

Silver Dependent Enantiodivergent Gold(I) Catalysed Asymmetric Intramolecular Hydroamination of Alkenes: A Theoretical Study

Abstract
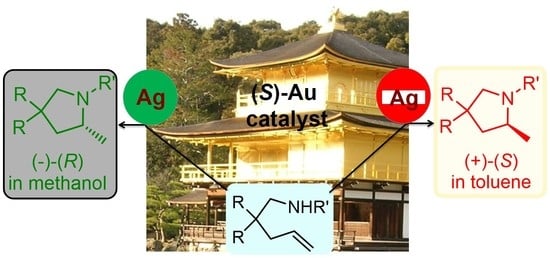
:
{kind=link}
{kind=link}
{kind=link}
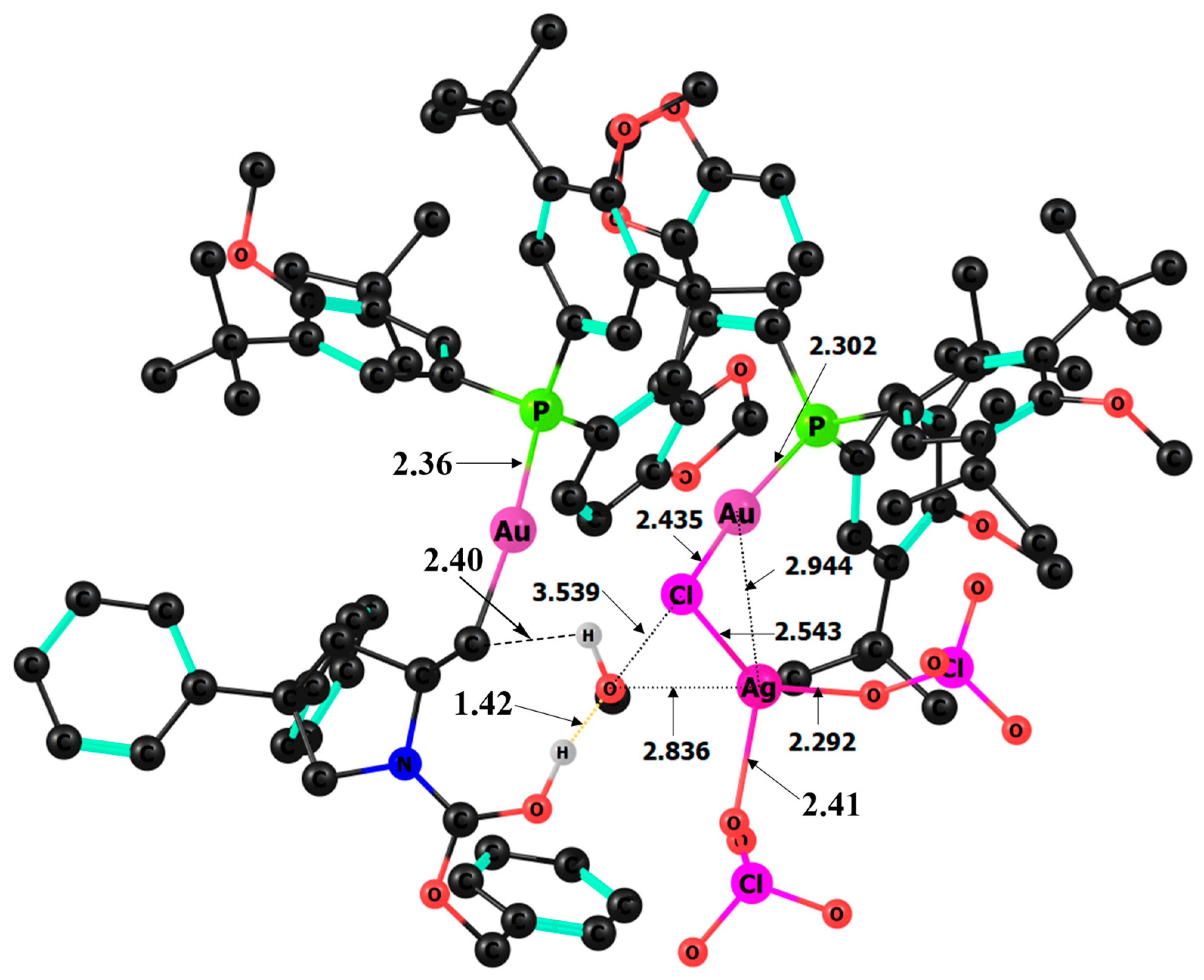{kind=link}
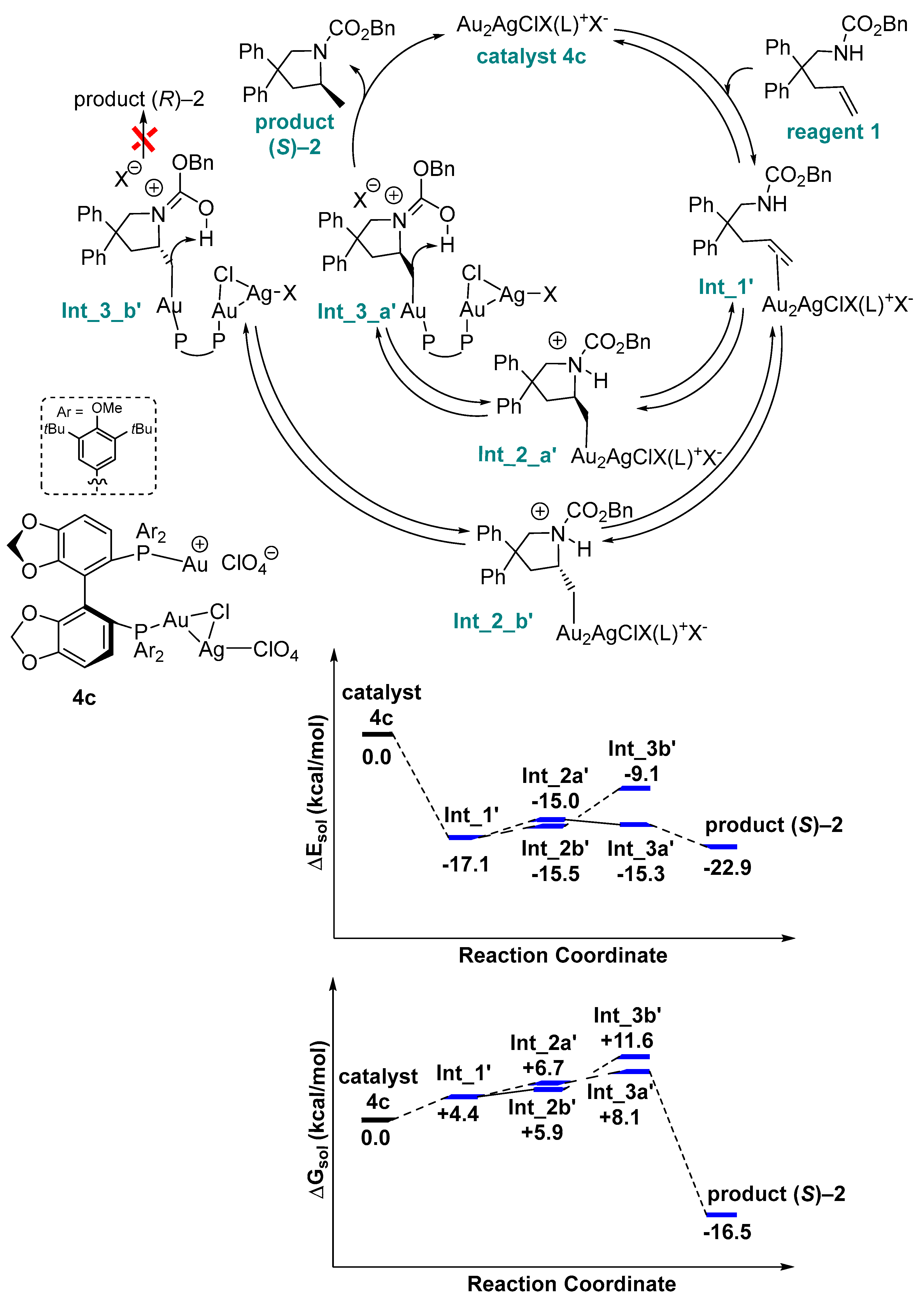{kind=link}
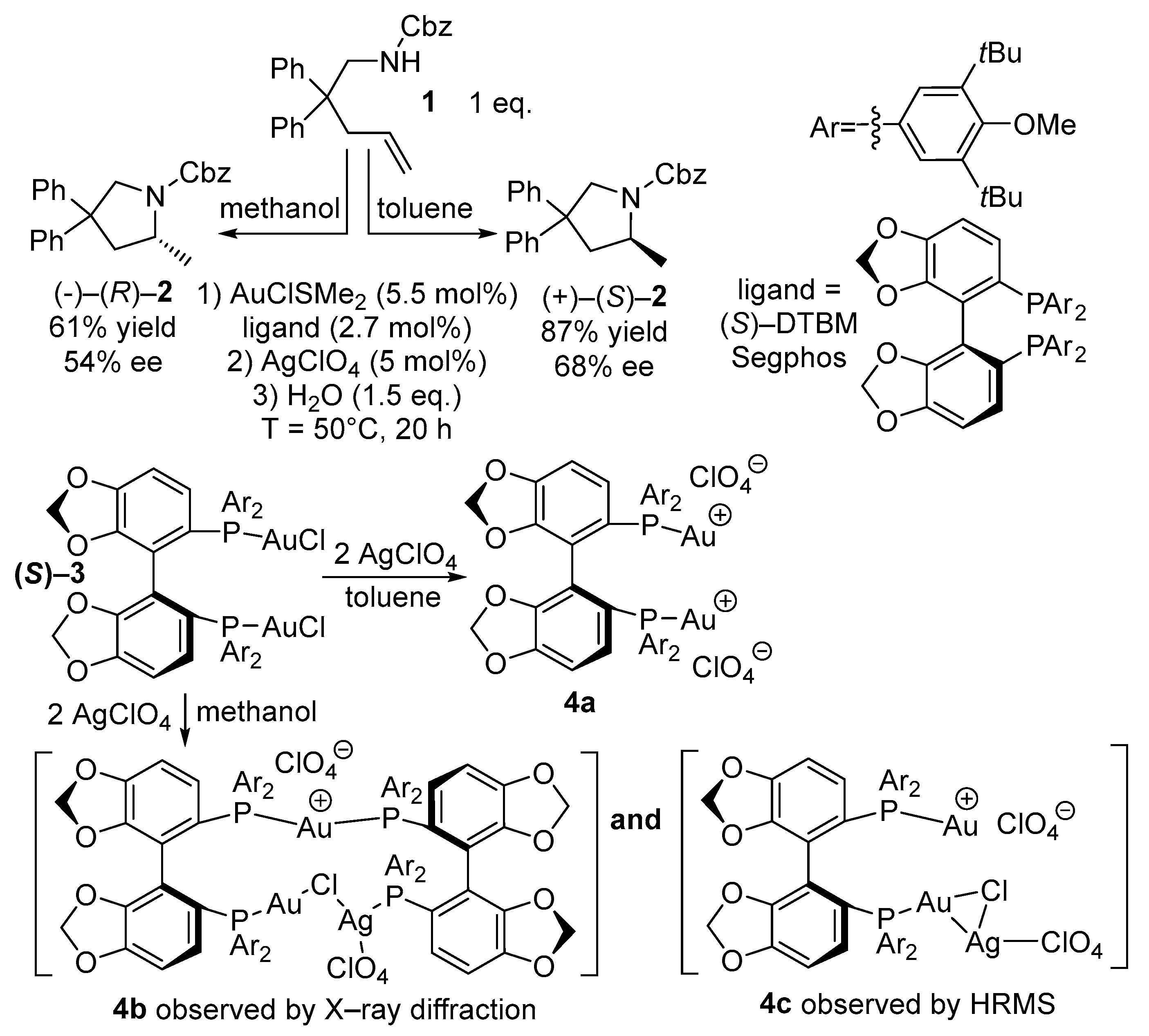{kind=link}
1. Introduction
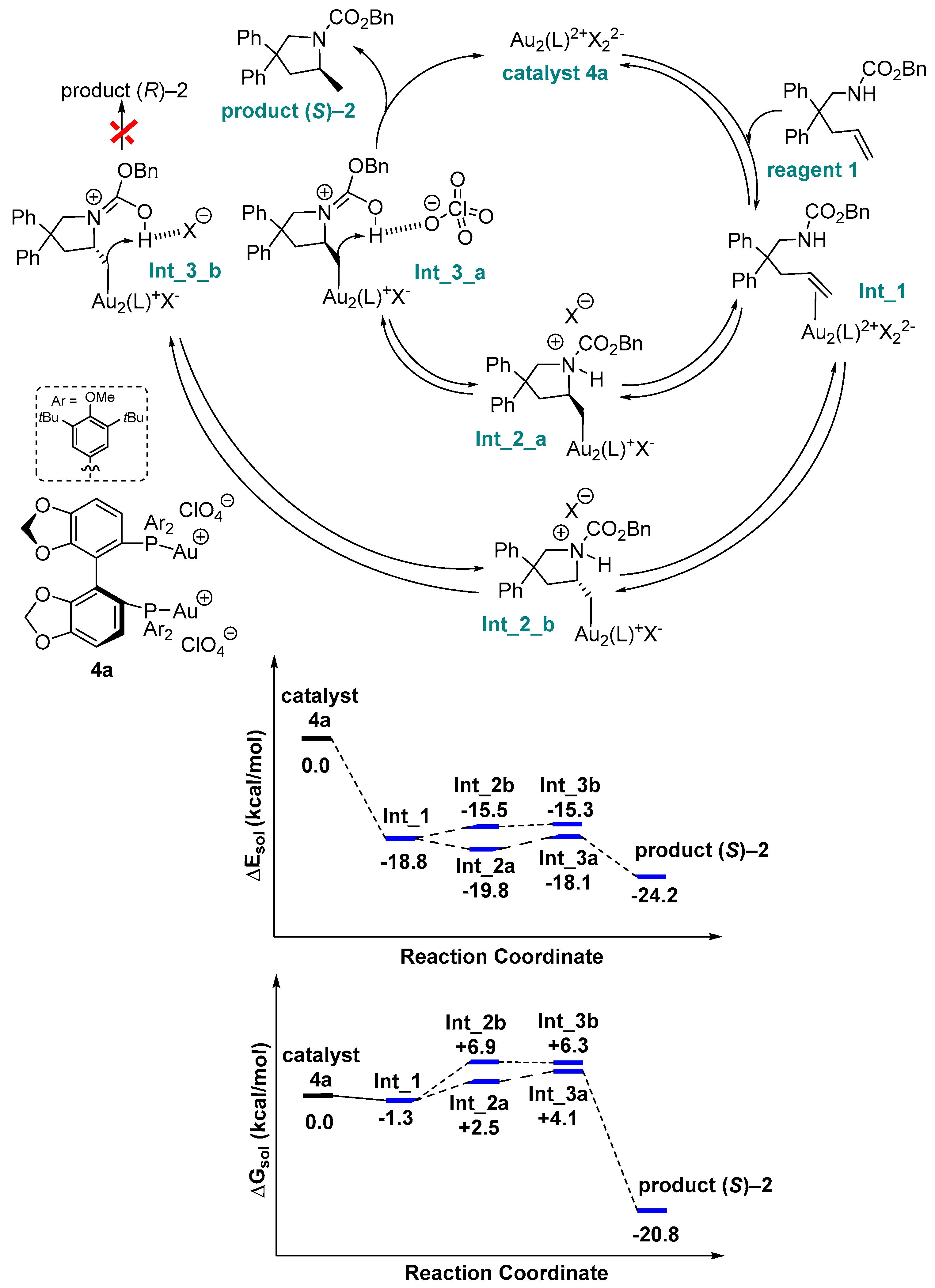
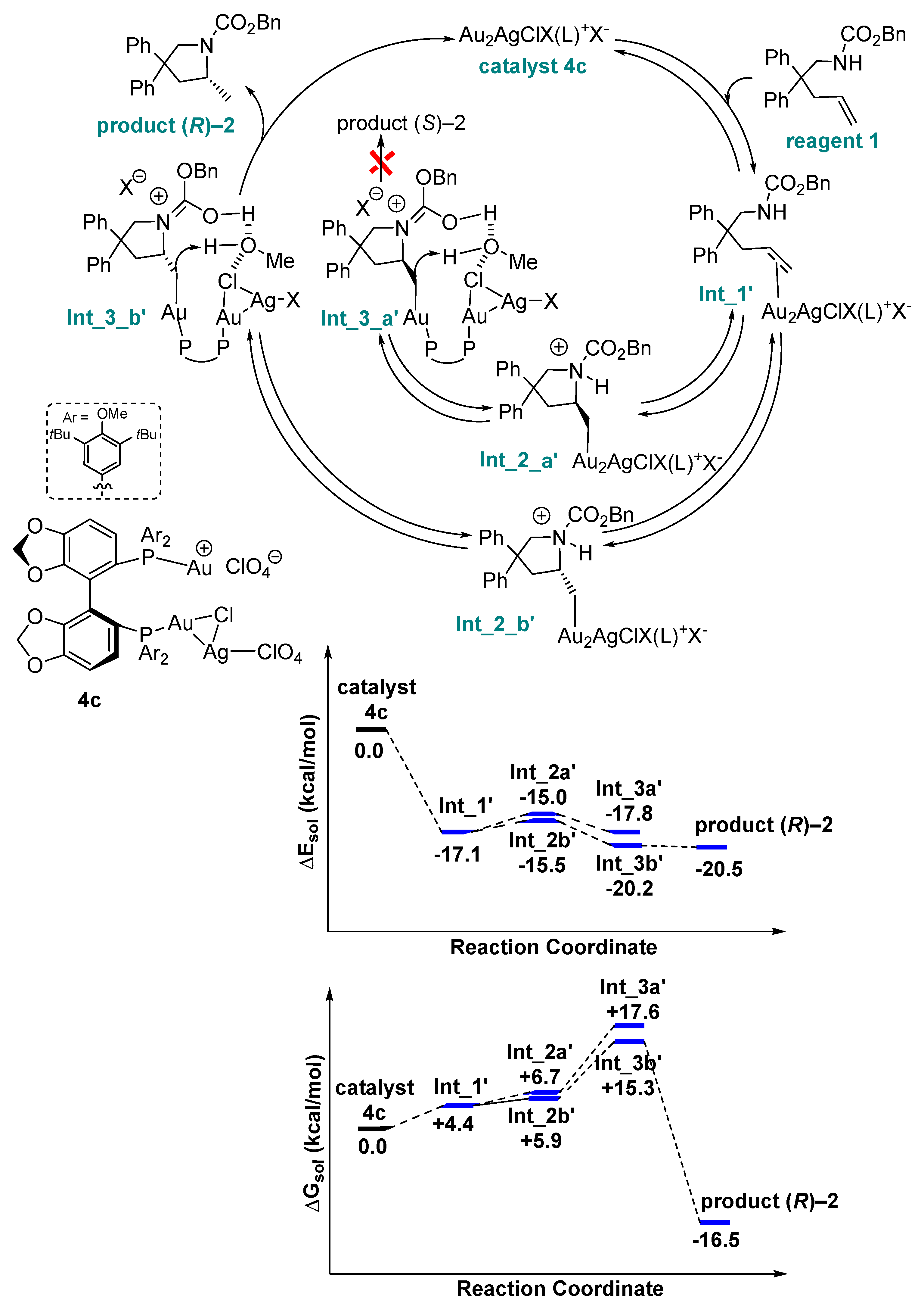
2. Results
3. Material and Methods
3.1. General Procedure for the Catalysis
3.2. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reznichenko, A.L.; Nawara-Hultzsch, A.J.; Hultzsch, K.C. Asymmetric hydroamination. Top. Curr. Chem. 2014, 343, 191–260. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Arndt, M.; Gooßen, K.; Heydt, H.; Gooßen, L.J. Late transition metal-catalyzed hydroamination and hydroamidation. Chem. Rev. 2015, 115, 2596–2697. [Google Scholar] [CrossRef] [PubMed]
- Michon, C.; Abadie, M.-A.; Medina, F.; Agbossou-Niedercorn, F. Recent metal-catalysed asymmetric hydroaminations of alkenes. J. Organomet. Chem. 2017, 847, 13–37. [Google Scholar] [CrossRef]
- Colonna, P.; Bezzenine, S.; Gil, R.; Hannedouche, J. Alkene Hydroamination via Earth-Abundant Transition Metal (Iron, Cobalt, Copper and Zinc) Catalysis: A Mechanistic Overview. Adv. Synth. Catal. 2020, 362, 1550–1563. [Google Scholar] [CrossRef]
- Praveen, C. Carbophilic activation of π-systems via gold coordination: Towards regioselective access of intermolecular addition products. Coord. Chem. Rev. 2019, 392, 1–34. [Google Scholar] [CrossRef]
- Lu, Z.; Hammond, G.B.; Xu, B. Improving Homogeneous Cationic Gold Catalysis through a Mechanism-Based Approach. Acc. Chem. Res. 2019, 52, 1275–1288. [Google Scholar] [CrossRef]
- Campeau, D.; Leon Rayo, D.F.; Mansour, A.; Muratov, K.; Gagosz, F. Gold-Catalyzed Reactions of Specially Activated Alkynes, Allenes, and Alkenes. Chem. Rev. 2021, 121, 8756–8867. [Google Scholar] [CrossRef]
- Zuccarello, G.; Escofet, I.; Caniparoli, U.; Echavarren, A.M. New-Generation Ligand Design for the Gold-Catalyzed Asymmetric Activation of Alkynes. ChemPlusChem 2021, 86, 1283–1296. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, S.D.; Widenhoefer, R.A. Intermolecular Hydroamination of Ethylene and 1-Alkenes with Cyclic Ureas Catalyzed by Achiral and Chiral Gold(I) Complexes. J. Am. Chem. Soc. 2009, 131, 5372–5373. [Google Scholar] [CrossRef] [Green Version]
- Kojima, M.; Mikami, K. Enantioselective Intramolecular Hydroamination of N-Alkenyl Ureas Catalyzed by tropos BIPHEP-Gold(I) Complexes with Au-Au Interaction. Synlett 2012, 23, 57–61. [Google Scholar] [CrossRef]
