
Organizing Multi-Enzyme Systems into Programmable Materials for Biocatalysis


Department of Biochemistry, Molecular Biology & Biophysics, University of Minnesota, Saint Paul, MN 55108, USA
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(4), 409; https://doi.org/10.3390/catal11040409
Submission received: 5 March 2021
/
Revised: 19 March 2021
/
Accepted: 21 March 2021
/
Published: 24 March 2021
(This article belongs to the Special Issue Biocatalysis and Whole-Cell Biotransformation in Biomanufacturing)
Abstract
:Significant advances in enzyme discovery, protein and reaction engineering have transformed biocatalysis into a viable technology for the industrial scale manufacturing of chemicals. Multi-enzyme catalysis has emerged as a new frontier for the synthesis of complex chemicals. However, the in vitro operation of multiple enzymes simultaneously in one vessel poses challenges that require new strategies for increasing the operational performance of enzymatic cascade reactions. Chief among those strategies is enzyme co-immobilization. This review will explore how advances in synthetic biology and protein engineering have led to bioinspired co-localization strategies for the scaffolding and compartmentalization of enzymes. Emphasis will be placed on genetically encoded co-localization mechanisms as platforms for future autonomously self-organizing biocatalytic systems. Such genetically programmable systems could be produced by cell factories or emerging cell-free systems. Challenges and opportunities towards self-assembling, multifunctional biocatalytic materials will be discussed.
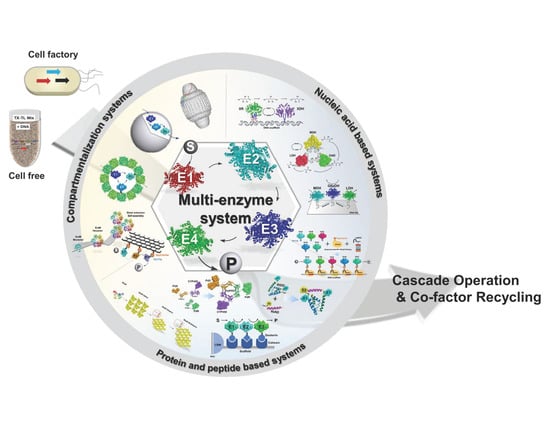
1. Introduction
Processes and products that take advantage of the catalytic activity and selectivity of enzymes and their mild reaction conditions have been developed since the beginning of the last century for a wide range of applications such as in the food, feed, detergent and textile industries. Yet, the broad use of biocatalytic processes for the industrial scale production of chemicals has, until about twenty years ago, been lacking from this list of applications. Over the past two decades, impressive advances have been made in enzyme discovery, protein and reaction engineering which have propelled the field of biocatalysis to become a viable alternative to synthetic chemistry for the industrial manufacturing of chemicals. Discovery and engineering pipelines created not only enzymes that catalyze new desired reactions but also biocatalysts able to meet critical industrial performance metrics such as space time yields, selectivity, and catalyst costs. The development of biocatalysis into a viable technology for scalable chemical synthesis has been discussed in detail in several excellent reviews dating back from about ten years ago until very recently [1,2,3,4,5]. In this review, we will look at opportunities for the design of next generation biocatalytic systems that will take advantage of current developments in synthetic biology and protein engineering. The design and operation of complex enzymatic cascade reactions is increasingly inspiring the development of new immobilization strategies borrowed from biological systems. Expanding upon these strategies and leveraging advances in cell-free expression systems has the potential of creating self-organizing systems for biocatalytic materials that in the future could be autonomously produced from DNA parts.
In parallel with biocatalyst discovery and engineering, enzymes have been combined in vitro into cascade reactions with the goal of designing sustainable synthetic routes that can replace existing chemical processes or create pathways to chemicals that are challenging to make synthetically. A plethora of cascade reactions have been designed where multiple enzyme catalysts operate simultaneously or sequentially in different configurations. From a cost perspective, one-pot systems where steps can be performed simultaneously are preferable due to reduced operational costs (eliminating intermediate isolation) as well as increased yields and selectivity because reactions can be driven to the final product. Progress in the design of biocatalytic cascade reactions has been covered in several comprehensive reviews [6,7,8,9,10,11,12]. Cell-free biocatalytic systems also offer opportunities for the integration of chemo-, photo- or electrocatalytic steps to access new chemistries and/or implement strategies for overcoming challenges and costs associated with co-factor use and recycling required for steps involving oxidoreductases [13,14].
The need to reduce operational costs of complex cascade reactions for industrial use has driven efforts aimed at increasing the operational performance of biocatalysts and overcoming costs associated with expensive co-factors [15]. Enzyme immobilization is a common strategy for improving the economics of a biocatalytic process by increasing enzyme stability, performance and allowing for biocatalyst reuse and easier product recovery [16,17,18,19,20]. While traditional methods of absorption or attachment to matrices or surfaces work well for the immobilization of one or two enzymes, the co-immobilization of multiple cascade enzymes is challenging. This is because it is often not possible to identify immobilization conditions that support the stability and activity of all involved catalysts while also benefitting the overall reaction efficiency of the cascade reaction. Biological systems, however, have solved this challenge by operating highly orchestrated enzyme cascades that are spatially organized as part of complex metabolic networks. In cells, the localization and clustering of enzymes in proximity with each other prevent escape and/or build-up of intermediates that could be unstable, inhibitory or transformed into unwanted side products. Sequestration, encapsulation, protein–protein interactions, and surface attachments increase enzyme stability and activity, prevent degradation and increase reaction efficiency by creating local co-factor and substrate pools. Inspired by these mechanisms, new types of co-immobilization strategies are therefore increasingly developed that emphasize spatial organization via diverse scaffolding and compartmentalization approaches [18,21,22,23,24,25,26,27,28,29].
We envision that current strategies of the scaffolding and compartmentalization of multi-enzyme systems could be expanded upon for the generation of transformational hierarchical biocatalytic materials that are genetically encoded for inexpensive fabrication by cell factories or, in the future, cell-free systems. Self-organization at the nano- to macro scale is a key characteristic of all biological systems that is genetically encoded. Adopting the principles and mechanisms of molecular self-organization could be used for the bottom–up design and low-cost fabrication of new types of biocatalytic materials. Advances in protein engineering and synthetic biology could ultimately lead to autonomous self-organizing cell-free biocatalytic reaction systems that operate cascade reactions with a similar complexity compared to engineered cellular biosynthetic pathways. Cell-free expression systems have already been developed for a range of applications, including for the proto-typing of metabolic pathways, biomanufacturing and biosynthesis [30,31,32,33,34,35,36]. By extending these approaches to cascade biocatalysis, genetically programmable cell-free systems could be created where the transcription–translation machinery obtained from, e.g., a cell lysate drives the production and self-organization of biocatalytic systems from supplied DNA parts. Although the development of such systems is currently far from realistic due to the lack of scalability of cell-free expression systems for industrial use, we will review the state-of-the art enzyme co-localization systems that are genetically encoded and could serve as platforms for the design of genetically programmable biocatalytic materials. We will discuss challenges and opportunities for the development of such types of material for future biocatalytic systems.
