Marosi, M.; Kis, Z.; Farkas, T.; Szatmári, I.; Fülöp, F.; Vécsei, L.; et al. Neuroprotection with a New Kynurenic Acid Analog in the Four-Vessel Occlusion Model of Ischemia. Eur. J. Pharm. Ther. 2011, 667, 182–187. [Google Scholar] [CrossRef]
- Gellért, L.; Varga, D.; Ruszka, M.; Toldi, J.; Farkas, T.; Szatmári, I.; Fülöp, F.; Vécsei, L.; Kis, Z. Behavioural Studies with a Newly Developed Neuroprotective KYNA-Amide. J. Neural Transm. 2012, 119, 165–172. [Google Scholar] [CrossRef]
- Kemp, J.A.; Foster, A.C.; Leeson, P.D.; Priestley, T.; Tridgett, R.; Iversen, L.L.; Woodruff, G.N. 7-Chlorokynurenic Acid Is a Selective Antagonist at the Glycine Modulatory Site of the N-Methyl-D-Aspartate Receptor Complex. Proc. Natl. Acad. Sci. USA 1988, 85, 6547–6550. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Sengupta, A.; Haque, N.; Grundke-Iqbal, I.; Iqbal, K. Memantine Inhibits and Reverses the Alzheimer Type Abnormal Hyperphosphorylation of Tau and Associated Neurodegeneration. FEBS Lett. 2004, 566, 261–269. [Google Scholar] [CrossRef]
- Molnár, K.; Lőrinczi, B.; Fazakas, C.; Szatmári, I.; Fülöp, F.; Kmetykó, N.; Berkecz, R.; Ilisz, I.; Krizbai, I.A.; Wilhelm, I.; et al. SZR-104, a Novel Kynurenic Acid Analogue with High Permeability through the Blood-Brain Barrier. Pharmaceutics 2021, 13, 61. [Google Scholar] [CrossRef]
- Tanaka, M.; Szabó, Á.; Lőrinczi, B.; Szatmári, I.; Fülöp, F.; Vécsei, L. Antidepressant-like Effects of Kynurenic Acid Analogues. Pharmacol. Rep. 2021, 22, 1623. [Google Scholar]
- Lukács, M.; Warfvinge, K.; Kruse, L.S.; Tajti, J.; Fülöp, F.; Toldi, J.; Vécsei, L.; Edvinsson, L. KYNA Analogue SZR72 Modifies CFA-Induced Dural Inflammation- Regarding Expression of PERK1/2 and IL-1β in the Rat Trigeminal Ganglion. J. Headache Pain 2016, 17, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mándi, Y.; Endrész, V.; Mosolygó, T.; Burián, K.; Lantos, I.; Fülöp, F.; Szatmári, I.; Lőrinczi, B.; Balog, A.; Vécsei, L. The Opposite Effects of Kynurenic Acid and Different Kynurenic Acid Analogs on Tumor Necrosis Factor-α (TNF-α) Production and Tumor Necrosis Factor-Stimulated Gene-6 (TSG-6) Expression. Front. Immunol. 2019, 10, 1406. [Google Scholar] [CrossRef] [PubMed]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.-H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR Restricts a Treg-Macrophage Suppressive Axis Induced by L-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef]
- García-Lara, L.; Pérez-Severiano, F.; González-Esquivel, D.; Elizondo, G.; Segovia, J. Absence of Aryl Hydrocarbon Receptors Increases Endogenous Kynurenic Acid Levels and Protects Mouse Brain against Excitotoxic Insult and Oxidative Stress. J. Neurosci. Res. 2015, 93, 1423–1433. [Google Scholar] [CrossRef]
- Tanaka, M.; Török, N.; Vécsei, L. Are 5-HT1 Receptor Agonists Effective Anti-Migraine Drugs? Expert Opin. Pharm. Ther. 2021, 1–5. [Google Scholar] [CrossRef]
- Grégoire, L.; Rassoulpour, A.; Guidetti, P.; Samadi, P.; Bédard, P.J.; Izzo, E.; Schwarcz, R.; Di Paolo, T. Prolonged Kynurenine 3-Hydroxylase Inhibition Reduces Development of Levodopa-Induced Dyskinesias in Parkinsonian Monkeys. Behav. Brain Res. 2008, 186, 161–167. [Google Scholar] [CrossRef]