- Sun, Y.W.; Xu, Q.; Shi, M. Synthesis of axially chiral gold complexes and their applications in asymmetric catalyses. Beilstein J. Org. Chem. 2013, 9, 2224–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.D.; Timmerman, J.C.; Widenhoefer, R.A. Enantioselective Intramolecular Hydroamination of Unactivated Alkenes Catalyzed by Mono- and Bis(gold) Phosphine Complexes. Adv. Synth. Catal. 2014, 356, 3187–3192. [Google Scholar] [CrossRef]
- Michon, C.; Abadie, M.-A.; Medina, F.; Agbossou-Niedercorn, F. Mononuclear gold catalysts for the asymmetric intramolecular hydroamination of alkenes. Catal. Today 2014, 235, 2–13. [Google Scholar] [CrossRef]
- Abadie, M.-A.; Medina, F.; Agbossou-Niedercorn, F.; Michon, C. Efficient gold(I) catalysed asymmetric hydroamination of alkenes. Chim. OGGI-Chem. Today 2014, 32, 19–21. [Google Scholar]
- Abadie, M.-A.; Trivelli, X.; Medina, F.; Capet, F.; Roussel, P.; Agbossou-Niedercorn, F.; Michon, C. Asymmetric Intramolecular Hydroamination of Alkenes in Mild and Wet Conditions—Structure and Reactivity of Cationic Binuclear Gold(I) Catalysts. ChemCatChem 2014, 6, 2235–2239. [Google Scholar] [CrossRef]
- Abadie, M.-A.; Trivelli, X.; Medina, F.; Duhal, N.; Kouach, M.; Linden, B.; Vandewalle, M.; Capet, F.; Roussel, P.; Del Rosal, I.; et al. Gold(I)-Catalysed Asymmetric Hydroamination of Alkenes: A Silver- and Solvent-Dependent Enantiodivergent Reaction. Chem. Eur. J. 2017, 23, 10777–10788. [Google Scholar] [CrossRef] [PubMed]
- Michon, C.; Medina, F.; Capet, F.; Roussel, P.; Agbossou-Niedercorn, F. Inter- and Intramolecular Hydroamination of Unactivated Alkenes Catalysed by a Combination of Copper and Silver Salts: The Unveiling of a Brønsted Acid Catalysis. Adv. Synth. Catal. 2010, 352, 3293–3305. [Google Scholar] [CrossRef]
- Medina, F.; Michon, C.; Agbossou-Niedercorn, F. Intermolecular Mono- and Dihydroamination of Activated Alkenes Using a Recoverable Gold Catalyst. Eur. J. Org. Chem. 2012, 2012, 6218–6227. [Google Scholar] [CrossRef]
- Michon, C.; Medina, F.; Abadie, M.-A.; Agbossou-Niedercorn, F. Asymmetric Intramolecular Hydroamination of Allenes using Mononuclear Gold Catalysts. Organometallics 2013, 32, 5589–5600. [Google Scholar] [CrossRef]
- Michon, C.; Gilbert, J.; Trivelli, X.; Nahra, F.; Cazin, C.S.J.; Agbossou-Niedercorn, F.; Nolan, S.P. Gold(I) catalysed regio- and stereoselective intermolecular hydroamination of internal alkynes: Towards functionalised azoles. Org. Biomol. Chem. 2019, 17, 3805–3811. [Google Scholar] [CrossRef]
- Bartók, M. Unexpected inversions in asymmetric reactions: Reactions with chiral metal complexes, chiral organocatalysts, and heterogeneous chiral catalysts. Chem. Rev. 2010, 110, 1663–1705. [Google Scholar] [CrossRef] [PubMed]
