
Differential Involvement of ACKR3 C-Tail in β-Arrestin Recruitment, Trafficking and Internalization

 ,
,  ,
,  , ,
, ,
Abstract
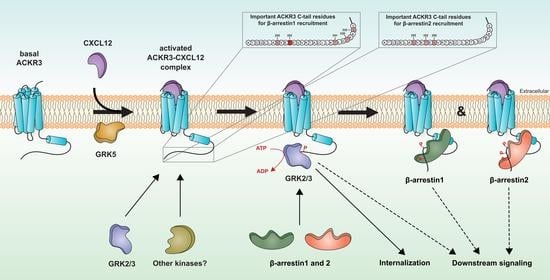
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Generation of ACKR3 Mutants
2.4. Bioluminescence Resonance Energy Transfer (BRET)-Based Recruitment Assay
2.5. Diffusion-Enhanced Resonance Energy Transfer (DERET)-Based Internalization Assay
2.6. Western Blot
3. Results
3.1. ACKR3 Recruitment of β-Arrestins
3.2. ACKR3 Recruitment of GRKs
3.3. ACKR3 Internalization upon Activation
3.4. Involvement C-Tail Phosphorylation Sites ACKR3 in Differential Recruitment of β-Arrestins
3.5. Involvement C-Tail Phosphorylation Sites ACKR3 in Internalization
3.6. Involvement β-Arrestins in ACKR3 Internalization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vacchini, A.; Locati, M.; Borroni, E.M. Overview and potential unifying themes of the atypical chemokine receptor family. J. Leukoc. Biol. 2016, 99, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulvmar, M.H.; Hub, E.; Rot, A. Atypical chemokine receptors. Exp. Cell Res. 2011, 317, 556–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, M.J.; Schlecht-Louf, G.; Neves, M.; van den Bor, J.; Penela, P.; Siderius, M.; Bachelerie, F.; Mayor, F., Jr. The CXCL12/CXCR4/ACKR3 axis in the tumor microenvironment: Signaling, crosstalk, and therapeutic targeting. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 541–563. [Google Scholar] [CrossRef]
- Gencer, S.; van der Vorst, E.P.C.; Aslani, M.; Weber, C.; Doring, Y.; Duchene, J. Atypical Chemokine Receptors in Cardiovascular Disease. Thromb. Haemost. 2019, 119, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Bonecchi, R.; Graham, G.J. Atypical chemokine receptors and their roles in the resolution of the inflammatory response. Front. Immunol. 2016, 7, 224. [Google Scholar] [CrossRef] [Green Version]
- Nibbs, R.J.; Graham, G.J. Immune regulation by atypical chemokine receptors. Nat. Rev. Immunol. 2013, 13, 815–829. [Google Scholar] [CrossRef]
- Neves, M.; Fumagalli, A.; van den Bor, J.; Marin, P.; Smit, M.J.; Mayor, F. The role of ACKR3 in breast, lung, and brain cancer. Mol. Pharmacol. 2019, 96, 819–825. [Google Scholar] [CrossRef]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Balabanian, K.; Lagane, B.; Infantino, S.; Chow, K.Y.; Harriague, J.; Moepps, B.; Arenzana-Seisdedos, F.; Thelen, M.; Bachelerie, F. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J. Biol. Chem. 2005, 280, 35760–35766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saaber, F.; Schutz, D.; Miess, E.; Abe, P.; Desikan, S.; Ashok Kumar, P.; Balk, S.; Huang, K.; Beaulieu, J.M.; Schulz, S.; et al. ACKR3 regulation of neuronal migration requires ACKR3 phosphorylation, but not beta-arrestin. Cell Rep. 2019, 26, 1473–1488.e1479. [Google Scholar] [CrossRef] [Green Version]
- Boldajipour, B.; Mahabaleshwar, H.; Kardash, E.; Reichman-Fried, M.; Blaser, H.; Minina, S.; Wilson, D.; Xu, Q.; Raz, E. Control of chemokine-guided cell migration by ligand sequestration. Cell 2008, 132, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chen, W.; Shen, J. CXCR7 targeting and its major disease relevance. Front. Pharmacol. 2018, 9, 641. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, A.; Heuninck, J.; Pizzoccaro, A.; Moutin, E.; Koenen, J.; Seveno, M.; Durroux, T.; Junier, M.P.; Schlecht-Louf, G.; Bachelerie, F.; et al. The atypical chemokine receptor 3 interacts with Connexin 43 inhibiting astrocytic gap junctional intercellular communication. Nat. Commun. 