2. Systems Platforms for Multi-Enzyme Self-Organization
Nucleic acid and protein or peptide-based systems can be genetically encoded and are therefore suitable for programming the bottom–up design of enzyme assemblies. The self-organization of biocatalysts can be achieved by genetically fusing interaction domains or peptides (referred to as “tags”) to the proteins. In the simplest case, enzymes can be cross-linked via high-affinity interaction tags placed at one or both termini of the proteins. Variation of the length and flexibility of the linker between the interaction tags, the type of interaction tags as well as the incorporation of different interaction pairs affords some control over the spatial organization and properties of the resulting multi-enzyme assembly. More control over biocatalyst organization along with the ability to incorporate additional functionalities is possible by immobilizing enzymes on self-assembling protein or nucleic acid scaffolds. This can be achieved either by direct genetic fusion of the scaffold building blocks to the enzymes or fusion of cognate interaction tags to enzymes and scaffold building blocks. Direct genetic fusion of enzymes to scaffold building blocks though will only work if the attached enzyme does not interfere with scaffold assembly. A more versatile strategy is the use of interaction tags fused to scaffold building blocks as they will less likely interfere with scaffold formation while also creating a highly flexible platform for enzyme immobilization. With such a scaffolding system, the organization of enzymes can be controlled by the spacing, orientation, types, and linkers of the displayed tags. Finally, enzymes can be co-localized by encapsulation in a proteinaceous compartment through designed targeting mechanisms. For future cell-free applications, the encapsulation of enzymes into lipid vesicles or polymersomes offers an interesting path forward for the creation of artificial cell-like systems for biocatalysis that could be genetically programmed. In the following sections, we will introduce these different systems and present relevant examples for enzyme immobilization, summarized in Table 1. We will move from nucleic acid-based systems to protein/peptide-based systems for enzyme cross-linking and scaffolding, followed by compartmentalization systems. Emphasis will be placed on in vitro applications with a few exceptions to discuss a particular aspect of a system or where in vitro examples are currently lacking.
3. Nucleic Acid Based Scaffolds
The Watson–Crick base pairing of DNA makes it naturally a highly engineerable material for the predictable design of nanoscale structures [56,57,58,59]. Over a decade ago, a technique dubbed DNA origami was used for the design of patterned DNA nanomaterials [60] and has since then been applied for the design of a multitude of higher order structures [61,62]. Many of these materials were designed for biomedical applications [63,64,65,66,67] but it was soon realized that DNA scaffolds could also be used for the spatial organization of proteins to create programmable, functional materials [68,69]. The precision with which the bottom–up assembly of DNA can be controlled together with the ability to position proteins on DNA made these DNA scaffolds an ideal platform for enzyme co-immobilization [70,71]. For example, DNA origami was used to immobilize enzymes onto hexagonal strips [72] or tiles of DNA [73]. In another examples, a co-factor was tethered to DNA between two proximally positioned enzymes to control co-factor channeling in a dual enzyme cascade [74,75]. The proximity control afforded by DNA origami has therefore been used to spatially organize multi-enzyme pathways with the goal of investigating the effect of distances and orientations of enzymes on catalysis [38,76]. Decreasing the distance between glucose oxidase (GOx) and horseradish peroxidase (HDP) to 10 nm increased the activity of the system up to 10-fold compared to the free enzyme system or when the enzymes were placed more than 20 nm apart [76]. Likewise, the scaffolding of xylose reductase (XR) and xylitol dehydrogenase (XDH) in close proximity to each other increased the catalytic efficiency of this NAD-co-factor coupled cascade for the conversion of xylose to xylulose [37]. A triangular DNA origami shape was designed for the assembly of malic dehydrogenase (MDH), oxaloacetate decarboxylase (OAD), and lactate dehydrogenase (LDH). In this example, clustering of the enzymes due to geometry of the assembly was more important for the overall activity of the cascade than the interenzyme distance [38,75] (Figure 1).
Although the term “substrate channeling” is often suggested to be responsible for the observed enhanced catalytic efficiencies of co-localized enzymes, this mechanism, however, is unlikely to be at play for engineered systems. Enzymes in the native metabolic enzyme cascade have co-evolved and enzymes in known metabolomes have evolved specific mechanisms that allow for the passage of molecules directly from one active site to the other [77]. The observed increased activities of engineered scaffolded systems, such those on DNA scaffolds, could be the result of the scaffold surface or other yet to be rigorously characterized factors [78]. Regardless of the exact mechanism, the above examples demonstrate that DNA scaffolding is a powerful system for the programmable organization of complex enzyme cascades with enhanced activities. However, there are several major drawbacks for the use of DNA in future biocatalytic systems, including limited robustness in cell-free reaction environments that will contain nucleases, the need to chemically modify enzymes for attachments (although this can be overcome by fusions with DNA binding domains) and the limited variability and engineerability of scaffold surface properties compared to proteins or peptide-based materials. Cost-efficient, large-scale production in the cell-free expression system may also be difficult.
4. Protein and Peptide-Based Systems for Enzyme Co-Localization
Compared to nucleic acids, peptide and protein building blocks offer by far the greatest versatility for the design of programmable functional materials because of their infinite chemical (sequence) and structural variability. They can also be readily produced (either as stand-alone building blocks or translationally fused to enzymes) by microbial cell factories or in the future, directly from DNA parts in cell-free systems. Furthermore, a significant and fast-growing knowledge base of natural and designed self-assembling and interacting proteins, protein domains and peptides can be leveraged for the design of self-organizing mechanisms and platforms for the fabrication of biocatalytic materials [79,80,81,82,83,84,85]. Although synthetic peptides as well as elastin-, collagen-, silk- or amyloid-like polypeptides and proteins have long been used for the creation various hydrogels, meshes and other biomaterials, these materials have mostly been designed for biomedical applications but not for biocatalysis [86,87,88,89]. Instead, peptide or protein domains, self-assembling helices or coiled-coils have been designed as fusion tags or scaffolds for enzyme assembly and immobilization. In these systems, self-assembly is mostly driven by non-covalent interactions, but a few mechanisms have been designed for high-affinity, covalent peptide conjugation.
4.1. Co-Localization Using Non-Covalent Interaction Domains and Tags
Numerous protein interaction domains exist in nature that could be used for the co-localization and scaffolding of enzymes if they interact with strong affinity and can be engineered for use as fusion tags [90] (Figure 2). One example of a designed, strong affinity peptide pairs are the 18 residue RIAD and 44 residue RIDD peptides derived from the dock and -lock peptide interacting family. The fusion of these tags to three menaquinone biosynthetic enzymes (MenF, D and H) enabled their hierarchical in vitro assembly into multi-enzyme complexes with increased catalytic efficiencies. The same approach was also validated for in vivo use with a three-enzyme complex for carotenoid production in Saccharomyces cerevisiae [39]. Other well-known examples of interacting domains are the PDZ, SH3 and GBD domains, which were used in a seminal paper by John Dueber for the scaffolding of three enzymes of the mevalonate biosynthetic pathway in Escherichia coli (E. coli), resulting in a 77-fold increase in metabolic flux [43]. Instead of arranging biosynthetic enzymes on a scaffold, itaconic acid production in E. coli was two- or four-fold improve by co-assembling two- or three enzymes into nanometer sized complexes through the direct genetic fusion of PDZ and SH3–protein–peptide interaction domains to the termini of the biocatalysts [40,41]. In another elegant example that takes advantage of the quaternary structures of enzymes for assembly, the PDZ domain was used to tether a dimeric formate dehydrogenase (FDH) (for co-factor recycling) to an octameric leucine dehydrogenase (LeuDH). The two multimer enzymes self-assembled into extended supramolecular structures for the production of l-tert-leucine in vitro and in E. coli. Compared to the free enzyme system, the biocatalytic structures had higher stability and three-fold increased activity [42].