- Escorihuela, J.; Burguete, M.I.; Luis, S.V. New advances in dual stereocontrol for asymmetric reactions. Chem. Soc. Rev. 2013, 42, 5595–5617. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Nájera, C.; Yus, M. Stereodivergent Catalysis. Chem. Rev. 2018, 118, 5080–5200. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Feng, X.; Liu, X. Reversal of enantioselectivity in chiral metal complex-catalyzed asymmetric reactions. Org. Biomol. Chem. 2019, 17, 6538–6550. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Naruse, S.; Takano, K.; Fukuda, K.; Katoh, A.; Inoue, Y. Unusual Temperature Dependence of Enantioselectivity in Asymmetric Reductions by Chiral NADH Models. Org. Lett. 2006, 8, 2067–2070. [Google Scholar] [CrossRef]
- Chan, V.S.; Chiu, M.; Bergman, R.G.; Toste, F.D. Development of Ruthenium Catalysts for the Enantioselective Synthesis of P-Stereogenic Phosphines via Nucleophilic Phosphido Intermediates. J. Am. Chem. Soc. 2009, 131, 6021–6032. [Google Scholar] [CrossRef]
- Sohtome, Y.; Tanaka, S.; Takada, K.; Yamaguchi, T.; Nagasawa, K. Solvent-dependent enantiodivergent Mannich-type reaction: Utilizing a conformationally flexible guanidine/bisthiourea organocatalyst. Angew. Chem. Int. Ed. 2010, 49, 9254–9257. [Google Scholar] [CrossRef]
- Chiarucci, M.; Mocci, R.; Syntrivanis, L.-D.; Cera, G.; Mazzanti, A.; Bandini, M. Merging synthesis and enantioselective functionalization of indoles by a gold-catalyzed asymmetric cascade reaction. Angew. Chem. Int. Ed. 2013, 52, 10850–10853. [Google Scholar] [CrossRef]
- Davies, P.W.; Martin, N. Counterion Effects in a Gold-Catalyzed Synthesis of Pyrroles from Alkynyl Aziridines. Org. Lett. 2009, 11, 2293–2296. [Google Scholar] [CrossRef]
- Fang, W.; Presset, M.; Guérinot, A.; Bour, C.; Bezzenine-Lafollée, S.; Gandon, V. Cationic gold(I)-catalyzed enantioselective hydroalkylation of unactivated alkenes: Influence of the chloride scavenger on the stereoselectivity. Org. Chem. Front. 2014, 1, 608–613. [Google Scholar] [CrossRef]
- Jia, M.; Bandini, M. Counterion Effects in Homogeneous Gold Catalysis. ACS Catal. 2015, 5, 1638–1652. [Google Scholar] [CrossRef]
- Jaroschik, F.; Simonneau, A.; Lemière, G.; Cariou, K.; Agenet, N.; Amouri, H.; Aubert, C.; Goddard, J.-P.; Lesage, D.; Malacria, M.; et al. Assessing Ligand and Counterion Effects in the Noble Metal Catalyzed Cycloisomerization Reactions of 1,6-Allenynes: A Combined Experimental and Theoretical Approach. ACS Catal. 2016, 6, 5146–5160. [Google Scholar] [CrossRef]
- Ilg, M.K.; Wolf, L.M.; Mantilli, L.; Farès, C.; Thiel, W.; Fürstner, A. A Striking Case of Enantioinversion in Gold Catalysis and Its Probable Origins. Chem. Eur. J. 2015, 21, 12279–12284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macchioni, A. Ion Pairing in Transition-Metal Organometallic Chemistry. Chem. Rev. 2005, 105, 2039–2074. [Google Scholar] [CrossRef] [PubMed]
- Pregosin, P.S. NMR spectroscopy and ion pairing: Measuring and understanding how ions interact. Pure Appl. Chem. 2009, 81, 615–633. [Google Scholar] [CrossRef] [Green Version]
- Kanno, O.; Kuriyama, W.; Wang, J.Z.; Toste, D.F. Regio- and enantioselective hydroamination of dienes by gold(I)/menthol cooperative catalysis. Angew. Chem. Int. Ed. 2011, 50, 9919–9922. [Google Scholar] [CrossRef] [Green Version]