2020, 11, 4855. [Google Scholar] [CrossRef]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Kang, D.S.; Benovic, J.L. beta-arrestins and G protein-coupled receptor trafficking. Handb. Exp. Pharmacol. 2014, 219, 173–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Koppen, C.J.; Jakobs, K.H. Arrestin-independent internalization of G protein-coupled receptors. Mol. Pharmacol. 2004, 66, 365–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiess, K.; Bagger, S.O.; Torz, L.J.; Jensen, K.H.R.; Walser, A.L.; Kvam, J.M.; Mogelmose, A.K.; Daugvilaite, V.; Junnila, R.K.; Hjorto, G.M.; et al. Arrestin-independent constitutive endocytosis of GPR125/ADGRA3. Ann. N. Y. Acad. Sci. 2019, 1456, 186–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Gastel, J.; Hendrickx, J.O.; Leysen, H.; Santos-Otte, P.; Luttrell, L.M.; Martin, B.; Maudsley, S. beta-arrestin based receptor signaling paradigms: Potential therapeutic targets for complex age-related disorders. Front. Pharmacol. 2018, 9, 1369. [Google Scholar] [CrossRef]
- Canals, M.; Scholten, D.J.; de Munnik, S.; Han, M.K.; Smit, M.J.; Leurs, R. Ubiquitination of CXCR7 controls receptor trafficking. PLoS ONE 2012, 7, e34192. [Google Scholar] [CrossRef] [Green Version]
- Luker, K.E.; Steele, J.M.; Mihalko, L.A.; Ray, P.; Luker, G.D. Constitutive and chemokine-dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene 2010, 29, 4599–4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montpas, N.; St-Onge, G.; Nama, N.; Rhainds, D.; Benredjem, B.; Girard, M.; Hickson, G.; Pons, V.; Heveker, N. Ligand-specific conformational transitions and intracellular transport are required for atypical chemokine receptor 3-mediated chemokine scavenging. J. Biol. Chem. 2018, 293, 893–905. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, V.V.; Gurevich, E.V. GPCR signaling regulation: The role of GRKs and arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of phosphorylation codes for arrestin recruitment by G protein-coupled receptors. Cell 2017, 170, 457–469.e413. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, F.; Muller, W.; Schutz, D.; Penfold, M.E.; Wong, Y.H.; Schulz, S.; Stumm, R. Rapid uptake and degradation of CXCL12 depend on CXCR7 carboxyl-terminal serine/threonine residues. J. Biol. Chem. 2012, 287, 28362–28377. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, A.; Zarca, A.; Neves, M.; Caspar, B.; Hill, S.J.; Mayor, F., Jr.; Smit, M.J.; Marin, P. CXCR4/ACKR3 phosphorylation and recruitment of interacting proteins: Key mechanisms regulating their functional status. Mol. Pharmacol. 2019, 96, 794–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liggett, S.B. Phosphorylation barcoding as a mechanism of directing GPCR signaling. Sci. Signal. 2011, 4, pe36. [Google Scholar] [CrossRef]
- Bahouth, S.W.; Nooh, M.M. Barcoding of GPCR trafficking and signaling through the various trafficking roadmaps by compartmentalized signaling networks. Cell Signal. 2017, 36, 42–55. [Google Scholar] [CrossRef]