One of the best known naturally occurring modular protein scaffolding system, however, are the cellulosomes from anaerobic cellulolytic bacteria such as Clostridium thermocellum. Different enzymes involved in cellulose breakdown along with carbohydrate binding domains are organized on an extracellular scaffolding structure (scaffoldin) via cohesin–dockerin interaction domains. The scaffoldin itself is also attached to the cell surface via a cohesin–dockerin domains. The modular nature of the cellulosome inspired the engineering of various designer cellulosomes as well as new multi-enzyme complexes displayed on cell surfaces [91,92]. For in vitro applications, a mini-scaffoldin system was designed for the co-localization of a three-enzyme systems composed of triosephosphate isomerase (TIM), aldolase (ALD), and fructose 1,6-biphosphatase (FBP) which yielded a 33-fold higher catalytic efficiency than the free enzymes [93,94]. In another application, cohesin–dockerin mediated scaffolding of four enzymes onto a chaperonin as a synthetic cellulosome (dubbed rosettazyme [95]) significantly increased cell free production of the platform chemical d-xylonic acid by more than 71% compared to the free enzyme system [44].
As shown with the synthetic cellulosome example described above, in principle any interacting protein domains or multimeric proteins can be engineered as a scaffolding system as long as (i) the domain/protein interactions are sufficiently strong, (ii) genetic protein fusions are tolerated without interrupting assembly and (iii) the resulting fusion proteins can be readily expressed recombinantly. An example of such a scaffolding system derived from a naturally occurring multimeric protein is the heterotrimeric, ring-shaped proliferating cellular nuclear antigen (PCNA) complex [96,97]. The PCNA complex provides a circular scaffold with C-termini exposed to one side. This scaffold was used to co-localize a cytochrome P450 (CYP450) together with its two redox partners ferredoxin (PdX), and ferredoxin reductases (PdR) for more efficient electron transfer [98]. This system was then expanded to incorporate a homodimeric phosphite dehydrogenase for NADPH co-factor regeneration. The fusion of this homodimeric enzyme to the N-termini of the PCNA complex subunits facilitates the assembly of supramolecular structures [99].
4.2. Co-Localization Using Covalent Conjugation Mechanisms
The SpyTag/SpyCatcher system and its derivatives have become the most versatile and widely used mechanism for covalent ligation of proteins for diverse applications. [100,101,102,103]. Howarth’s group developed this technology by designing a split version of the Streptococcus pyogene fibronectin-binding adhesin by separating an intramolecular isopeptide bond formed between a reactive Lys and Asp residues [104]. The resulting split system consists of a short 13 amino acid SpyTag containing the reactive Asp and a 116 amino acid SpyCatcher domain that contains the Lys residue. Upon interactions, the two polypeptides spontaneously link together by forming an intermolecular isopeptide bond. Bond formation occurs within minutes with high affinity under a wide range of temperatures (4 to 37 °C), pH values (pH 5–8) and buffer conditions. The original split system was subsequently engineered to create versions with faster reactivity and for protein complex formation at low protein concentration [105,106]. In addition, an orthogonal SnoopTag/SnoopCatcher system was derived from the Streptococcus pneumoniae adhesion protein, which when combined with the SpyTag/SpyCatcher system facilitated the directed assembly of polyprotein complexes [107]. Most recently, a ligation incompetent SpyDock domain was engineered for reversible binding which was used for the purification of SpyTag-proteins [108]. In this work, the authors also fused the SpyTag to self-assembling helical peptides to facilitate the oligomerization of SpyCatcher fused proteins.
The combination of high affinity protein ligation with the predictable self-assembling properties of designed alpha-helices [109] is a powerful strategy for the higher-order assembly of biocatalytic materials. Another benefit of the Tag/Catcher system is the small size of the SpyTag which allows it to be not only placed at the termini of a protein, but also displayed at other surface accessible regions of proteins. The SpyCatcher domain can be readily fused to the termini of proteins with different linkers such as the frequently used flexible and protease resistant Gly/Ser linkers [110]. A complete list of greater than 800 variations and hundreds of publications of the Tag/Catcher systems is compiled by the Howard laboratory in a SpyBank described in [106].
The high-affinity Tag/Catcher system was quickly adopted for the immobilization and co-localization of biocatalysts. Enzyme complexes can be readily formed in vitro or in vivo by genetically fusing SpyTag and/or SpyCatcher to the terminal residues of enzymes. For example, SpyTag/Catcher tagging of maltooligosyl trehalose synthase (MTS) and maltooligosyl trehalose trehalohydrolase (MTH) created a two-enzyme complex for faster in vitro trehalose conversion than the untagged enzyme system [45]. In another example, instead of assembling leucine dehydrogenase (LeuDH) and formate dehydrogenase (FDH) [42] into supramolecular structures using the PDZ interacting protein–peptide system described above, the SpyTag/SpyCatcher system was combined with elastin-like peptide (ELP) fusions for enzyme co-assembly and temperature-dependent reversible hydrogel formation for purification [48]. The extension of the SpyCatcher domain with an ELP fusion allowed for the easy purification of the protein system. The assembled enzymes showed up to 32-fold higher conversion rates compared to the free enzyme system [48].
The SpyTag/Catcher system is equally useful for in vivo assembly of enzymes, as was shown for linking three enzymes of the mevalonate pathway together with Spy/SnoopTag/Catcher fusions. The combined enzymes increased carotenoid production rates in engineered E. coli up to five-fold [49]. The system has also been applied in combination with other high-affinity tag systems—the well-known streptavidin system and the Halo-tag system for the in vitro self-immobilization of a three-enzyme system on microbeads. Immobilized (R)-selective alcohol dehydrogenase (ADH) and (S)-selective methylglyoxal reductase (Gre2P) catalyzed the enantioselective reduction of the prochiral 5-nitrononane-2,8-dione while NADPH co-factor recycling was performed with glucose 1-dehydrogenase (GDH) [47]. Cascade reactions were run for 14 days in a microfluidic reactor system with high conversion yields (>95%) and stereoselectivity (d.r. > 99%).
In an interesting approach, nanometric spacing of the five enzymes of the violacein biosynthetic pathway (VioA–E) on a DNA scaffold was recently achieved by conjugated SpyTagged Vio enzymes to SpyCatcher fused dCas9 (dead Cas9) (Figure 3). Each purified enzyme-dCas9 complex was then complexed with a guide RNA (sgRNA) for binding to five targeting sequences on a DNA scaffold. The best scaffolded system was three times more efficient than the free system. Interestingly, scaffolding of the enzymes in the vicinity of each other rather than in order was important for the increased efficiency [50].
Another system available for protein ligation that does not require the chemical modification of proteins is the bacterial transpeptidase sortase. This enzyme attaches secreted surface protein to the bacterial cell wall envelop [111,112]. It recognizes a C-terminal sorting motif (Leu-Pro-X-Thr-Gly, LPXTG, where X = any amino acid) and cleaves the TG (Thr-Gly in the sorting motif)-peptide and transfers the peptide via an acyl–enzyme intermediate to the N-terminal amine group of an oligo–glycine modified acceptor protein. Engineered variants of sortase A from Staphylococcus aureus with improved activity and orthogonal sorting motif recognition have been used for a wide range of ligation activities [113,114,115], including for hydrogel cross-linking [116], protein conjugation on a cyclodextrin scaffold [117], and co-immobilization of a glucose oxidase (GOx) and horseradish peroxidase (HRP) [118]. Recently, sortase-mediated ligation was used to co-localize two cellulases on the surface of a designed protein cage to create a cellulolytic cage with enhanced cellulose degradation activity compared to the free enzyme system [119]. Compared to the autocatalytic ligation of the Tag/Catcher system, the sortase ligation approach requires an additional enzyme and the N- and C-terminal placement of tags, which makes a less versatile system. However, it does offer more control over protein ligation through the enzymatic step involved.