- Lutz, F.; Igarashi, T.; Kinoshita, T.; Asahina, M.; Tsukiyama, K.; Kawasaki, T.; Soai, K. Mechanistic Insights in the Reversal of Enantioselectivity of Chiral Catalysts by Achiral Catalysts in Asymmetric Autocatalysis. J. Am. Chem. Soc. 2008, 130, 2956–2958. [Google Scholar] [CrossRef]
- Nojiri, A.; Kumagai, N.; Shibasaki, M. Linking structural dynamics and functional diversity in asymmetric catalysis. J. Am. Chem. Soc. 2009, 131, 3779–3784. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Z.; Chen, D.; Liu, X.; Lin, L.; Feng, X. Highly Enantioselective Michael Addition of Pyrazolin-5-ones Catalyzed by Chiral Metal/N,N′-Dioxide Complexes: Metal-Directed Switch in Enantioselectivity. Angew. Chem. Int. Ed. 2011, 50, 4928–4932. [Google Scholar] [CrossRef]
- Lu, G.; Yoshino, T.; Morimoto, H.; Matsunaga, S.; Shibasaki, M. Stereodivergent direct catalytic asymmetric Mannich-type reactions of α-isothiocyanato ester with ketimines. Angew. Chem. Int. Ed. 2011, 50, 4382–4385. [Google Scholar] [CrossRef]
- Noble-Terán, M.E.; Buhse, T.; Cruz, J.M.; Coudret, C.; Micheau, J.C. Nonlinear Effects in Asymmetric Synthesis: A Practical Tool for the Discrimination between Monomer and Dimer Catalysis. ChemCatChem 2016, 8, 1836–1845. [Google Scholar] [CrossRef]
- Satyanarayana, T.; Abraham, S.; Kagan, H.B. Nonlinear effects in asymmetric catalysis. Angew. Chem. Int. Ed. 2009, 48, 456–494. [Google Scholar] [CrossRef]
- Hu, J.-Y.; Zhang, J.; Wang, G.-X.; Sun, H.-L.; Zhang, J.-L. Constructing a Catalytic Cycle for C-F to C-X (X = O, S, N) Bond Transformation Based on Gold-Mediated Ligand Nucleophilic Attack. Inorg. Chem. 2016, 55, 2274–2283. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Simon, C.; Garcia-Borras, M.; Gomez, L.; Garcia-Bosch, I.; Osuna, S.; Swart, M.; Luis, J.P.; Rovira, C.; Almeida, M.; Imaz, I.; et al. Self-Assembled Tetragonal Prismatic Molecular Cage Highly Selective for Anionic π Guests. Chem. Eur. J. 2013, 19, 1445–1456. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Li, Y. 1,3-Cationic Alkylidene Migration of Nonclassical Carbocation: A Density Functional Theory Study on Gold(I)-Catalyzed Cycloisomerization of 1,5-Enynes Containing Cyclopropene Moiety. J. Am. Chem. Soc. 2014, 136, 1505–1513. [Google Scholar] [CrossRef]
- Herrero-Gómez, E.; Nieto-Oberhuber, C.; López, S.; Benet-Buchholz, J.; Echavarren, A.M. Cationic η1/η2-gold(I) complexes of simple arenes. Angew. Chem. Int. Ed. 2006, 45, 5455–5459. [Google Scholar] [CrossRef]
- Chifotides, H.T.; Dunbar, K.R. Anion-π Interactions in Supramolecular Architectures. Acc. Chem. Res. 2013, 46, 894–906. [Google Scholar] [CrossRef]
