
Post-Transcriptional Regulation of PARP7 Protein Stability Is Controlled by Androgen Signaling

 and
and
Abstract
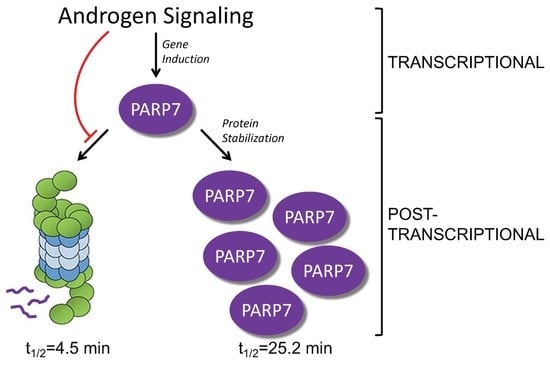
:
1. Introduction
2. Materials and Methods
2.1. Plasmid DNA
2.2. Cell Lines
2.3. Chemical Reagents
2.4. Antibodies
2.5. Immunoblotting
2.6. Protein Half-Life Determination
2.7. RT-qPCR
2.8. Immunofluorescence Microscopy
2.9. Mice and Histology
2.10. Statistical Analysis
2.11. Ethics Statement
3. Results
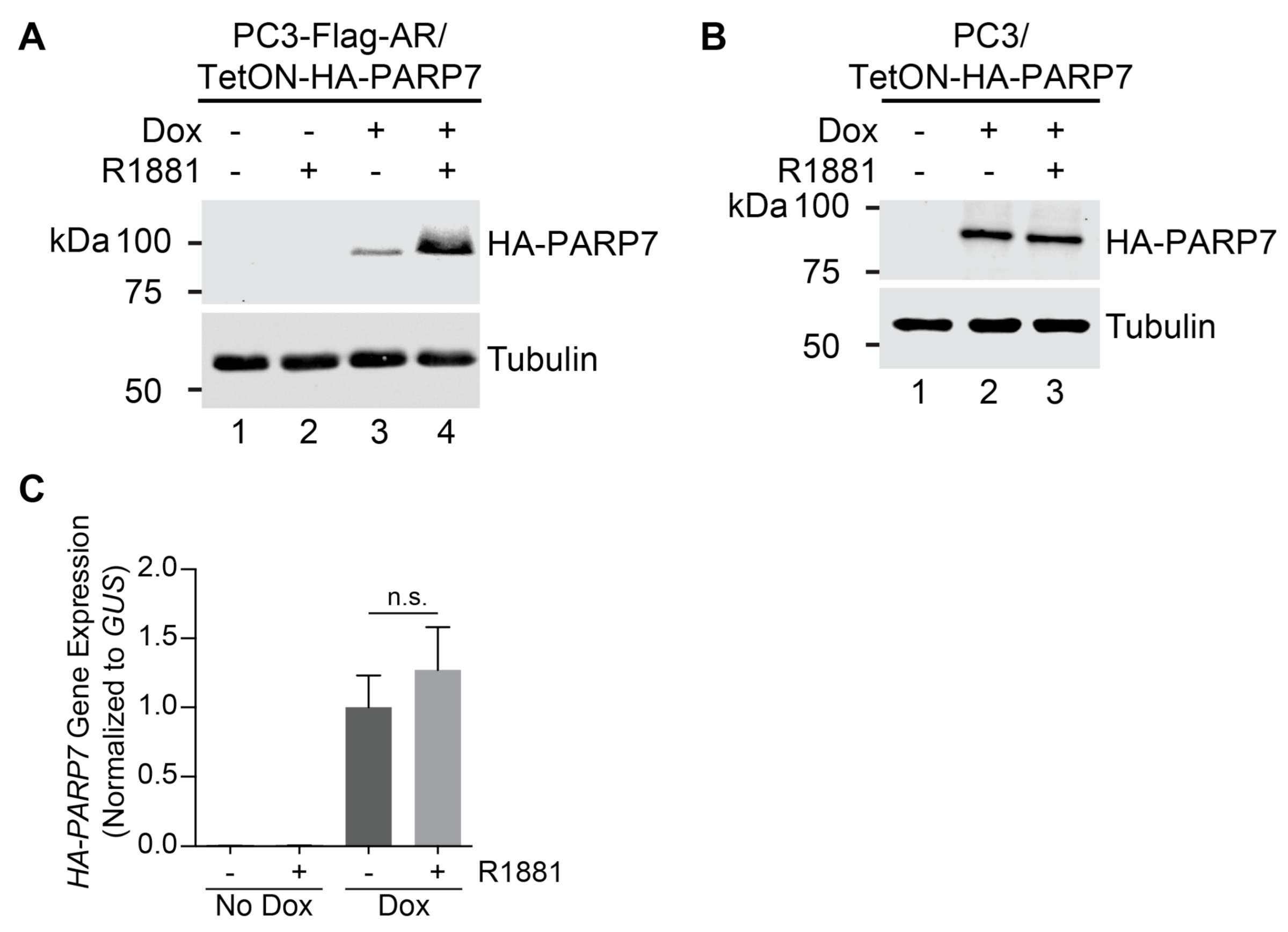
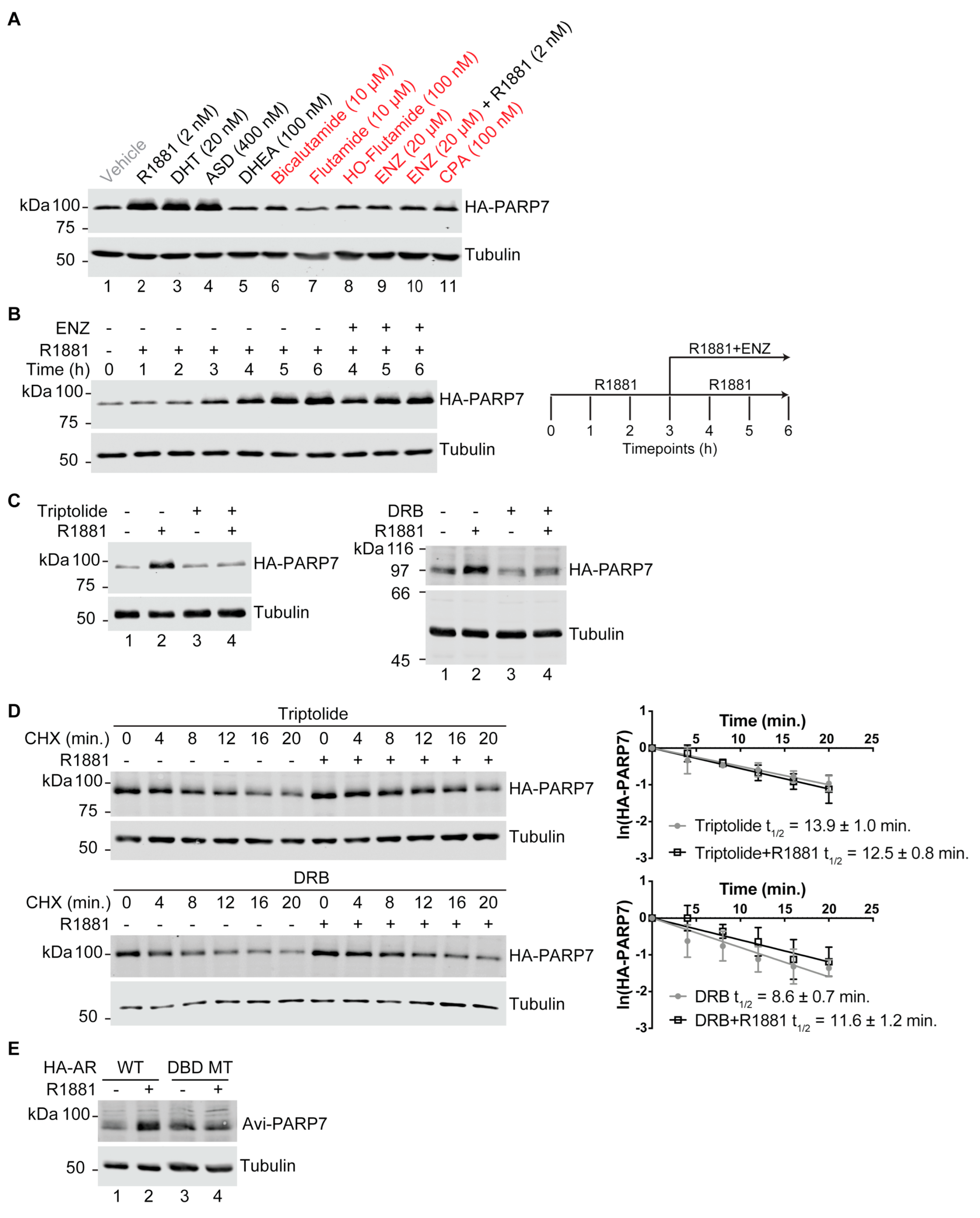
3.1. PARP7 Is Regulated by a Post-Transcriptional Mechanism that Requires AR
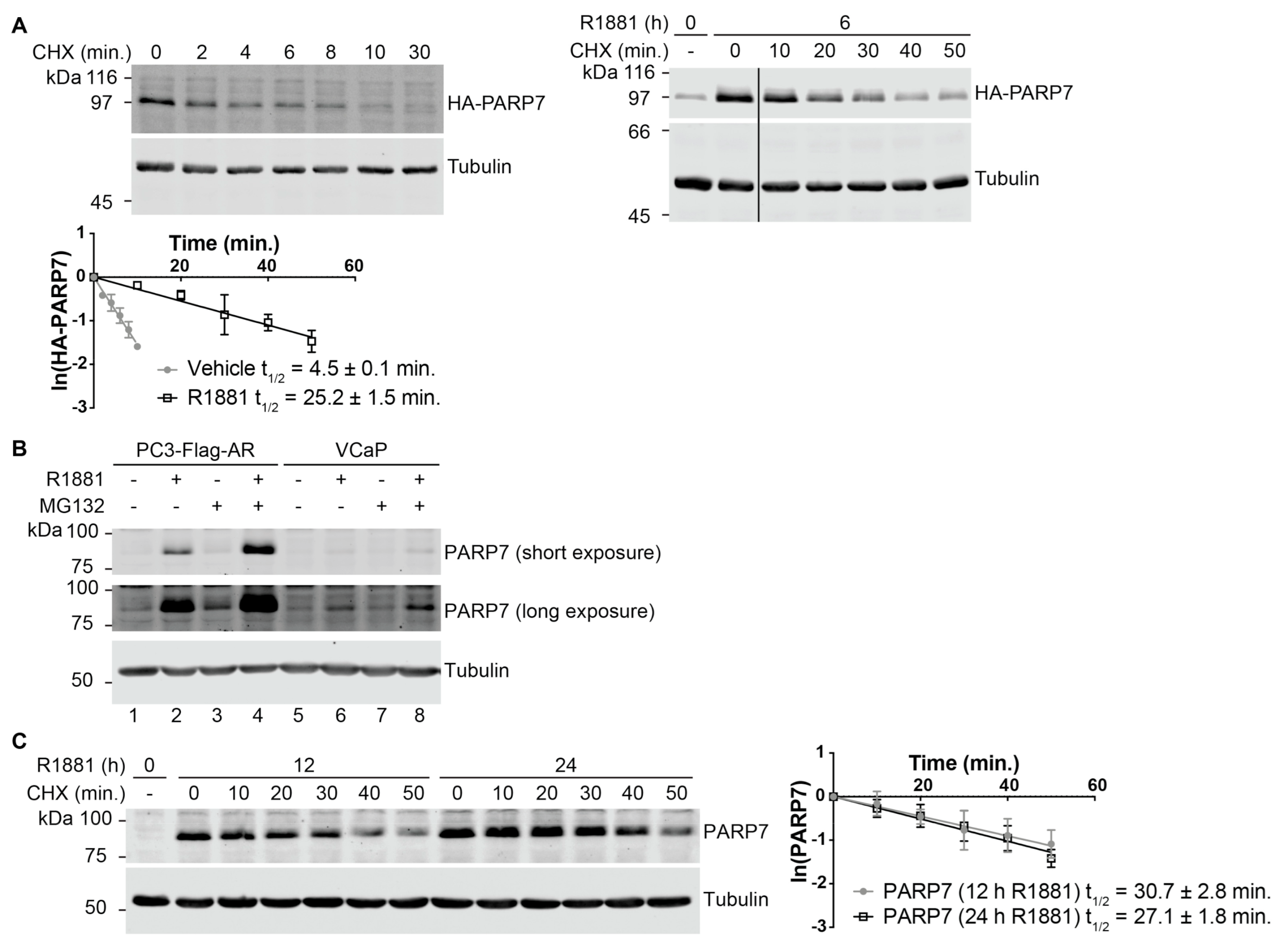
3.2. PARP7 Is a Short-Lived Protein that Is Stabilized by AR Signaling
3.3. Multiple Domains Contribute to the Rapid Turnover of PARP7
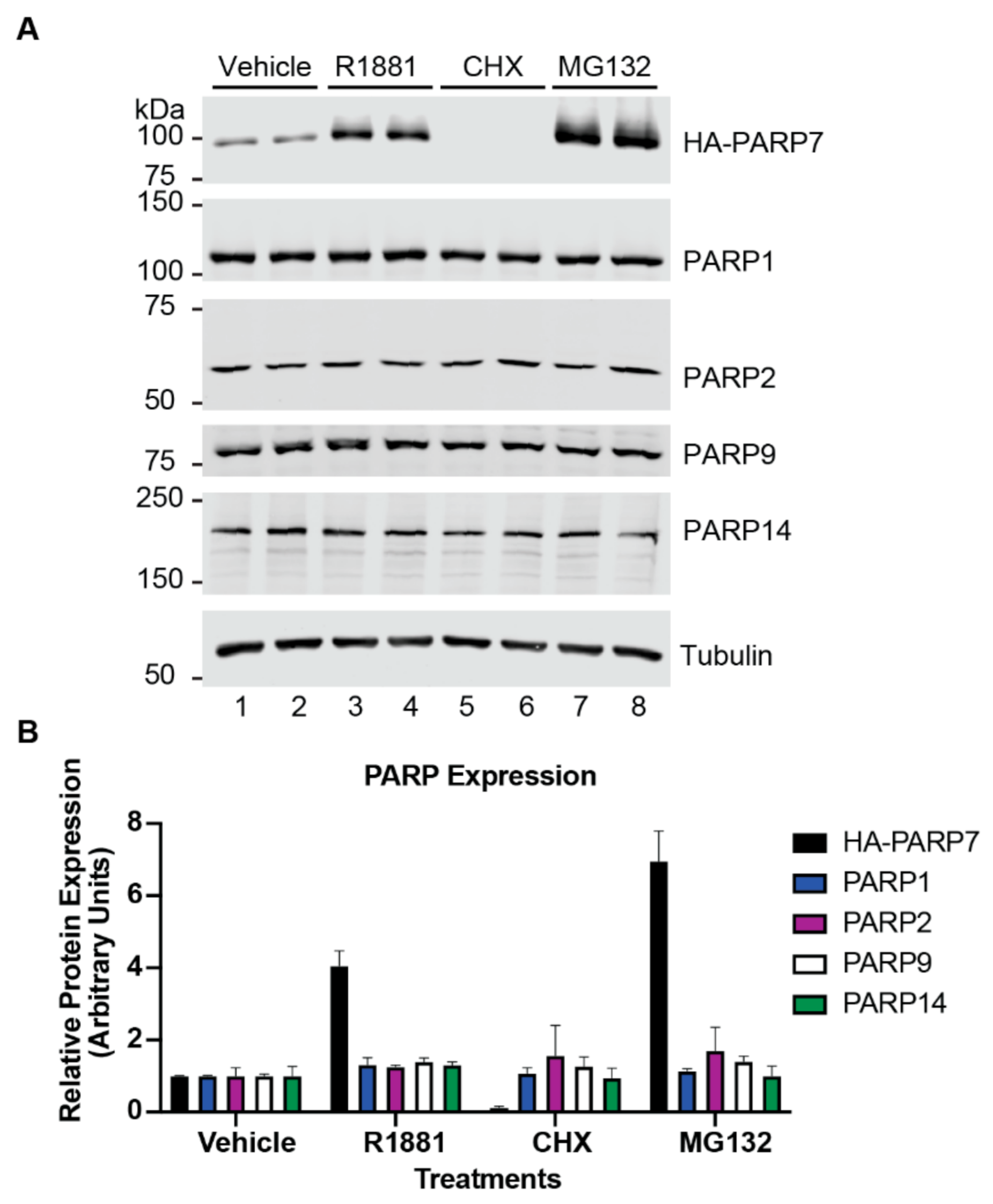
3.4. Rapid Turnover and Androgen-Dependent Stabilization Are Not General Characteristics of PARPs
3.5. AR-Dependent Stabilization of PARP7 Drives Its Nuclear Accumulation
3.6. AR-Dependent Transcription Is Necessary for PARP7 Stabilization
3.7. Characterizing In Vivo Role of PARP7 in Mouse Prostate
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bock, F.J.; Chang, P. New Directions in Poly(ADP-Ribose) Polymerase Biology. FEBS J. 2016, 283, 4017–4031. [Google Scholar] [CrossRef] [Green Version]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-Ribosylation: Recent Advances Linking Molecular Functions to Biological Outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef] [Green Version]