- Ceresoli-Borroni, G.; Guidetti, P.; Amori, L.; Pellicciari, R.; Schwarcz, R. Perinatal Kynurenine 3-Hydroxylase Inhibition in Rodents: Pathophysiological Implications. J. Neurosci. Res. 2007, 85, 845–854. [Google Scholar] [CrossRef]
- Giorgini, F.; Huang, S.-Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Thomas, M.A.R.; Tararina, M.; Wu, H.-Q.; Schwarcz, R.; Muchowski, P.J. Targeted Deletion of Kynurenine 3-Monooxygenase in Mice: A New Tool for Studying Kynurenine Pathway Metabolism in Periphery and Brain. J. Biol. Chem. 2013, 288, 36554–36566. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Ayala, T.; Sathyasaikumar, K.V.; Uys, J.D.; Pérez-de-la-Cruz, V.; Pidugu, L.S.; Schwarcz, R. N-Acetylcysteine Inhibits Kynurenine Aminotransferase II. Neuroscience 2020, 444, 160–169. [Google Scholar] [CrossRef]
Figure 1.
The main pathways of tryptophan (TRP) metabolism.

Figure 2.
The course of the kynurenine pathway in various populations of the central nervous system cells. 3-HAA: 3-hydroxyanthranilic acid, 3-HAO: 3-hydroxyanthranilic acid 3,4-dioxygenase, 3-HKYN: 3-hydorxykynurenine, AA: anthranilic acid, AMO: aminocarboxymuconatesemialdehyde decarboxylase, IDO: indoleamine 2,3-dioxygenase, KAT: kynurenine aminotransferase, KMO: kynurenine 3-monooxygenase, KYN: kynurenine, KYNA: kynurenic acid, KYNU: kynureninase, LAT-1: large neutral amino acid transporter 1, NAD+: oxidized form of nicotinamide adenine dinucleotide, OATs: organic anion transporters, PA: picolinic acid, QA: quinolinic acid, TDO: tryptophan 2,3-dioxygenase, TRP: tryptophan, XA: xanthurenic acid.
Figure 2.
The course of the kynurenine pathway in various populations of the central nervous system cells. 3-HAA: 3-hydroxyanthranilic acid, 3-HAO: 3-hydroxyanthranilic acid 3,4-dioxygenase, 3-HKYN: 3-hydorxykynurenine, AA: anthranilic acid, AMO: aminocarboxymuconatesemialdehyde decarboxylase, IDO: indoleamine 2,3-dioxygenase, KAT: kynurenine aminotransferase, KMO: kynurenine 3-monooxygenase, KYN: kynurenine, KYNA: kynurenic acid, KYNU: kynureninase, LAT-1: large neutral amino acid transporter 1, NAD+: oxidized form of nicotinamide adenine dinucleotide, OATs: organic anion transporters, PA: picolinic acid, QA: quinolinic acid, TDO: tryptophan 2,3-dioxygenase, TRP: tryptophan, XA: xanthurenic acid.

Figure 3.
The involvement of 3-hydroxykynurenine, 3-hydroxyanthranilic acid, and quinolinic acid in the development of oxidative damages in the central nervous system cells. 3-HAA: 3-hydroxyanthranilic acid 3,4-dioxygenase, 3-HKYN: 3-hydroxykynurenine, acetyl-CoA: acetyl coenzyme A; Ca2+: calcium, NAD+: oxidized form of nicotinamide adenine dinucleotide, NADH: reduced form of nicotinamide adenine dinucleotide, NMDA: N-methyl-D-aspartate, nNOS: neuronal nitric oxide synthases, NO: nitric oxide, O2•−: superoxide, OONO−: peroxynitrite, QA: quinolinic acid, ROS: reactive oxygen species, TCA cycle: tricarboxylic acid cycle, Ψm: the mitochondrial membrane potential.
Figure 3.