- Dougherty, D.A. The Cation-π Interaction. Acc. Chem. Res. 2013, 46, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Mahadevi, A.S.; Sastry, G.N. Cation-π interaction: Its role and relevance in chemistry, biology, and material science. Chem. Rev. 2013, 113, 2100–2138. [Google Scholar] [CrossRef]
- Weber, D.; Gagné, M.R. Dinuclear gold-silver resting states may explain silver effects in gold(I)-catalysis. Org. Lett. 2009, 11, 4962–4965. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Cai, R.; Sharma, S.; Jirak, J.; Thummanapelli, S.K.; Akhmedov, N.G.; Zhang, H.; Liu, X.; Petersen, J.L.; Shi, X. Silver effect in gold(I) catalysis: An overlooked important factor. J. Am. Chem. Soc. 2012, 134, 9012–9019. [Google Scholar] [CrossRef]
- Zhu, Y.; Day, C.S.; Zhang, L.; Hauser, K.J.; Jones, A.C. A Unique Au-Ag-Au Triangular Motif in a Trimetallic Halonium Dication: Silver Incorporation in a Gold(I) Catalyst. Chem. Eur. J. 2013, 19, 12264–12271. [Google Scholar] [CrossRef] [PubMed]
- Homs, A.; Escofet, I.; Echavarren, A.M. On the silver effect and the formation of chloride-bridged digold complexes. Org. Lett. 2013, 15, 5782–5785. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Lu, M.; Dong, B.; Chen, H.; Shi, X. Silver-Catalyzed Alkyne Activation: The Surprising Ligand Effect. Adv. Synth. Catal. 2014, 356, 692–696. [Google Scholar] [CrossRef]
- Fang, W.; Presset, M.; Guérinot, A.; Bour, C.; Bezzenine-Lafollée, S.; Gandon, V. Silver-Free Two-Component Approach in Gold Catalysis: Activation of [LAuCl] Complexes with Derivatives of Copper, Zinc, Indium, Bismuth, and Other Lewis Acids. Chem. Eur. J. 2014, 20, 5439–5446. [Google Scholar] [CrossRef] [PubMed]
- Zhdanko, A.; Maier, M.E. Explanation of Silver Effects in Gold(I)-Catalyzed Hydroalkoxylation of Alkynes. ACS Catal. 2015, 5, 5994–6004. [Google Scholar] [CrossRef]
- Xu, G.; Liu, K.; Dai, Z.; Sun, J. Gold/silver-catalyzed controllable regioselective vinylcarbene insertion into O-H bonds. Org. Biomol. Chem. 2017, 15, 2345–2348. [Google Scholar] [CrossRef]
- Veguillas, M.; Rosair, G.M.; Bebbington, M.W.P.; Lee, A.-L. Silver Effect in Regiodivergent Gold-Catalyzed Hydroaminations. ACS Catal. 2019, 9, 2552–2557. [Google Scholar] [CrossRef] [Green Version]
- Franchino, A.; Montesinos-Magraner, M.; Echavarren, A.M. Silver-Free Catalysis with Gold(I) Chloride Complexes. Bull. Chem. Soc. Jpn. 2021, 94, 1099–1117. [Google Scholar] [CrossRef]
- Kim, U.B.; Jung, D.J.; Jeon, H.J.; Rathwell, K.; Lee, S. Synergistic Dual Transition Metal Catalysis. Chem. Rev. 2020, 120, 13382–13433. [Google Scholar] [CrossRef]
- Bayler, A.; Bauer, A.; Schmidbaur, H. Synthesis and Structure of Binuclear Single-Bridged Bis[(phosphane)gold(I)]halogenonium Complexes. Chem. Ber. 1997, 130, 115–118. [Google Scholar] [CrossRef]
- Hamel, A.; Mitzel, N.W.; Schmidbaur, H. Metallophilicity: The dimerization of bis[(triphenylphosphine)gold(I)]chloronium cations. J. Am. Chem. Soc. 2001, 123, 5106–5107. [Google Scholar] [CrossRef] [PubMed]