- Hottiger, M.O.; Hassa, P.O.; Lüscher, B.; Schüler, H.; Koch-Nolte, F. Toward a Unified Nomenclature for Mammalian ADP-Ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Barkauskaite, E.; Jankevicius, G.; Ahel, I. Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation. Mol. Cell 2015, 58, 935–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hottiger, M.O. SnapShot: ADP-Ribosylation Signaling. Mol. Cell 2015, 58, 1134–1134.e1. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Dantzer, F.; Ame, J.-C.; De Murcia, G. Poly(ADP-Ribose): Novel Functions for an Old Molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Matic, I.; Uchima, L.; Rood, J.; Zaja, R.; Hay, R.T.; Ahel, I.; Chang, P. Family-Wide Analysis of Poly(ADP-Ribose) Polymerase Activity. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, L.; Mikoč, A.; Ahel, I. ADP-Ribosylation: New Facets of an Ancient Modification. FEBS J. 2017, 284, 2932–2946. [Google Scholar] [CrossRef]
- Kunze, F.A.; Hottiger, M.O. Regulating Immunity via ADP-Ribosylation: Therapeutic Implications and Beyond. Trends Immunol. 2019, 40, 159–173. [Google Scholar] [CrossRef]
- Palazzo, L.; Mikolčević, P.; Mikoč, A.; Ahel, I. ADP-Ribosylation Signalling and Human Disease. Open Biol. 2019, 9, 190041. [Google Scholar] [CrossRef] [Green Version]
- Slade, D. Mitotic Functions of Poly(ADP-Ribose) Polymerases. Biochem. Pharmacol. 2019, 167, 33–43. [Google Scholar] [CrossRef]
- Szántó, M.; Bai, P. The Role of ADP-Ribose Metabolism in Metabolic Regulation, Adipose Tissue Differentiation, and Metabolism. Genes Dev. 2020, 34, 321–340. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; Bono, J.S. A Decade of Clinical Development of PARP Inhibitors in Perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, J. AHR Toxicity and Signaling: Role of TIPARP and ADP-Ribosylation. Curr. Opin. Toxicol. 2017, 2, 50–57. [Google Scholar] [CrossRef]