4.3. Self-Assembling Protein Arrays and Nanostructures as Scaffolds
Higher-order scaffolds and nanostructures assembled from self-associating protein building blocks could deliver exceptional platforms for the design of programmable and hierarchical biocatalytic materials. The self-association of hundreds to thousands of building blocks into highly robust protein assemblies would create large surface areas that could be genetically engineered for functionalization and autonomous organization into hierarchical structures with properties of choice.
Native or designed self-assembling building blocks could seed the design of such materials. The proteinaceous shells of viruses, for example, represent such highly ordered and self-organized nanostructures whose assembly has been studied extensively [120,121]. Other examples of natural nanostructures are the protein S-layers (cell envelope surface layers) made by many prokaryotes [122,123] and the different types of proteinaceous nano- and microcompartments made by prokaryotic and eukaryotic cells [124,125,126,127,128,129] discussed in the next section. In addition, impressive progress has been made in the design and construction of self-assembling protein nanostructures that can form the foundation of new types of materials for biocatalysis [83,109,130,131,132,133,134,135]. Recent publications demonstrate the design of diverse protein nanostructures including cages, layers, crystals, and filaments [136,137,138,139,140,141], the engineering of a preexisting cage into various structures [142,143,144,145], the assembly of computationally designed icosahedral nanostructures [137,138], 2D-arrays, crystals [146,147], and protein filaments [81], as well as the design of metal-coordination, disulfide bridges, and surface electrostatics for the creation of functional 2D-lattices and cages [146,148,149,150,151,152].
Yet, despite the diversity of available protein nanostructures, only a limited number of arrays and scaffolds have so far been explored for in vitro biocatalysis and specifically, enzyme co-localization. For example, the ubiquitous paracrystalline S-layers made by prokaryotes seem to make an excellent platform for the design of biocatalytic materials. S-layers have been used as nanomaterial for various applications and biofunctionalized with different proteins, including enzymes, either by chemical modification or by genetic engineering [122]. However, so far, these scaffolds have not yet been explored for the co-localization of enzyme cascades for in vitro biocatalysis, which may be due to their low sequence conservation, complex, and varied assembly properties that make engineering more challenging. In recent work though, the S-layer protein SbsB from Geobacilllus stearothermophilus was engineered to display SnoopTag and SpyTag peptides for conjugation with Spy/SnoopCatcher fused fluorescent proteins. The incorporation of bifunctional Catcher protein constructs as cross-linkers then enabled self-assembly into 3D-stacks, suggesting that S-layers could be developed into programmable biocatalytic materials [153] (Figure 4).
Our group has taken advantage of the robust self-assembling properties of the bacterial microcompartment shell (BMC, see next section) protein EutM identified during work on engineering the ethanolamine BMC of Salmonella enterica [154,155,156]. The EutM shell protein monomer rapidly self-assembles into highly robust hexameric arrays (Figure 4). Important for material development, this protein can be readily recombinantly produced, isolated, and functionalized to display genetic fusions at either side of the protein arrays. We have used this scaffolding system for the co-localization of a self-sufficient, dual enzyme cascade composed of a NAD+-dependent alcohol dehydrogenase (ADH) and a NADH-dependent amine dehydrogenase (AmDH) for chiral amine production [46]. The SpyTag/Catcher system was used for enzyme attachment. By varying the molar ratio of biocatalyst to scaffold building blocks, the spacing of the catalyst was optimized and the final system reached >90% conversion twice as fast as the free enzyme system. Subsequently, we have expanded this system by building a scaffold toolbox from EutM homologs to create scaffolds with different surface electrostatics, architectures, and hybrid compositions that can be tailored towards the individual requirement of a biocatalyst or system [157,158,159].
5. Compartmentalization Systems
The co-localization of enzyme systems into protein or lipid compartments is another strategy to create high catalyst densities for increased pathway flux, to sequester toxic intermediates in cellular systems and to provide dedicated co-factor pools [160,161]. Only protein compartments, though, can be engineered and programmed for autonomous assembly and material fabrication. In addition, pores in their shells allow for the exchange of molecules and can be engineered for altered selectivity. Membrane-based systems, however, will be useful for the design of future artificial biocatalytic cell-like systems when combined with cell-free expression systems. They would need to be optimized for operational robustness and the efficient transport of molecules across their membranes. Both types of compartments have been used for enzyme catalysis and examples for in vitro applications and will be discussed next.
5.1. Encapsulation in Proteinaceous Compartments, Cages and Virus-Like Particles
Diverse biological and engineered protein compartments and cages have been repurposed for biomedical applications, as composite nanomaterials, for bioremediation and biosensing and to a much lesser extent, for in vitro enzyme catalysis and as in vivo nanoreactors [124,162,163,164]. Biological compartments large enough for multi-enzyme encapsulation include the eukaryotic major vault particles, bacterial microcompartments (~50–200 nm), encapsulins (~20–40 nm), and lumazine synthase (~15 nm) (Figure 5). Cargo proteins can be encapsulated into these compartments either with native encapsulation peptides identified on native cargo or scaffolding proteins that interact with the luminal face of shell proteins, or through engineered mechanisms such as the SpyTag/Catcher system, coiled-coil peptide interactions, fusion to shell or scaffolding proteins or designed charge complementary interactions (reviewed in [124]).
Vault particles are an intriguing class of very large compartments that are ubiquitously found in most eukaryotic organisms. Their function is still unknown, but they can be recombinantly produced (mostly in insect cells) and have been engineered for cargo protein encapsulation and surface display. Vaults have mostly been used for biomedical applications but have not yet been explored for biocatalysis. They do not have pores but the joint where the two halves of the vault meet opens and closes dynamically for cargo and molecule exchange [124]. Bacterial microcompartments are present in a large number of bacteria. They are known to sequester catabolic pathways that generate a reactive aldehyde intermediate, or encapsulate RuBisCO (ribulose-1,5-bisphosphate carboxylase) and carbonic anhydrase for CO2 fixation [127,128,165]. The engineering of these compartments has also focused mostly on in vivo applications due to their requirement for in vivo co-assembly of shells and cargo protein. Recent advances in structural characterizations and protein engineering, however, has led to the design of BMC-like protein cages [166], engineered shells for controlled in vitro assembly [167], and opening a path for in vitro applications. The smaller encapsulin compartments can be readily assembled in vitro and they have been developed into various nanomaterials and for in vivo biosynthesis [126], but have also not received much attention for in vitro biocatalysis application. Their simpler homomeric organization compared to the large vaults and heteromeric microcompartments and the fact that they can be more readily assembled in vitro would make them good platforms for enzyme encapsulation. Most recently, different biocatalysts including a heme-containing catalase, flavin adenine dinucleotide (FAD)-containing Bayer–Villiger monooxygenase (CHMO) and carbohydrate oxidase (mChito) and a metal-containing peroxidase (SviDyP) were encapsulated in these shells. Except for CHMO, the other enzymes were functional and substrates up to a certain molecular weight were able to pass through the pores. However, NADPH required for CHMO was too large for the pores [168]. This can be remedied by enlarging the pore diameter, which was carried out for the prototypical encapsulin from Thermotoga maritima [169]. Even smaller compartments are formed by the lumazine synthase from Aquifex aeolicus, which self-assembles into small capsids that readily form in vitro and are highly engineerable. Hilvert’s group has extensively engineered these cages for a variety of applications [142,144,145,170]. For example, they designed an artificial carboxysome by engineering the co-encapsulation or RuBisCO and carbonic anhydrase and although co-localization of the two enzymes did not increase overall enzyme activity, it protected the catalytic system from proteolytic inactivation [143].