- Schmidbaur, H.; Hamel, A.; Mitzel, N.W.; Schier, A.; Nogai, S. Cluster self-assembly of di[gold(I)]halonium cations. Proc. Natl. Acad. Sci. USA 2002, 99, 4916–4921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Prabhavathy, J.; Yip, J.H.K.; Koh, L.L.; Tan, G.K.; Vittal, J.J. First Examples of AuI-X-AgI Halonium Cations (X = Cl and Br). J. Am. Chem. Soc. 2003, 125, 8452–8453. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.G.; Rominger, F.; Straub, B.F. Isolated Silver Intermediate of Gold Precatalyst Activation. Eur. J. Inorg. Chem. 2012, 2012, 2863–2867. [Google Scholar] [CrossRef]
- Simonneau, A.; Jaroschik, F.; Lesage, D.; Karanik, M.; Guillot, R.; Malacria, M.; Tabet, J.-C.; Goddard, J.-P.; Fensterbank, L.; Gandon, V.; et al. Tracking gold acetylides in gold(I)-catalyzed cycloisomerization reactions of enynes. Chem. Sci. 2011, 2, 2417–2422. [Google Scholar] [CrossRef]
- Zhdanko, A.; Maier, M.E. Synthesis of gem-Diaurated Species from Alkynols. Chem. Eur. J. 2013, 19, 3932–3942. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.; Shcherbachenko, E.; Roithová, J. Investigation of Geminally Diaurated Arene Complexes in the Gas Phase. Organometallics 2015, 34, 3979–3987. [Google Scholar] [CrossRef]
- Linden, H.B. Liquid injection field desorption ionization: A new tool for soft ionization of samples including air sensitive catalysts and non-polar hydrocarbons. Eur. J. Mass Spectrom. 2004, 10, 459–468. [Google Scholar] [CrossRef]
- Gross, J.H.; Nieth, N.; Linden, H.B.; Blumbach, U.; Richter, F.J.; Tauchert, M.E.; Tompers, R.; Hofmann, P. Liquid injection field desorption/ionization of reactive transition metal complexes. Anal. Bioanal. Chem. 2006, 386, 52–58. [Google Scholar] [CrossRef] [PubMed]
- LaLonde, R.L.; Brenzovich, W.E.; Benitez, D.; Tkatchouk, E.; Kelley, K.; Goddard, W.A., III; Toste, F.D. Alkylgold complexes by the intramolecular aminoauration of unactivated alkenes. Chem. Sci. 2010, 1, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.A.; Ciancaleoni, G.; Zuccaccia, D.; Bistoni, G.; Belpassi, L.; Tarantelli, F.; Belanzoni, P. Strong Electron-Donating Ligands Accelerate the Protodeauration Step in Gold(I)-Catalyzed Reactions: A Quantitative Understanding of the Ligand Effect. Organometallics 2016, 35, 2275–2285. [Google Scholar] [CrossRef]
- Couce-Rios, A.; Lledós, A.; Fernández, I.; Ujaque, G. Origin of the Anti-Markovnikov Hydroamination of Alkenes Catalyzed by L-Au(I) Complexes: Coordination Mode Determines Regioselectivity. ACS Catal. 2019, 9, 848–858. [Google Scholar] [CrossRef]
- Gubler, J.; Radić, M.; Stöferle, Y.; Chen, P. 2-Aminoalkylgold Complexes: The Putative Intermediate in Au-Catalyzed Hydroamination of Alkenes Does Not Protodemetalate. Chem. Eur. J. 2022, 28, e202200332. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Y.; Guo, Z.; Dong, S.S.; Li, X.-H.; Che, C.-M. Highly Efficient and Diastereoselective Gold(I)-Catalyzed Synthesis of Tertiary Amines from Secondary Amines and Alkynes: Substrate Scope and Mechanistic Insights. Chem. Eur. J. 2011, 17, 12932–12945. [Google Scholar] [CrossRef]