- Klammer, M.; Kaminski, M.; Zedler, A.; Oppermann, F.; Blencke, S.; Marx, S.; Muller, S.; Tebbe, A.; Godl, K.; Schaab, C. Phosphosignature predicts dasatinib response in non-small cell lung cancer. Mol. Cell. Proteom. 2012, 11, 651–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Haar Petersen, M.; Ibanez-Vea, M.; Lassen, P.S.; Larsen, M.R.; Palmisano, G. simultaneous enrichment of cysteine-containing peptides and phosphopeptides using a cysteine-specific phosphonate adaptable tag (CysPAT) in combination with titanium dioxide (TiO2) chromatography. Mol. Cell Proteom. 2016, 15, 3282–3296. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ren, Z.; Kang, X.; Zhang, L.; Li, X.; Wang, Y.; Xue, T.; Shen, Y.; Liu, Y. Identification of tyrosine-phosphorylated proteins associated with metastasis and functional analysis of FER in human hepatocellular carcinoma cells. BMC Cancer 2009, 9, 366. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Curto, E.; Inoue, A.; Jenkins, L.; Raihan, S.Z.; Prihandoko, R.; Tobin, A.B.; Milligan, G. Targeted elimination of G proteins and arrestins defines their specific contributions to both intensity and duration of G protein-coupled receptor signaling. J. Biol. Chem. 2016, 291, 27147–27159. [Google Scholar] [CrossRef] [Green Version]
- Milligan, G.; Inoue, A. Genome editing provides new insights into receptor-controlled signalling pathways. Trends. Pharmacol. Sci. 2018, 39, 481–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adlere, I.; Sun, S.; Zarca, A.; Roumen, L.; Gozelle, M.; Viciano, C.P.; Caspar, B.; Arimont, M.; Bebelman, J.P.; Briddon, S.J.; et al. Structure-based exploration and pharmacological evaluation of N-substituted piperidin-4-yl-methanamine CXCR4 chemokine receptor antagonists. Eur. J. Med. Chem. 2019, 162, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Verweij, E.W.E.; Al Araaj, B.; Prabhata, W.R.; Prihandoko, R.; Nijmeijer, S.; Tobin, A.B.; Leurs, R.; Vischer, H.F. Differential role of serines and threonines in intracellular loop 3 and C-terminal tail of the histamine H4 receptor in beta-arrestin and G protein-coupled receptor kinase interaction, internalization, and signaling. ACS Pharmacol. Transl. Sci. 2020, 3, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Lan, T.H.; Kuravi, S.; Lambert, N.A. Internalization dissociates beta2-adrenergic receptors. PLoS ONE 2011, 6, e17361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, T.H.; Liu, Q.; Li, C.; Wu, G.; Lambert, N.A. Sensitive and high resolution localization and tracking of membrane proteins in live cells with BRET. Traffic 2012, 13, 1450–1456. [Google Scholar] [CrossRef] [Green Version]
- de Wit, R.H.; Mujic-Delic, A.; van Senten, J.R.; Fraile-Ramos, A.; Siderius, M.; Smit, M.J. Human cytomegalovirus encoded chemokine receptor US28 activates the HIF-1alpha/PKM2 axis in glioblastoma cells. Oncotarget 2016, 7, 67966–67985. [Google Scholar] [CrossRef] [Green Version]
- Pfleger, K.D.; Eidne, K.A. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 2006, 3, 165–174. [Google Scholar] [CrossRef]
- Luker, K.E.; Gupta, M.; Steele, J.M.; Foerster, B.R.; Luker, G.D. Imaging ligand-dependent activation of CXCR7. Neoplasia 2009, 11, 1022–1035. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Jiang, X.; Shen, K.; Fischer, C.C.; Wedegaertner, P.B. The regulator of G protein signaling (RGS) domain of G protein-coupled receptor kinase 5 (GRK5) regulates plasma membrane localization and function. Mol. Biol. Cell. 2014, 25, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.M.; Yeow, R.Y.; Schoenau, C.; Huber, J.; Tesmer, J.J. Molecular mechanism of selectivity among G protein-coupled receptor kinase 2 inhibitors. Mol. Pharmacol. 2011, 80, 294–303. [Google Scholar] [CrossRef] [Green Version]