Another frequently engineered group of compartments for biomedical and biotechnological applications are virus-like particles (VLPs) that self-assemble in vitro from one or two different structural capsid proteins (e.g., from the P22 or MS2 bacteriophages, hepatitis B virus (HBV) or Qβ Levivirus) [162]. P22 VLP’s are a versatile system that assembles from hundreds of coat protein and a correspondingly, slightly lower number of scaffolding protein copies into a highly porous shell. Genetic fusion of cargo proteins to a truncated version of the scaffolding protein is used to direct their encapsulation into the capsid lumen. In a recent example, two enzymes for isobutanol synthesis from α-ketoisovalerate were separately encapsulated into P22 VLPs [171]. The exterior surface display of a negatively charged peptide tag was then used to co-assemble the two catalytic VLPs into a supramolecular cage lattice in the presence of polyamidoamine dendrimers. Co-localization of the two nano-reactors in this lattice and the subsequent compacting of the matrix led to significantly enhanced isobutanol production compared to the uncondensed VLP system. The only example so far for the in vitro co-localization of a multi-enzyme system into VLPs is that of a translationally fused construct of a beta-glucosidase (CelB), ATP-dependent galactokinase (GALK) and glucokinase (GLUK) encapsulated in P22 VLPs. However, co-localization had no effect on the catalytic efficiency of the enzyme system [51]. The in vivo co-localization of a two-enzyme indigo pathway in SpyTag modified viral capsids and the subsequent purification of the enzyme-loaded capsids for in vitro characterization also did not show any increased activity compared to the free system. However, encapsulation greatly enhanced the storage stability of the enzymes [172].
From the above examples, it is not apparent that the co-encapsulation of non-native cargo enzymes into compartments has an effect on the reaction efficiency of multi-enzyme systems. Encapsulation does benefit the enzyme’s stability and protect it from degradation. However, especially in the case for VLPs, high catalyst densities may be required to achieve similar effects on conversion rates seen with scaffolded enzyme systems. One issue of VLPs and other compartmentalized systems is that the cargo proteins often remain attached to the shell interiors due to the encapsulation mechanisms used, limiting dense cargo packaging. To alleviate this issue, a calcium-dependent cargo release system was recently engineered for HBV VLPs [173]. Other controllable protein affinity interactions, e.g., the above mentioned SpyTag/SpyDock system, could be similarly adopted for this purpose.
5.2. Encapsulation in Lipid Vesicles and Polymersomes
The encapsulation of proteins into membrane vesicles is a relatively straightforward approach for enzyme co-localization. However, control over encapsulation and vesicle formation can be difficult. Liposome-based systems are also mechanically less stable compared to other heterogenous systems for enzyme co-localization. More robust compartments can be fabricated from amphiphilic block copolymers [174,175]. These synthetic polymersomes lack the intrinsic membrane permeability of lipid vesicles, and permeability and other membrane functionalities of biological vesicles must therefore be achieved by integrating different membrane pores and channels. Polymersomes have been successfully used as permeable nanobioreactors with encapsulated enzymes [176,177,178] and for multi-enzymatic reactions [53,54,55]. For example, by taking advantage of the different environments present in a polymersome system, a three-enzyme system was spatially separated to increase compatibility by localizing them to the membrane (Candida antarctica lipase B, CalB), into the lumen (glucose oxidase, GOx), and on the exterior surface (horseradish peroxidase, HRP) for the efficient conversion of glucose acetate to gluconolactone [53]. A different approach for separating incompatible steps was pursued by creating separate compartments within polymersomes for each enzyme of a three-enzyme cascade (composed of phenylacetone monooxygenase (PAMO), CalB, and alcohol dehydrogenase (ADH) for the conversion of 7-((4-oxopentyl)oxy)-3H-phenoxazin-3-one to resorufin [54]. A more complex enzyme system that separates incompatible reaction steps and facilitates selective mass transfer across polymersome membranes via a channel pore, was developed for a three-step enzymatic reaction that convert N-acyl-d-glucosamine (GlgNAc) to CMP-N-acetylneuraminate (CMP-Neu5Ac) [55]. Here, the first reaction step is localized to the lumen to prevent inhibition of the N-acyl-d-glucosamine 2-epimerase (AGE) by cytidine triphosphate (CTP) which is required for the last reaction step catalyzed by CMP-sialic acid synthetase (CSS). CSS, together with the second enzyme of the cascade, N-acetylneuraminate lyase (NAL), are attached to the exterior polymersome surface [55].
Controlling enzyme cargo encapsulation in membrane vesicles can be challenging without the natural or engineered targeting mechanisms of protein compartments. This can, however, be achieved by the genetic fusion of high-affinity binding to proteins embedded in the vesicle membrane. For example, Silver’s group repurposed a membrane protein of the ϕ6 bacteriophage for controlled cargo-loading and lipid scaffold generation in E. coli [52]. The genetic fusion of two enzymes for indigo production to the C-terminus of the P9 membrane protein facilitated cargo sequestration into synthetic lipid containing scaffolds in E. coli, resulting in enhanced indigo production. While this system has not been designed for in vitro use, the membrane protein fusion construct could be useful for directing cargo encapsulation into membrane vesicles in vitro. Efficient packaging of a phosphotriesterase (PTE) into outer membrane vesicles was demonstrated with a SpyTag fusion at the luminal side of an engineered OmpA porin protein and a SpyCatcher fusion to the PTE cargo protein [179]. Together, the above examples illustrate the versatility of polymersomes for the design of biocatalytic systems, which could be incorporated into future cell-free systems with protein-based components (enzymes and scaffolds) synthesized from DNA.
6. Conclusions and Future Directions
Ten years ago, we reviewed the emerging field of multi-enzyme biocatalysis [180]. We suggested that new nanoscale enzyme co-localization strategies, including genetically engineered scaffolding and confinement systems, will be important for increasing reaction rates and efficiencies of longer enzyme cascades. Eight years later, we concluded that the spatial organization of multi-enzyme systems until recently had long been ignored by the field in industrial biocatalysis due to prioritizing the optimization of enzyme properties [27]. Rapid progress in synthetic biology, de novo protein design and cell-free approaches now have created a robust foundation for the design of genetically engineered enzyme assemblies for integration and upscaling in industrial processes. In this review, we have focused primarily on genetically programmable systems for cell-free biocatalysis. Although several new scaffolds, compartments and cages for the nano-scale co-localization and confinement of enzymes have been designed over the past few years, relatively few of those systems have yet been applied for in vitro multi-step enzyme catalysis. Given the current push for fundamental and applied research aimed at the design and engineering of genetically encoded biomaterials in the US, we anticipate that these efforts will also accelerate the design of new types of programmable biocatalytic biomaterials and future biocatalytic processes envisioned in Figure 6.
Controlling protein self-assembly across multiple length scales into bulk materials that can be produced at scale for industrial application is a major challenge. The bottom–up design of such materials must encompass for the ability to integrate multiple functions (enzyme immobilization, stabilization, co-factor recycling, surface binding, etc.) along with required mechanical properties (porosity, compressive strength, etc.). In addition, materials must maintain their integrity and function for a prolonged time under operational conditions. Nature has already solved many of these issues by producing highly robust, self-assembling protein-based materials and scaffolds, including biomineralized composite materials with exceptional mechanical properties [181]. Elucidating and adopting the design principles of these biological materials for the fabrication of hierarchical, multifunctional materials has great potential for the development of future cell-free biomanufacturing platforms. Advances in protein design already provide many building blocks and tools for the design of such materials. The development of new biomaterials design workflows that incorporate a predictive computational design and streamlined prototyping of material properties will be important to further accelerate functional biomaterial design.