- Katari, M.; Rao, M.N.; Rajaraman, G.; Ghosh, P. Computational Insight into a Gold(I) N-Heterocyclic Carbene Mediated Alkyne Hydroamination Reaction. Inorg. Chem. 2012, 51, 5593–5604. [Google Scholar] [CrossRef]
- Alvarado, E.; Badaj, A.C.; Larocque, T.G.; Lavoie, G.G. N-Heterocyclic Carbenes and Imidazole-2-thiones as Ligands for the Gold(I)-Catalysed Hydroamination of Phenylacetylene. Chem. Eur. J. 2012, 18, 12112–12121. [Google Scholar] [CrossRef]
- Brooner, R.E.M.; Windenhoefer, R.A. Cationic, two-coordinate gold π complexes. Angew. Chem. Int. Ed. 2013, 52, 11714–11724. [Google Scholar] [CrossRef]
- Kovács, G.; Ujaque, G.; Lledós, A. The Reaction Mechanism of the Hydroamination of Alkenes Catalyzed by Gold(I)-Phosphine: The Role of the Counterion and the N-Nucleophile Substituents in the Proton-Transfer Step. J. Am. Chem. Soc. 2008, 130, 853–864. [Google Scholar] [CrossRef]
- Appelhans, L.N.; Zuccaccia, D.; Kovacevic, A.; Chianese, A.R.; Miecznikowski, J.R.; Macchioni, A.; Clot, E.; Eisenstein, O.; Crabtree, R.H. An anion-dependent switch in selectivity results from a change of C-H activation mechanism in the reaction of an imidazolium salt with IrH5(PPh3)2. J. Am. Chem. Soc. 2005, 127, 16299–16311. [Google Scholar] [CrossRef]
- Davies, D.L.; Donald, S.M.A.; Macgregor, S.A. Computational study of the mechanism of cyclometalation by palladium acetate. J. Am. Chem. Soc. 2005, 127, 13754–13755. [Google Scholar] [CrossRef] [PubMed]
- García-Cuadrado, D.; Braga, A.A.C.; Maseras, F.; Echavarren, A.M. Proton abstraction mechanism for the palladium-catalyzed intramolecular arylation. J. Am. Chem. Soc. 2006, 128, 1066–1067. [Google Scholar] [CrossRef] [PubMed]
- Basallote, M.G.; Besora, M.; Castillo, C.E.; Fernández-Trujillo, M.J.; Lledós, A.; Maseras, F.; Máñez, M.A. Crucial role of anions on the deprotonation of the cationic dihydrogen complex trans-[FeH(η2-H2)(dppe)2]+. J. Am. Chem. Soc. 2007, 129, 6608–6618. [Google Scholar] [CrossRef] [PubMed]
- Mishra, H.; Enami, S.; Nielsen, R.J.; Hoffmann, M.R.; Goddard, W.A., III; Colussi, A.J. Anions dramatically enhance proton transfer through aqueous interfaces. Proc. Natl. Acad. Sci. USA 2012, 109, 10228–10232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munz, D.; Webster-Gardiner, M.; Fu, R.; Strassner, T.; Goddard, W.A., III; Gunnoe, T.B. Proton or Metal? The H/D Exchange of Arenes in Acidic Solvents. ACS Catal. 2015, 5, 769–775. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Shen, W.; Li, L.; Li, M. Gold(I)-Catalyzed Cycloaddition of 1-(1-Alkynyl)cyclopropyl Ketones with Nucleophiles To Yield Substituted Furans: A DFT Study. Organometallics 2009, 28, 3129–3139. [Google Scholar] [CrossRef]