- Gomez, A.; Bindesbøll, C.; Satheesh, S.V.; Grimaldi, G.; Hutin, D.; MacPherson, L.; Ahmed, S.; Tamblyn, L.; Cho, T.; Nebb, H.I.; et al. Characterization of TCDD-Inducible Poly-ADP-Ribose Polymerase (TIPARP/ARTD14) Catalytic Activity. Biochem. J. 2018, 475, 3827–3846. [Google Scholar] [CrossRef] [Green Version]
- MacPherson, L.; Tamblyn, L.; Rajendra, S.; Bralha, F.; McPherson, J.P.; Matthews, J. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Poly(ADP-Ribose) Polymerase (TiPARP, ARTD14) Is a Mono-ADP-Ribosyltransferase and Repressor of Aryl Hydrocarbon Receptor Transactivation. Nucleic Acids Res. 2013, 41, 1604–1621. [Google Scholar] [CrossRef] [PubMed]
- Bindesbøll, C.; Tan, S.; Bott, D.; Cho, T.; Tamblyn, L.; MacPherson, L.; Grønning-Wang, L.; Nebb, H.I.; Matthews, J. TCDD-Inducible Poly-ADP-Ribose Polymerase (TIPARP/PARP7) Mono-ADP-Ribosylates and Co-Activates Liver X Receptors. Biochem. J. 2016, 473, 899–910. [Google Scholar] [CrossRef]
- Atasheva, S.; Akhrymuk, M.; Frolova, E.I.; Frolov, I. New PARP Gene with an Anti-Alphavirus Function. J. Virol. 2012, 86, 8147–8160. [Google Scholar] [CrossRef] [Green Version]
- Atasheva, S.; Frolova, E.I.; Frolov, I. Interferon-Stimulated Poly(ADP-Ribose) Polymerases Are Potent Inhibitors of Cellular Translation and Virus Replication. J. Virol. 2014, 88, 2116–2130. [Google Scholar] [CrossRef] [Green Version]
- Kozaki, T.; Komano, J.; Kanbayashi, D.; Takahama, M.; Misawa, T.; Satoh, T.; Takeuchi, O.; Kawai, T.; Shimizu, S.; Matsuura, Y.; et al. Mitochondrial Damage Elicits a TCDD-Inducible Poly(ADP-Ribose) Polymerase-Mediated Antiviral Response. Proc. Natl. Acad. Sci. USA 2017, 114, 2681–2686. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Horimoto, H.; Kameyama, T.; Hayakawa, S.; Yamato, H.; Dazai, M.; Takada, A.; Kida, H.; Bott, D.; Zhou, A.C.; et al. Constitutive Aryl Hydrocarbon Receptor Signaling Constrains Type I Interferon–Mediated Antiviral Innate Defense. Nat. Immunol. 