The involvement of 3-hydroxykynurenine, 3-hydroxyanthranilic acid, and quinolinic acid in the development of oxidative damages in the central nervous system cells. 3-HAA: 3-hydroxyanthranilic acid 3,4-dioxygenase, 3-HKYN: 3-hydroxykynurenine, acetyl-CoA: acetyl coenzyme A; Ca2+: calcium, NAD+: oxidized form of nicotinamide adenine dinucleotide, NADH: reduced form of nicotinamide adenine dinucleotide, NMDA: N-methyl-D-aspartate, nNOS: neuronal nitric oxide synthases, NO: nitric oxide, O2•−: superoxide, OONO−: peroxynitrite, QA: quinolinic acid, ROS: reactive oxygen species, TCA cycle: tricarboxylic acid cycle, Ψm: the mitochondrial membrane potential.


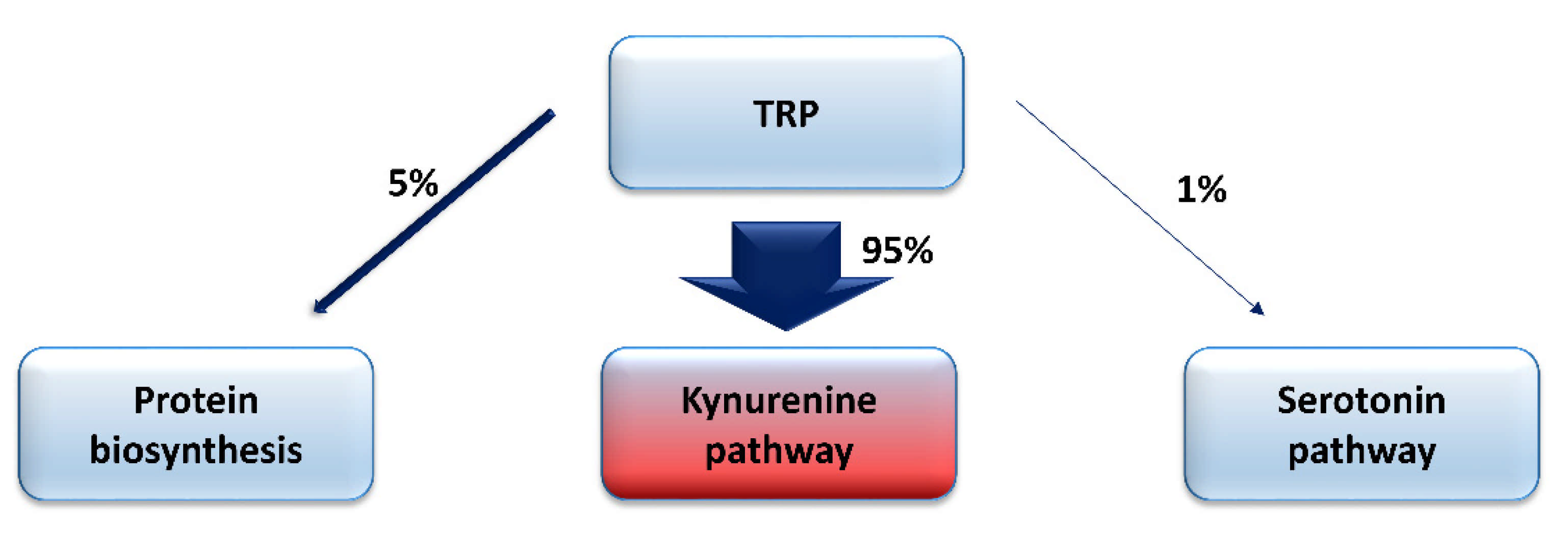{kind=link}
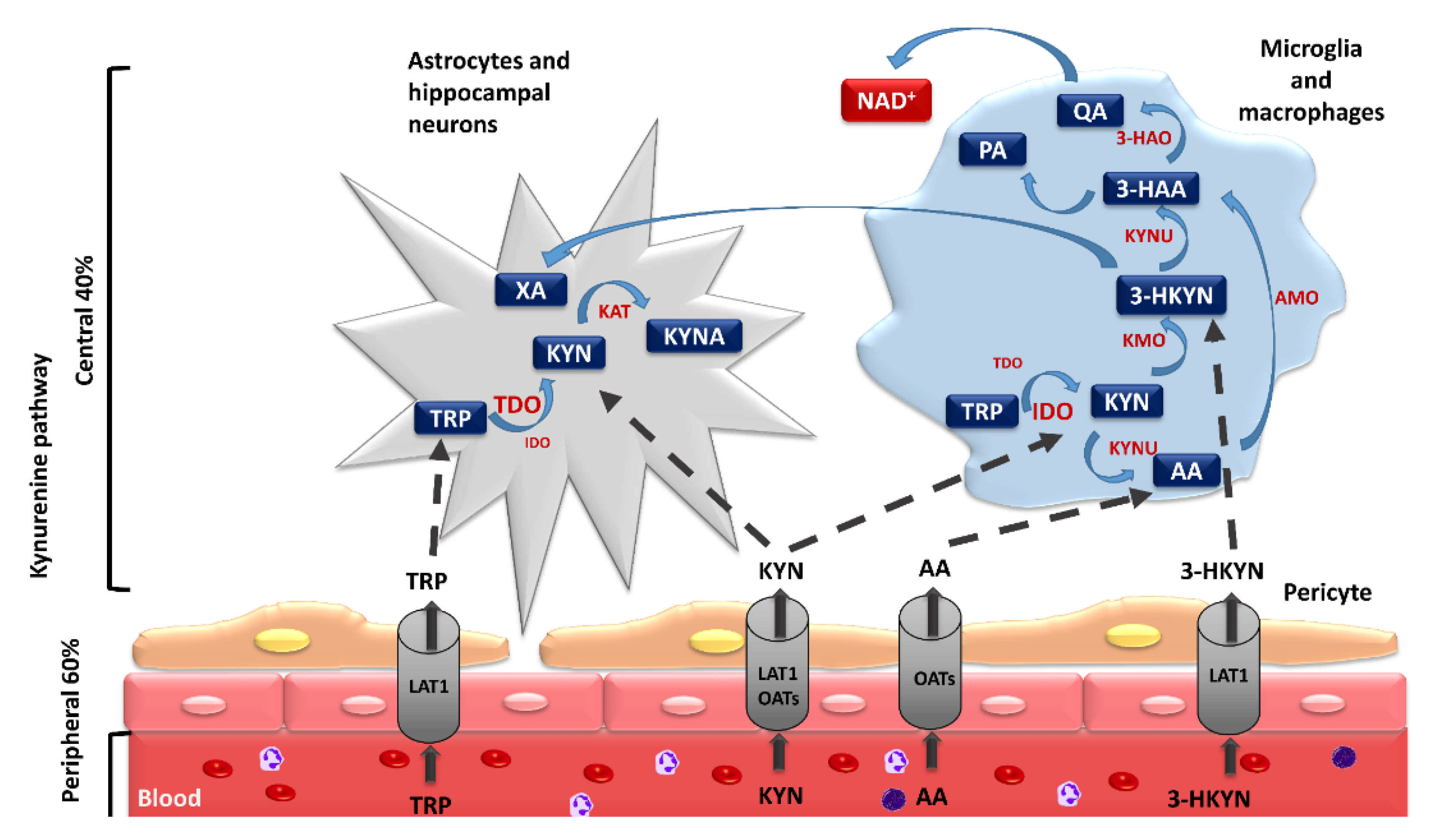{kind=link}
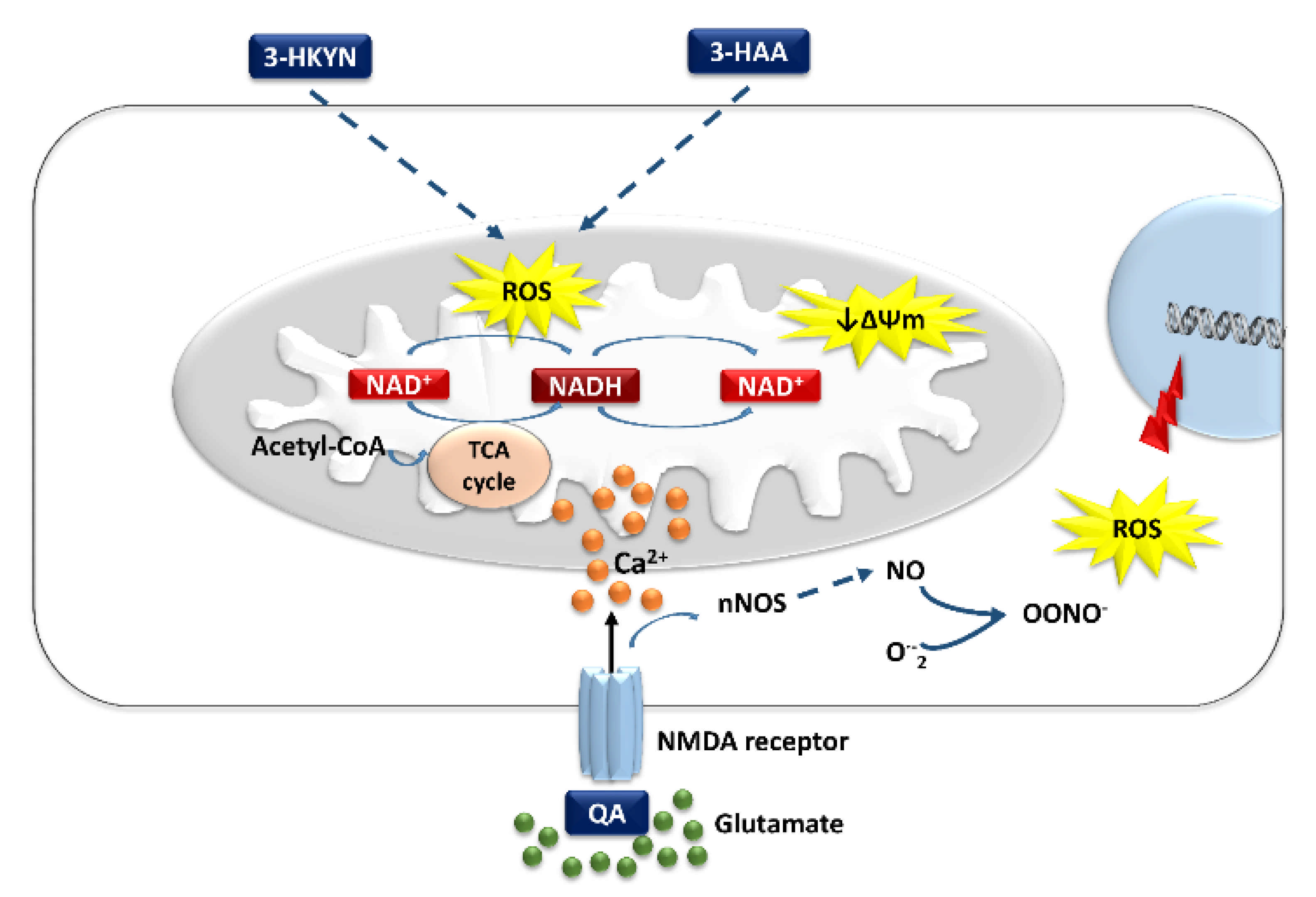{kind=link}
Table 1.
Impact of kynurenine pathway metabolites on oxidative stress during the development of neurodegenerative processes.
Table 1.