Author Contributions
M.-J.S. and C.S.-D. wrote this review article. All authors have read and agreed to the published version of the manuscript.
Funding
Research on the development of self-assembling protein materials in the Schmidt-Dannert Laboratory has been supported by Defense Threat Reduction Agency Grant HDTRA-15-0004, and is currently supported by: Defense Advanced Research Projects Agency Contract HR0011-17-0038, National Science Foundation CBET-1916030, MnDRIVE and the University of Minnesota’s Biocatalysis Initiative through the BioTechnology Institute.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. Engl. 2021, 60, 88–119. [Google Scholar] [CrossRef]
- Bornscheuer, U.T. The fourth wave of biocatalysis is approaching. Philos. Trans. A Math. Phys. Eng. Sci. 2018, 376. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U.T. Biocatalysis: Successfully Crossing Boundaries. Angew. Chem. Int. Ed. Engl. 2016, 55, 4372–4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldon, R.A.; Brady, D. Broadening the Scope of Biocatalysis in Sustainable Organic Synthesis. Chemsuschem 2019, 12, 2859–2881. [Google Scholar] [CrossRef] [PubMed]
- Ricca, E.; Brucher, B.; Schrittwieser, J.H. Multi-Enzymatic Cascade Reactions: Overview and Perspectives. Adv. Synth. Catal. 2011, 353, 2239–2262. [Google Scholar] [CrossRef]
- France, S.P.; Hepworth, L.J.; Turner, N.J.; Flitsch, S.L. Constructing Biocatalytic Cascades: In Vitro and in Vivo Approaches to de Novo Multi-Enzyme Pathways. ACS Catal. 2017, 7, 710–724. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Velikogne, S.; Hall, M.; Kroutil, W. Artificial Biocatalytic Linear Cascades for Preparation of Organic Molecules. Chem. Rev. 2018, 118, 270–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperl, J.M.; Sieber, V. Multienzyme Cascade Reactions-Status and Recent Advances. ACS Catal. 2018, 8, 2385–2396. [Google Scholar] [CrossRef]
- Gandomkar, S.; Zadlo-Dobrowolska, A.; Kroutil, W. Extending Designed Linear Biocatalytic Cascades for Organic Synthesis. ChemCatChem 2019, 11, 225–243. [Google Scholar] [CrossRef] [Green Version]
- Kuska, J.; O’Reilly, E. Engineered biosynthetic pathways and biocatalytic cascades for sustainable synthesis. Curr. Opin. Chem. Biol. 2020, 58, 146–154. [Google Scholar] [CrossRef]
- Losada-Garcia, N.; Cabrera, Z.; Urrutia, P.; Garcia-Sanz, C.; Andreu, A.; Palomo, J.M. Recent Advances in Enzymatic and Chemoenzymatic Cascade Processes. Catalysts 2020, 10, 1258. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lim, K.; Minteer, S.D. Cascaded Biocatalysis and Bioelectrocatalysis: Overview and Recent Advances. Annu. Rev. Phys. Chem. 2021. [Google Scholar] [CrossRef]
- Rudroff, F.; Mihovilovic, M.D.; Groger, H.; Snajdrova, R.; Iding, H.; Bornscheuer, U.T. Opportunities and challenges for combining chemo- and biocatalysis. Nat. Catal. 2018, 1, 12–22. [Google Scholar] [CrossRef]
- Dias Gomes, M.; Woodley, J.M. Considerations when Measuring Biocatalyst Performance. Molecules 2019, 24, 3573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Zhang, X.; Yuan, H.; Huang, D.; Wang, R.; Liu, H.; Wang, T. Research progress and the biotechnological applications of multienzyme complex. Appl. Microbiol. Biotechnol. 2021, 105, 1759–1777. [Google Scholar] [CrossRef]
- Mohamad, N.R.; Marzuki, N.H.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Chen, X.; Zheng, R.; Zheng, Y. Immobilization of Multi-Enzymes on Support Materials for Efficient Biocatalysis. Front. Bioeng. Biotechnol. 2020, 8, 660. [Google Scholar] [CrossRef]
- Wahab, R.A.; Elias, N.; Abdullah, F.; Ghoshal, S.K. On the taught new tricks of enzymes immobilization: An all-inclusive overview. React. Funct. Polym. 2020, 152. [Google Scholar] [CrossRef]
- Romero-Fernandez, M.; Paradisi, F. Protein immobilization technology for flow biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.Z.; Li, C.H.; Jiao, X.B.; Jia, S.R.; Jiang, Y.J.; Bilal, M.; Cui, J.D. Recent progress in multienzymes co-immobilization and multienzyme system applications. Chem. Eng. J. 2019, 373, 1254–1278. [Google Scholar] [CrossRef]
- Hwang, E.T.; Lee, S. Multienzymatic Cascade Reactions via Enzyme Complex by Immobilization. ACS Catal. 2019, 9, 4402–4425. [Google Scholar] [CrossRef]
- Petroll, K.; Kopp, D.; Care, A.; Bergquist, P.L.; Sunna, A. Tools and strategies for constructing cell-free enzyme pathways. Biotechnol. Adv. 2019, 37, 91–108. [Google Scholar] [CrossRef]
- Berckman, E.A.; Hartzell, E.J.; Mitkas, A.A.; Sun, Q.; Chen, W. Biological Assembly of Modular Protein Building Blocks as Sensing, Delivery, and Therapeutic Agents. Annu. Rev. Chem. Biomol. Eng. 2020, 11, 35–62. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, P.; de Carvalho, C.C.C.R. Multi-Enzyme Systems in Flow Chemistry. Processes 2021, 9, 225. [Google Scholar] [CrossRef]
- Bugada, L.F.; Smith, M.R.; Wen, F. Engineering Spatially Organized Multienzyme Assemblies for Complex Chemical Transformation. ACS Catal. 2018, 8, 7898–7906. [Google Scholar] [CrossRef] [Green Version]
- Schmid-Dannert, C.; Lopez-Gallego, F. Advances and opportunities for the design of self-sufficient and spatially organized cell-free biocatalytic systems. Curr. Opin. Chem. Biol. 2019, 49, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Ellis, G.A.; Klein, W.P.; Lasarte-Aragones, G.; Thakur, M.; Walper, S.A.; Medintz, I.L. Artificial Multienzyme Scaffolds: Pursuing in Vitro Substrate Channeling with an Overview of Current Progress. ACS Catal. 2019, 9, 10812–10869. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wu, Y.; Zhang, S.; Tian, Y.; Yang, D.; Jiang, Z. Bioinspired construction of multi-enzyme catalytic systems. Chem. Soc. Rev. 2018, 47, 4295–4313. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Sheng, J.; Feng, X. Mini-review: In vitro Metabolic Engineering for Biomanufacturing of High-value Products. Comput. Struct. Biotechnol. J. 2017, 15, 161–167. [Google Scholar] [CrossRef]
- Tinafar, A.; Jaenes, K.; Pardee, K. Synthetic Biology Goes Cell-Free. BMC Biol. 2019, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Khambhati, K.; Bhattacharjee, G.; Gohil, N.; Braddick, D.; Kulkarni, V.; Singh, V. Exploring the Potential of Cell-Free Protein Synthesis for Extending the Abilities of Biological Systems. Front. Bioeng. Biotechnol. 2019, 7, 248. [Google Scholar] [CrossRef] [PubMed]