2016, 17, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Li, Z.; Huang, Y.-Z.; Zhang, X.; Dai, X.-Y.; Shi, L.; Xi, P.-W.; Wei, J.-F.; Ding, Q. TCDD-Inducible Poly-ADP-Ribose Polymerase (TIPARP), A Novel Therapeutic Target of Breast Cancer. Available online: https://www.dovepress.com/tcdd-inducible-poly-adp-ribose-polymerase-tiparp-a-novel-therapeutic-t-peer-reviewed-article-CMAR (accessed on 7 April 2020).
- Zhang, L.; Cao, J.; Dong, L.; Lin, H. TiPARP Forms Nuclear Condensates to Degrade HIF-1α and Suppress Tumorigenesis. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef]
- Goode, E.L.; Chenevix-Trench, G.; Song, H.; Ramus, S.J.; Notaridou, M.; Lawrenson, K.; Widschwendter, M.; Vierkant, R.A.; Larson, M.C.; Kjaer, S.K.; et al. A Genome-Wide Association Study Identifies Susceptibility Loci for Ovarian Cancer at 2q31 and 8q24. Nat. Genet. 2010, 42, 874–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palavalli Parsons, L.H.; Lea, J.S. PARP 7 Has a Significant Role in Overall Survival of Patients with Ovarian Cancer. Gynecol. Oncol. 2018, 149, 10. [Google Scholar] [CrossRef]
- Bolton, E.C.; So, A.Y.; Chaivorapol, C.; Haqq, C.M.; Li, H.; Yamamoto, K.R. Cell- and Gene-Specific Regulation of Primary Target Genes by the Androgen Receptor. Genes Amp Dev. 2007, 21, 2005–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jividen, K.; Kedzierska, K.Z.; Yang, C.-S.; Szlachta, K.; Ratan, A.; Paschal, B.M. Genomic Analysis of DNA Repair Genes and Androgen Signaling in Prostate Cancer. BMC Cancer 2018, 18, 960. [Google Scholar] [CrossRef] [Green Version]