- Schmick, M.; Vartak, N.; Papke, B.; Kovacevic, M.; Truxius, D.C.; Rossmannek, L.; Bastiaens, P.I.H. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 2014, 157, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, E.; Severin, F.; Backer, J.M.; Hyman, A.A.; Zerial, M. Rab5 regulates motility of early endosomes on microtubules. Nat. Cell Biol. 1999, 1, 376–382. [Google Scholar] [CrossRef]
- Lebrand, C.; Corti, M.; Goodson, H.; Cosson, P.; Cavalli, V.; Mayran, N.; Faure, J.; Gruenberg, J. Late endosome motility depends on lipids via the small GTPase Rab7. EMBO J. 2002, 21, 1289–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, J.G.; Stow, J.L. Rab11 in recycling endosomes regulates the sorting and basolateral transport of E-cadherin. Mol. Biol. Cell 2005, 16, 1744–1755. [Google Scholar] [CrossRef]
- Tjelle, T.E.; Brech, A.; Juvet, L.K.; Griffiths, G.; Berg, T. Isolation and characterization of early endosomes, late endosomes and terminal lysosomes: Their role in protein degradation. J. Cell Sci. 1996, 109, 2905–2914. [Google Scholar]
- Brown, N.E.; Blumer, J.B.; Hepler, J.R. Bioluminescence resonance energy transfer to detect protein-protein interactions in live cells. Methods Mol. Biol. 2015, 1278, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Levoye, A.; Zwier, J.M.; Jaracz-Ros, A.; Klipfel, L.; Cottet, M.; Maurel, D.; Bdioui, S.; Balabanian, K.; Prezeau, L.; Trinquet, E.; et al. A broad G protein-coupled receptor internalization assay that combines SNAP-Tag labeling, diffusion-enhanced resonance energy transfer, and a highly emissive terbium cryptate. Front. Endocrinol. (Lausanne) 2015, 6, 167. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12 / CXCR4 / CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabel, B.A.; Wang, Y.; Lewen, S.; Berahovich, R.D.; Penfold, M.E.; Zhang, P.; Powers, J.; Summers, B.C.; Miao, Z.; Zhao, B.; et al. Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J. Immunol. 2009, 183, 3204–3211. [Google Scholar] [CrossRef] [Green Version]
- Coggins, N.L.; Trakimas, D.; Chang, S.L.; Ehrlich, A.; Ray, P.; Luker, K.E.; Linderman, J.J.; Luker, G.D. CXCR7 controls competition for recruitment of beta-arrestin 2 in cells expressing both CXCR4 and CXCR7. PLoS ONE 2014, 9, e98328. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Reyes-Alcaraz, A.; Yong, H.J.; Nguyen, L.P.; Park, H.K.; Inoue, A.; Lee, C.S.; Seong, J.Y.; Hwang, J.I. CXCR7: A beta-arrestin-biased receptor that potentiates cell migration and recruits beta-arrestin2 exclusively through Gbetagamma subunits and GRK2. Cell Biosci. 2020, 10, 134. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Wang, J.; Plouffe, B.; Smith, J.S.; Yamani, L.; Kaur, S.; Jean-Charles, P.Y.; Gauthier, C.; Lee, M.H.; Pani, B.; et al. Manifold roles of beta-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiina, T.; Arai, K.; Tanabe, S.; Yoshida, N.; Haga, T.; Nagao, T.; Kurose, H. Clathrin box in G protein-coupled receptor kinase 2. J. Biol. Chem. 2001, 276, 33019–33026. [Google Scholar] [CrossRef] [Green Version]
- Diviani, D.; Lattion, A.L.; Abuin, L.; Staub, O.; Cotecchia, S. The adaptor complex 2 directly interacts with the alpha 1b-adrenergic receptor and plays a role in receptor endocytosis. J. Biol. Chem. 2003, 278, 19331–19340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penela, P.; Ribas, C.; Sanchez-Madrid, F.; Mayor, F., Jr. G protein-coupled receptor kinase 2 (GRK2) as a multifunctional signaling hub. Cell Mol. Life Sci. 2019, 76, 4423–4446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi-Agnihotri, H.; Chaturvedi, M.; Baidya, M.; Stepniewski, T.M.; Pandey, S.; Maharana, J.; Srivastava, A.; Caengprasath, N.; Hanyaloglu, A.C.; Selent, J.; et al. Distinct phosphorylation sites in a prototypical GPCR differently orchestrate beta-arrestin interaction, trafficking, and signaling. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Mueller, W.; Schutz, D.; Nagel, F.; Schulz, S.; Stumm, R. Hierarchical organization of multi-site phosphorylation at the CXCR4 C terminus. PLoS ONE 2013, 8, e64975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, K.; Li, Z.; Xu, M.; Du, J.; Lv, Z.; Zhang, Y. A novel protein kinase A-independent, beta-arrestin-1-dependent signaling pathway for p38 mitogen-activated protein kinase activation by beta2-adrenergic receptors. J. Biol. Chem. 2008, 283, 29028–29036. [Google Scholar] [CrossRef] [Green Version]
- Vibhuti, A.; Gupta, K.; Subramanian, H.; Guo, Q.; Ali, H. Distinct and shared roles of beta-arrestin-1 and beta-arrestin-2 on the regulation of C3a receptor signaling in human mast cells. PLoS ONE 2011, 6, e19585. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.K.; Westfield, G.H.; Xiao, K.; Reis, R.I.; Huang, L.Y.; Tripathi-Shukla, P.; Qian, J.; Li, S.; Blanc, A.; Oleskie, A.N.; et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 2014, 512, 218–222. [Google Scholar] [CrossRef]
- Kumari, P.; Srivastava, A.; Ghosh, E.; Ranjan, R.; Dogra, S.; Yadav, P.N.; Shukla, A.K. Core engagement with beta-arrestin is dispensable for agonist-induced vasopressin receptor endocytosis and ERK activation. Mol. Biol. Cell. 2017, 28, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Yoon, H.J.; Park, J.Y.; Baidya, M.; Dwivedi-Agnihotri, H.; Maharana, J.; Chaturvedi, M.; Chung, K.Y.; Shukla, A.K.; Lee, H.H. Crystal Structure of beta-Arrestin 2 in Complex with CXCR7 Phosphopeptide. Structure 2020, 28, 1014–1023.e1014. [Google Scholar] [CrossRef]
- Zhan, X.; Gimenez, L.E.; Gurevich, V.V.; Spiller, B.W. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J. Mol. Biol. 2011, 406, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sensoy, O.; Moreira, I.S.; Morra, G. Understanding the differential selectivity of arrestins toward the phosphorylation state of the receptor. ACS Chem. Neurosci. 2016, 7, 1212–1224. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BRET Results pEC50 ± SEM (n = 3) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| GRK2 | GRK3 | GRK5 | GRK6 | β-Arrestin1 | β-Arrestin2 | K-Ras | Rab5a | Rab7a | Rab11 |
| 8.2 ± 0.1 | 8.1 ± 0.0 | 8.3 ± 0.2 | ND * | 8.1 ± 0.1 | 8.2 ± 0.0 | 8.9 ± 0.1 | 9.1 ± 0.1 | ND * | 9.1 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarca, A.; Perez, C.; van den Bor, J.; Bebelman, J.P.; Heuninck, J.; de Jonker, R.J.F.; Durroux, T.; Vischer, H.F.; Siderius, M.; Smit, M.J. Differential Involvement of ACKR3 C-Tail in β-Arrestin Recruitment, Trafficking and Internalization. Cells 2021, 10, 618. https://doi.org/10.3390/cells10030618
Zarca A, Perez C, van den Bor J, Bebelman JP, Heuninck J, de Jonker RJF, Durroux T, Vischer HF, Siderius M, Smit MJ. Differential Involvement of ACKR3 C-Tail in β-Arrestin Recruitment, Trafficking and Internalization. Cells. 2021; 10(3):618. https://doi.org/10.3390/cells10030618
Chicago/Turabian StyleZarca, Aurélien, Claudia Perez, Jelle van den Bor, Jan Paul Bebelman, Joyce Heuninck, Rianna J. F. de Jonker, Thierry Durroux, Henry F. Vischer, Marco Siderius, and Martine J. Smit. 2021. "Differential Involvement of ACKR3 C-Tail in β-Arrestin Recruitment, Trafficking and Internalization" Cells 10, no. 3: 618. https://doi.org/10.3390/cells10030618