- Noireaux, V.; Liu, A.P. The New Age of Cell-Free Biology. Annu. Rev. Biomed. Eng. 2020, 22, 51–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowie, J.U.; Sherkhanov, S.; Korman, T.P.; Valliere, M.A.; Opgenorth, P.H.; Liu, H. Synthetic Biochemistry: The Bio-inspired Cell-Free Approach to Commodity Chemical Production. Trends Biotechnol. 2020, 38, 766–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubbe, W.S.; Rasor, B.J.; Kruger, A.; Jewett, M.C.; Karim, A.S. Cell-free styrene biosynthesis at high titers. Metab. Eng. 2020, 61, 89–95. [Google Scholar] [CrossRef]
- Liu, W.Q.; Wu, C.; Jewett, M.C.; Li, J. Cell-free protein synthesis enables one-pot cascade biotransformation in an aqueous-organic biphasic system. Biotechnol. Bioeng. 2020, 117, 4001–4008. [Google Scholar] [CrossRef] [PubMed]
- Ngo, T.A.; Nakata, E.; Saimura, M.; Morii, T. Spatially Organized Enzymes Drive Cofactor-Coupled Cascade Reactions. J. Am. Chem. Soc. 2016, 138, 3012–3021. [Google Scholar] [CrossRef]
- Liu, M.; Fu, J.; Qi, X.; Wootten, S.; Woodbury, N.W.; Liu, Y.; Yan, H. A Three-Enzyme Pathway with an Optimised Geometric Arrangement to Facilitate Substrate Transfer. Chembiochem 2016, 17, 1097–1101. [Google Scholar] [CrossRef]
- Kang, W.; Ma, T.; Liu, M.; Qu, J.; Liu, Z.; Zhang, H.; Shi, B.; Fu, S.; Ma, J.; Lai, L.T.F.; et al. Modular enzyme assembly for enhanced cascade biocatalysis and metabolic flux. Nat. Commun. 2019, 10, 4248. [Google Scholar] [CrossRef]
- Yang, Z.; Gao, X.; Xie, H.; Wang, F.; Ren, Y.; Wei, D. Enhanced itaconic acid production by self-assembly of two biosynthetic enzymes in Escherichia coli. Biotechnol. Bioeng. 2017, 114, 457–462. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, H.; Wang, Y.; Ren, Y.; Wei, D. Manufacturing Multienzymatic Complex Reactors In Vivo by Self-Assembly To Improve the Biosynthesis of Itaconic Acid in Escherichia coli. ACS Synth. Biol. 2018, 7, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yang, S.; Zhao, C.; Ren, Y.; Wei, D. Artificial multienzyme supramolecular device: Highly ordered self-assembly of oligomeric enzymes in vitro and in vivo. Angew. Chem. Int. Ed. Engl. 2014, 53, 14027–14030. [Google Scholar] [CrossRef]
- Dueber, J.E.; Wu, G.C.; Malmirchegini, G.R.; Moon, T.S.; Petzold, C.J.; Ullal, A.V.; Prather, K.L.; Keasling, J.D. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 2009, 27, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Kibblewhite, R.E.; Paavola, C.D.; Orts, W.J.; Wagschal, K. Production of D-Xylonic Acid from Hemicellulose Using Artificial Enzyme Complexes. J. Microbiol. Biotechn. 2017, 27, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Tang, S.; Xu, Q.; Huang, H.; Jiang, L. SpyCatcher/SpyTag-Mediated Self-Assembly of a Supramolecular Complex for Improved Biocatalytic Production of Trehalose. Appl. Biochem. Microbiol. 2019, 55, 596–602. [Google Scholar] [CrossRef]
- Zhang, G.Q.; Quin, M.B.; Schmidt-Dannert, C. Self-Assembling Protein Scaffold System for Easy in Vitro Coimmobilization of Biocatalytic Cascade Enzymes. ACS Catal. 2018, 8, 5611–5620. [Google Scholar] [CrossRef]
- Peschke, T.; Skoupi, M.; Burgahn, T.; Gallus, S.; Ahmed, I.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Fusion Enzymes for Compartmentalized Biocatalysis. ACS Catal. 2017, 7, 7866–7872. [Google Scholar] [CrossRef]
- Jiang, W.; Zeng, W. Construction of a Self-Purification and Self-Assembly Coenzyme Regeneration System for the Synthesis of Chiral Drug Intermediates. ACS Omega 2021, 6, 1911–1916. [Google Scholar] [CrossRef]
- Qu, J.; Cao, S.; Wei, Q.; Zhang, H.; Wang, R.; Kang, W.; Ma, T.; Zhang, L.; Liu, T.; Wing-Ngor Au, S.; et al. Synthetic Multienzyme Complexes, Catalytic Nanomachineries for Cascade Biosynthesis In Vivo. ACS Nano 2019, 13, 9895–9906. [Google Scholar] [CrossRef]
- Lim, S.; Kim, J.; Kim, Y.; Xu, D.; Clark, D.S. CRISPR/Cas-directed programmable assembly of multi-enzyme complexes. Chem. Commun. (Camb.) 2020, 56, 4950–4953. [Google Scholar] [CrossRef]
- Patterson, D.P.; Schwarz, B.; Waters, R.S.; Gedeon, T.; Douglas, T. Encapsulation of an Enzyme Cascade within the Bacteriophage P22 Virus-Like Particle. ACS Chem. Biol. 2014, 9, 359–365. [Google Scholar] [CrossRef]
- Myhrvold, C.; Polka, J.K.; Silver, P.A. Synthetic Lipid-Containing Scaffolds Enhance Production by Colocalizing Enzymes. ACS Synth. Biol. 2016, 5, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, S.F.; Nallani, M.; Cornelissen, J.J.; Nolte, R.J.; van Hest, J.C. A Three-enzyme cascade reaction through positional assembly of enzymes in a polymersome nanoreactor. Chemistry 2009, 15, 1107–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, R.J.; Marguet, M.; Marais, S.; Fraaije, M.W.; van Hest, J.C.; Lecommandoux, S. Cascade Reactions in Multicompartmentalized Polymersomes. Angew. Chem. Int. Ed. Engl. 2014, 53, 146–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klermund, L.; Poschenrieder, S.T.; Castiglione, K. Biocatalysis in Polymersomes: Improving Multienzyme Cascades with Incompatible Reaction Steps by Compartmentalization. ACS Catal. 2017, 7, 3900–3904. [Google Scholar] [CrossRef]
- Seeman, N.C. DNA in a material world. Nature 2003, 421, 427–431. [Google Scholar] [CrossRef]
- Linko, V.; Dietz, H. The enabled state of DNA nanotechnology. Curr. Opin. Biotechnol. 2013, 24, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. Nanomaterials based on DNA. Annu. Rev. Biochem. 2010, 79, 65–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.J.; Wang, Y.Q.; Wang, J.; Li, Z.H.; Yuan, Q. Emerging Biomimetic Applications of DNA Nanotechnology. ACS Appl. Mater. Interfaces 2019, 11, 13859–13873. [Google Scholar] [CrossRef]
- Rothemund, P.W. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Bush, J.; Singh, S.; Vargas, M.; Oktay, E.; Hu, C.H.; Veneziano, R. Synthesis of DNA Origami Scaffolds: Current and Emerging Strategies. Molecules 2020, 25, 3386. [Google Scholar] [CrossRef]
- Hong, F.; Zhang, F.; Liu, Y.; Yan, H. DNA Origami: Scaffolds for Creating Higher Order Structures. Chem. Rev. 2017, 117, 12584–12640. [Google Scholar] [CrossRef]