- Kelley, J.B.; Paschal, B.M. Fluorescence-Based Quantification of Nucleocytoplasmic Transport. Methods 2019, 157, 106–114. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamaguchi, M.T.; Ohteki, T.; Sasaki, T.; Kaisho, T.; Kimura, Y.; Yoshida, R.; Wakeham, A.; Higuchi, T.; Fukumoto, M.; et al. T Cell-Specific Loss of Pten Leads to Defects in Central and Peripheral Tolerance. Immunity 2001, 14, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Wu, J.; Huang, J.; Powell, W.C.; Zhang, J.; Matusik, R.J.; Sangiorgi, F.O.; Maxson, R.E.; Sucov, H.M.; Roy-Burman, P. Generation of a Prostate Epithelial Cell-Specific Cre Transgenic Mouse Model for Tissue-Specific Gene Ablation. Mech. Dev. 2001, 101, 61–69. [Google Scholar] [CrossRef]
- Raymond, C.S.; Soriano, P. High-Efficiency FLP and PhiC31 Site-Specific Recombination in Mammalian Cells. PLoS ONE 2007, 2, e162. [Google Scholar] [CrossRef] [PubMed]
- Bjerke, G.A.; Yang, C.-S.; Frierson, H.F.; Paschal, B.M.; Wotton, D. Activation of Akt Signaling in Prostate Induces a TGFβ-Mediated Restraint on Cancer Progression and Metastasis. Oncogene 2014, 33, 3660–3667. [Google Scholar] [CrossRef] [Green Version]
- Bjerke, G.A.; Pietrzak, K.; Melhuish, T.A.; Frierson, H.F.; Paschal, B.M.; Wotton, D. Prostate Cancer Induced by Loss of Apc Is Restrained by TGFβ Signaling. PLoS ONE 2014, 9, e92800. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gong, S.; Roy-Burman, P.; Lee, P.; Culig, Z. Current Mouse and Cell Models in Prostate Cancer Research. Endocr. Relat. Cancer 2013, 20. [Google Scholar] [CrossRef] [Green Version]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and Characterization of a Human Prostatic Carcinoma Cell Line (PC-3). Investig. Urol. 1979, 17, 16–23. [Google Scholar] [PubMed]
- Emanuele, M.J.; Elia, A.E.H.; Xu, Q.; Thoma, C.R.; Izhar, L.; Leng, Y.; Guo, A.; Chen, Y.-N.; Rush, J.; Hsu, P.W.-C.; et al. Global Identification of Modular Cullin-RING Ligase Substrates. Cell 2011, 147, 459–474. [Google Scholar] [CrossRef] [Green Version]