Impact of kynurenine pathway metabolites on oxidative stress during the development of neurodegenerative processes.
| Metabolite | Role | References | |
|---|---|---|---|
| In Vitro | In Vivo | ||
| Tryptophan (TRP) | - Possesses redox properties - Reacts readily with hydroxyl radical and singlet oxygen to form multiple oxidation products - Induces intracellular oxidized form of NAD (NAD+) synthesis | - Induces oxidative stress, an imbalance between free radical content and the capacity of the scavenging systems, in the cerebral cortex of the rats | [83,84,85,86,87,88,89] |
| |||
| Kynurenine (KYN) | - The neuroprotective effect presented by KYN is at least partially dependent on its scavenging activity - Decreases ROS production lipid peroxidation in rat brain homogenates below basal levels - Reacts with hydroxyl radical and peroxynitrite, generating kynurenic acid in these reactions - Can scavenge hydrogen peroxide and superoxide, and in effect reduce oxidative damage - Its excess induces apoptosis through ROS generation, since its co-incubation with an antioxidant blocks cytochrome c release and caspase 3 activation downregulating this apoptosis pathway | - Reacts with hydroxyl radical and peroxynitrite, generating kynurenic acid in these reactions | [11,12,13,90,91,92,93,94,95] |
| |||
| Kynurenic acid (KYNA) | - Can scavenge ROS in an N-methyl-D-aspartate receptor- and nicotinic receptor-independent way - In the concentration above 100 µM, it can abolish ROS production induced by iron(II) sulfate - Its excess can have detrimental effects on the function of the CNS cells | [12,13,96,97,98] | |
| |||
| 3-Hydroxykynurenine (3-HKYN) | - Interacts with cellular xanthine oxidase, can generate superoxide radicals, hydrogen peroxide, and hydroxyl radical in amounts capable of inducing internucleosomal DNA damage - Participates in latter phases of cellular ROS production in response to inflammation; the early ROS production is not related to the mitochondrial alteration induced by 3-HKYN - Can decrease lipid peroxidation and reduced GSH oxidation in brain cortex homogenates - Promotes NAD+ synthesis at concentrations below 100 nM - In concentrations above 100 nM, it causes a decrease in intracellular NAD+ level and an increase in extracellular lactate dehydrogenase (LDH) activity - Exerts toxic effects through mechanisms that include impairment of cellular energy metabolism, which are not related to early ROS production - Acts with pro-oxidant actions at low (5–20 nM) concentrations, causes cell death in primary neuronal cultures prepared from rat striatum by the generation of hydrogen peroxide and hydroxyl radical, and can work as an antioxidant in higher (100 nM) concentrations and scavenge hydroxyl radicals and peroxynitrite | - Stimulates synthesis and activation of the Nrf2, the transcription factor, causing a partial increase in cell resistance against oxidative stress - Decreases mitochondrial membrane potential both in vitro in rat cultured cortical astrocytes and in vivo after intrastriatal injection into Wistar rats - Induces glutathione GST and SOD activities in rat brains | [58,99,100,101,102,103,104,105,106] |
| |||
| 3-hydroxyanthranilic acid (3-HAA) | - Can scavenge hydroxyl radical and peroxynitrite - Can form complexes with iron(II), inhibiting its autoxidation, reducing ROS formation - Decreases mitochondrial membrane potential in vitro in rat cortical astrocyte cultures - Exerts toxic effects through mechanisms that include impairment of cellular energy metabolism, which are not related to early ROS production - Can decrease lipid peroxidation and reduced GS oxidation in brain cortex homogenates - Is prone to self-oxidation - Inhibits the mitochondrial respiratory chain reactions | - Decreases mitochondrial membrane potential in vivo after intrastriatal injection into Wistar rats - Induces glutathione GST and SOD activities, generating superoxide radicals, H2O2, and cinnabarinic acid | [11,103,107,108,109] |
| |||
| Anthranilic acid (AA) Xanthurenic acid (XA) | - Can form complexes with iron inhibiting its autoxidation - Blocks citric acid cycle and the respiratory chain complexes I–III, interfering with mitochondrial function - Has an anti-inflammatory effect by forming a complex with copper and acting as a hydroxyl radical inactivating ligand | [109,110,111,112] | |
| |||
| - Demonstrated both pro- and antioxidative properties | [113,114] | ||
| |||
| Picolinic acid (PIC) | - has non-selective metal ion chelating and neuroprotective abilities | [115] | |
| |||
| Quinolinic acid (QA) | - Enhances Ca2+-induced ROS formation - Increases lipid and protein peroxidation - Forms complexes with iron, which induce the formation of hydroxyl radicals - Enhances ROS synthesis, inducing nitric oxide synthases and intracellular poly(ADP-ribose) polymerase activity and extracellular LDH activation | - Stimulates synthesis and activation of the Nrf2, the transcription factor, and causes a partial increase in cell resistance against oxidative stress - Can modify the synthesis and activity of certain endogenous antioxidants in the brain, such as reduced GS and SOD - Increases the severity of the lipid and protein peroxidation induced by 3-nitropropionic acid, and probably other pro-oxidants, dependent on the cellular Ca2+ level - Can form complexes with iron(II), which enhance the formation of hydroxyl radicals - Can generate toxic peroxide radicals and peroxynitrite, both after intrastratial and intrahippocampal injection | [11,93,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131] |
| |||
| Nicotinamide adenine dinucleotide (NAD) | - Works as an electron carrier and cofactor in some redox reactions - Protects cells against energy depletion | [61,62,63,64] | |
| |||
Table 2.