- Krauter, C.M.; Hashmi, A.S.K.; Pernpointner, M. A New Insight into Gold(I)-Catalyzed Hydration of Alkynes: Proton Transfer. ChemCatChem 2010, 2, 1226–1230. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Robertson, C.C.; Wright, J.S.; Carrington, E.J.; Perutz, R.N.; Hunter, C.A.; Brammer, L. Hydrogen bonding vs. halogen bonding: The solvent decides. Chem. Sci. 2017, 8, 5392–5398. [Google Scholar] [CrossRef] [Green Version]
- Sutar, R.L.; Huber, S.M. Catalysis of Organic Reactions through Halogen Bonding. ACS Catal. 2019, 9, 9622–9639. [Google Scholar] [CrossRef]
- Perchloric Acid as Well as All Organic and Organometallic Perchlorate Salts Are Often Explosive and Are Thus Highly Dangerous. Available online: http://ehs.berkeley.edu/lessons-learned/lesson-learned-chemical-explosion-causes-eye-injury (accessed on 7 November 2022).
- TURBOMOLE V7.1 2016, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 7 November 2022).
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3869. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 1995, 240, 283–290. [Google Scholar] [CrossRef]
- Sierka, M.; Hogekamp, A.; Ahlrichs, R. Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys. 2003, 118, 9136–9148. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kamalakannan, S.; Prakash, M.; Chambaud, G.; Hochlaf, M. Adsorption of Hydro-phobic and Hydrophilic Ionic Liquids at the Au(111) Surface. ACS Omega 2018, 3, 18039–18051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.-J.; Lee, J.; Windus, T.L.; Thiel, P.A.; Evans, J.W. Sulfur-enhanced dynamics of coinage metal(111) surfaces: Step edges versus terraces as locations for metal-sulfur complex formation. Surf. Sci. 2018, 676, 2–8. [Google Scholar] [CrossRef]
- Pounder, A.; Tam, W.; Chen, L.D. The Mechanism and Origin of Enantioselectivity in the RhodiumCatalyzed Asymmetric Ring-Opening Reactions of Oxabicyclic Alkenes with Organoboronic Acids: A DFT Investigation. Organometallics 2021, 40, 1588–1597. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dixit, R.; Sharma, H.; Agbossou-Niedercorn, F.; Vanka, K.; Michon, C. Silver Dependent Enantiodivergent Gold(I) Catalysed Asymmetric Intramolecular Hydroamination of Alkenes: A Theoretical Study. Catalysts 2022, 12, 1392. https://doi.org/10.3390/catal12111392
Dixit R, Sharma H, Agbossou-Niedercorn F, Vanka K, Michon C. Silver Dependent Enantiodivergent Gold(I) Catalysed Asymmetric Intramolecular Hydroamination of Alkenes: A Theoretical Study. Catalysts. 2022; 12(11):1392. https://doi.org/10.3390/catal12111392
Chicago/Turabian StyleDixit, Ruchi, Himanshu Sharma, Francine Agbossou-Niedercorn, Kumar Vanka, and Christophe Michon. 2022. "Silver Dependent Enantiodivergent Gold(I) Catalysed Asymmetric Intramolecular Hydroamination of Alkenes: A Theoretical Study" Catalysts 12, no. 11: 1392. https://doi.org/10.3390/catal12111392