- Roper, S.J.; Chrysanthou, S.; Senner, C.E.; Sienerth, A.; Gnan, S.; Murray, A.; Masutani, M.; Latos, P.; Hemberger, M. ADP-Ribosyltransferases Parp1 and Parp7 Safeguard Pluripotency of ES Cells. Nucleic Acids Res. 2014, 42, 8914–8927. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Knecht, K.; Birzin, E.; Fisher, J.; Wilkinson, H.; Mojena, M.; Moreno, C.T.; Schmidt, A.; Harada, S.; Freedman, L.P.; et al. Direct Agonist/Antagonist Functions of Dehydroepiandrosterone. Endocrinology 2005, 146, 4568–4576. [Google Scholar] [CrossRef] [Green Version]
- Gonit, M.; Zhang, J.; Salazar, M.D.; Cui, H.; Shatnawi, A.; Trumbly, R.; Ratnam, M. Hormone Depletion-Insensitivity of Prostate Cancer Cells Is Supported by the AR without Binding to Classical Response Elements. Mol. Endocrinol. 2011, 25, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobaccaro, J.M.; Poujol, N.; Chiche, L.; Lumbroso, S.; Brown, T.R.; Sultan, C. Molecular Modeling and in Vitro Investigations of the Human Androgen Receptor DNA-Binding Domain: Application for the Study of Two Mutations. Mol. Cell. Endocrinol. 1996, 116, 137–147. [Google Scholar] [CrossRef]
- Schmahl, J.; Raymond, C.S.; Soriano, P. PDGF Signaling Specificity Is Mediated through Multiple Immediate Early Genes. Nat. Genet. 2007, 39, 52–60. [Google Scholar] [CrossRef]
- Shen, M.M.; Abate-Shen, C. Molecular Genetics of Prostate Cancer: New Prospects for Old Challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [Green Version]