Impact of enzymes of the kynurenine pathway on oxidative stress during the development of neurodegenerative processes.
Table 2.
Impact of enzymes of the kynurenine pathway on oxidative stress during the development of neurodegenerative processes.
| Enzyme | Role | References | |
|---|---|---|---|
| In Vitro | In Vivo | ||
| Tryptophan 2,3-dioxygenase (TDO, EC 1.13.11.11) | - Is necessary to maintain proper NAD+ intracellular level | - Can increase inflammation-associated oxidative stress level, leading to increase in depression, anxiety-related behavior, AD, and HD symptoms severity | [11,68,151,163,199,200] |
| Indoleamine 2,3-dioxygenase (IDO, EC 1.13.11.17 | - Can utilize superoxide anion radical as both a substrate and a co-factor - Is necessary to maintain proper NAD+ intracellular level - Its activation increases cell viability during inflammation and decreases in astrocytes death ratio after exposure to hydrogen peroxide | [11,160,192,194,195,196,197,198] | |
| Kynurenine 3-monooxygenase (KMO, EC 1.14.13.9) | - In inflammatory conditions its amount increases in mitochondrial mass - Its moderate activation increases NAD+ synthesis and promotes favorable bioenergetics processes - Its prolonged induction decreases the mitochondrial spare-respiratory capacity and increases ROS production - Its chronic overactivity leads to energy stores depletion and induces cell damage - Its functional interactions with PINK1, PRKN, and DRP1 implicate KMO in mechanisms associated with mitochondrial dysfunction in neurodegeneration, such as defects in mitochondrial morphology and mitophagy | - In a Drosophila S2R+ cell genome-wide RNAi screen, the KMO homologue cinnabar was identified as a modulator of mitochondrial morphology and the recruitment of the familial Parkinsonism-related protein Parkin to depolarized mitochondria | [160,163,201,202,203] |
| Kynurenine aminotransferases (KAT, EC 2.6.1.7) | - Its inhibition, although it decreases brain level of antioxidative KYNA, may enhance cognitive abilities | [73,204,205,206,207] | |
| 3-hydroxyanthranilic acid 3,4-dioxygenase (3-HAO, EC 1.13.1.5) | - Its inhibition leads to the decrease in QA synthesis and alleviates functional deficits in the experimental model of spinal cord injury | [208] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mor, A.; Tankiewicz-Kwedlo, A.; Krupa, A.; Pawlak, D. Role of Kynurenine Pathway in Oxidative Stress during Neurodegenerative Disorders. Cells 2021, 10, 1603. https://doi.org/10.3390/cells10071603
AMA Style
Mor A, Tankiewicz-Kwedlo A, Krupa A, Pawlak D. Role of Kynurenine Pathway in Oxidative Stress during Neurodegenerative Disorders. Cells. 2021; 10(7):1603. https://doi.org/10.3390/cells10071603
Chicago/Turabian StyleMor, Adrian, Anna Tankiewicz-Kwedlo, Anna Krupa, and Dariusz Pawlak. 2021. "Role of Kynurenine Pathway in Oxidative Stress during Neurodegenerative Disorders" Cells 10, no. 7: 1603. https://doi.org/10.3390/cells10071603
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.