- Diehl, J.A.; Zindy, F.; Sherr, C.J. Inhibition of Cyclin D1 Phosphorylation on Threonine-286 Prevents Its Rapid Degradation via the Ubiquitin-Proteasome Pathway. Genes Dev. 1997, 11, 957–972. [Google Scholar] [CrossRef] [Green Version]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen Synthase Kinase-3β Regulates Cyclin D1 Proteolysis and Subcellular Localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacker, K.; Benke, S.; Agerer, B.; Scinicariello, S.; Budroni, V.; Versteeg, G.A. A Repetitive Acidic Region Contributes to the Extremely Rapid Degradation of the Cell-Context Essential Protein TRIM52. Sci. Rep. 2019, 9, 7901. [Google Scholar] [CrossRef] [PubMed]
- Berra, E.; Roux, D.; Richard, D.E.; Pouysségur, J. Hypoxia-Inducible Factor-1α (HIF-1α) Escapes O2-Driven Proteasomal Degradation Irrespective of Its Subcellular Localization: Nucleus or Cytoplasm. EMBO Rep. 2001, 2, 615–620. [Google Scholar] [CrossRef] [Green Version]
- Moroz, E.; Carlin, S.; Dyomina, K.; Burke, S.; Thaler, H.T.; Blasberg, R.; Serganova, I. Real-Time Imaging of HIF-1α Stabilization and Degradation. PLoS ONE 2009, 4, e5077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwami, K.; Wang, J.Y.; Jain, R.; McCormack, S.; Johnson, L.R. Intestinal Ornithine Decarboxylase: Half-Life and Regulation by Putrescine. Am. J. Physiol.-Gastrointest. Liver Physiol. 1990, 258, G308–G315. [Google Scholar] [CrossRef] [PubMed]
- Erdős, G.; Dosztányi, Z. Analyzing Protein Disorder with IUPred2A. Curr. Protoc. Bioinform. 2020, 70, e99. [Google Scholar] [CrossRef] [Green Version]
- Van der Lee, R.; Lang, B.; Kruse, K.; Gsponer, J.; Sánchez de Groot, N.; Huynen, M.A.; Matouschek, A.; Fuxreiter, M.; Babu, M.M. Intrinsically Disordered Segments Affect Protein Half-Life in the Cell and during Evolution. Cell Rep. 2014, 8, 1832–1844. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Zhang, N.-Y.; Zurawel, A.; Hansen, K.C.; Liu, C.-W. Degradation of Some Polyubiquitinated Proteins Requires an Intrinsic Proteasomal Binding Element in the Substrates. J. Biol. Chem. 2010, 285, 4771–4780. [Google Scholar] [CrossRef] [Green Version]
- Riccio, A.A.; Cingolani, G.; Pascal, J.M. PARP-2 Domain Requirements for DNA Damage-Dependent Activation and Localization to Sites of DNA Damage. Nucleic Acids Res. 2016, 44, 1691–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, S.; Mickanin, C.; Feng, Y.; Charlat, O.; Michaud, G.A.; Schirle, M.; Shi, X.; Hild, M.; Bauer, A.; et al. RNF146 Is a Poly(ADP-Ribose)-Directed E3 Ligase That Regulates Axin Degradation and Wnt Signalling. Nat. Cell Biol. 2011, 13, 623. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PARP7 | t1/2 (Vehicle) | t1/2 (R1881) |
|---|---|---|
| Endogenous PARP7 | N/A 1 | 12 h R1881: 30.7 ± 2.8 24 h R1881: 27.1 ± 1.8 |
| HA-PARP7 WT | 4.5 ± 0.1 | 25.2 ± 1.5 |
| C243A | 99.1 ± 7.4 | 74.9 ± 8.8 |
| C251A | 99.0 ± 10.5 | 90.3 ± 9.2 |
| H532A | 40.7 ± 3.5 | 42.0 ± 2.5 |
| Y564A | 34.0 ± 1.6 | 33.0 ± 1.4 |
| ∆WWE | 14.2 ± 0.8 | 14.0 ± 0.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamata, T.; Yang, C.-S.; Melhuish, T.A.; Frierson Jr., H.F.; Wotton, D.; Paschal, B.M. Post-Transcriptional Regulation of PARP7 Protein Stability Is Controlled by Androgen Signaling. Cells 2021, 10, 363. https://doi.org/10.3390/cells10020363
Kamata T, Yang C-S, Melhuish TA, Frierson Jr. HF, Wotton D, Paschal BM. Post-Transcriptional Regulation of PARP7 Protein Stability Is Controlled by Androgen Signaling. Cells. 2021; 10(2):363. https://doi.org/10.3390/cells10020363
Chicago/Turabian StyleKamata, Teddy, Chun-Song Yang, Tiffany A. Melhuish, Henry F. Frierson Jr., David Wotton, and Bryce M. Paschal. 2021. "Post-Transcriptional Regulation of PARP7 Protein Stability Is Controlled by Androgen Signaling" Cells 10, no. 2: 363. https://doi.org/10.3390/cells10020363