- Goldberg, M.; Langer, R.; Jia, X. Nanostructured materials for applications in drug delivery and tissue engineering. J. Biomater. Sci. Polym. Ed. 2007, 18, 241–268. [Google Scholar] [CrossRef] [Green Version]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, T.A.; Raman, S.; Dey, R.; Burkhard, P. Nanoscale assemblies and their biomedical applications. J. R. Soc. Interface 2013, 10, 20120740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, J.J.; Elisseeff, J.H. Mimicking biological functionality with polymers for biomedical applications. Nature 2016, 540, 386–394. [Google Scholar] [CrossRef]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef]
- Fruk, L.; Muller, J.; Weber, G.; Narvaez, A.; Dominguez, E.; Niemeyer, C.M. DNA-directed immobilization of horseradish peroxidase-DNA conjugates on microelectrode arrays: Towards electrochemical screening of enzyme libraries. Chemistry 2007, 13, 5223–5231. [Google Scholar] [CrossRef] [PubMed]
- Cheglakov, Z.; Weizmann, Y.; Braunschweig, A.B.; Wilner, O.I.; Willner, I. Increasing the complexity of periodic protein nanostructures by the rolling-circle-amplified synthesis of aptamers. Angew. Chem. Int. Ed. Engl. 2008, 47, 126–130. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, J.B.; Clark, D.S.; Glover, D.J. Functional Applications of Nucleic Acid-Protein Hybrid Nanostructures. Trends Biotechnol. 2020, 38, 976–989. [Google Scholar] [CrossRef]
- Fu, J.; Wang, Z.; Liang, X.H.; Oh, S.W.; St Iago-McRae, E.; Zhang, T. DNA-Scaffolded Proximity Assembly and Confinement of Multienzyme Reactions. Top. Curr. Chem. 2020, 378, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilner, O.I.; Weizmann, Y.; Gill, R.; Lioubashevski, O.; Freeman, R.; Willner, I. Enzyme cascades activated on topologically programmed DNA scaffolds. Nat. Nanotechnol. 2009, 4, 249–254. [Google Scholar] [CrossRef]
- Timm, C.; Niemeyer, C.M. Assembly and purification of enzyme-functionalized DNA origami structures. Angew. Chem. Int. Ed. Engl. 2015, 54, 6745–6750. [Google Scholar] [CrossRef]
- Fu, J.; Yang, Y.R.; Johnson-Buck, A.; Liu, M.; Liu, Y.; Walter, N.G.; Woodbury, N.W.; Yan, H. Multi-enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat. Nanotechnol. 2014, 9, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Ke, G.; Liu, M.; Jiang, S.; Qi, X.; Yang, Y.R.; Wootten, S.; Zhang, F.; Zhu, Z.; Liu, Y.; Yang, C.J.; et al. Directional Regulation of Enzyme Pathways through the Control of Substrate Channeling on a DNA Origami Scaffold. Angew. Chem. Int. Ed. Engl. 2016, 55, 7483–7486. [Google Scholar] [CrossRef]
- Fu, J.; Liu, M.; Liu, Y.; Woodbury, N.W.; Yan, H. Interenzyme substrate diffusion for an enzyme cascade organized on spatially addressable DNA nanostructures. J. Am. Chem. Soc. 2012, 134, 5516–5519. [Google Scholar] [CrossRef] [Green Version]
- Sweetlove, L.J.; Fernie, A.R. The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nat. Commun. 2018, 9, 2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tsitkov, S.; Hess, H. Proximity does not contribute to activity enhancement in the glucose oxidase–horseradish peroxidase cascade. Nat. Commun. 2016, 7, 13982. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.B.; Mo, S.S.; Liu, M.W.; Liu, L.; Yu, L.L.; Wang, C.X. Rationally Designed Protein Building Blocks for Programmable Hierarchical Architectures. Front. Chem. 2020, 8, 23. [Google Scholar] [CrossRef]
- Gerbelli, B.B.; Vassiliades, S.V.; Rojas, J.E.U.; Pelin, J.; Mancini, R.S.N.; Pereira, W.S.G.; Aguilar, A.M.; Venanzi, M.; Cavalieri, F.; Giuntini, F.; et al. Hierarchical Self-Assembly of Peptides and its Applications in Bionanotechnology. Macromol. Chem. Phys. 2019, 220, 22. [Google Scholar] [CrossRef]
- Shen, H.; Fallas, J.A.; Lynch, E.; Sheffler, W.; Parry, B.; Jannetty, N.; Decarreau, J.; Wagenbach, M.; Vicente, J.J.; Chen, J.; et al. De novo design of self-assembling helical protein filaments. Science 2018, 362, 705–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapenta, F.; Aupic, J.; Strmsek, Z.; Jerala, R. Coiled coil protein origami: From modular design principles towards biotechnological applications. Chem. Soc. Rev. 2018, 47, 3530–3542. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Arai, R. Design and construction of self-assembling supramolecular protein complexes using artificial and fusion proteins as nanoscale building blocks. Curr. Opin. Biotechnol. 2017, 46, 57–65. [Google Scholar] [CrossRef]
- Magnotti, E.; Conticello, V. Two-Dimensional Peptide and Protein Assemblies. Adv. Exp. Med. Biol. 2016, 940, 29–60. [Google Scholar] [CrossRef]
- Drobnak, I.; Ljubetic, A.; Gradisar, H.; Pisanski, T.; Jerala, R. Designed Protein Origami. Adv. Exp. Med. Biol. 2016, 940, 7–27. [Google Scholar] [CrossRef]
- Yeboah, A.; Cohen, R.I.; Rabolli, C.; Yarmush, M.L.; Berthiaume, F. Elastin-like polypeptides: A strategic fusion partner for biologics. Biotechnol. Bioeng. 2016, 113, 1617–1627. [Google Scholar] [CrossRef]
- Miranda-Nieves, D.; Chaikof, E.L. Collagen and Elastin Biomaterials for the Fabrication of Engineered Living Tissues. ACS Biomater. Sci. Eng. 2017, 3, 694–711. [Google Scholar] [CrossRef]
- Wang, Y.; Katyal, P.; Montclare, J.K. Protein-Engineered Functional Materials. Adv. Healthc. Mater. 2019, e1801374. [Google Scholar] [CrossRef]
- Das, S.; Jacob, R.S.; Patel, K.; Singh, N.; Maji, S.K. Amyloid Fibrils: Versatile Biomaterials for Cell Adhesion and Tissue Engineering Applications. Biomacromolecules 2018, 19, 1826–1839. [Google Scholar] [CrossRef] [PubMed]
- Behrendorff, J.B.Y.H.; Borras-Gas, G.; Pribil, M. Synthetic Protein Scaffolding at Biological Membranes. Trends Biotechnol. 2020, 38, 432–446. [Google Scholar] [CrossRef] [PubMed]
- Alves, V.D.; Fontes, C.; Bule, P. Cellulosomes: Highly Efficient Cellulolytic Complexes. Subcell. Biochem. 2021, 96, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Artzi, L.; Bayer, E.A.; Moraïs, S. Cellulosomes: Bacterial nanomachines for dismantling plant polysaccharides. Nat. Rev. Microbiol. 2017, 15, 83–95. [Google Scholar] [CrossRef]
- You, C.; Myung, S.; Zhang, Y.H. Facilitated substrate channeling in a self-assembled trifunctional enzyme complex. Angew. Chem. Int. Ed. Engl. 2012, 51, 8787–8790. [Google Scholar] [CrossRef]
- You, C.; Zhang, Y.H. Self-assembly of synthetic metabolons through synthetic protein scaffolds: One-step purification, co-immobilization, and substrate channeling. ACS Synth. Biol. 2013, 2, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Mitsuzawa, S.; Kagawa, H.; Li, Y.; Chan, S.L.; Paavola, C.D.; Trent, J.D. The rosettazyme: A synthetic cellulosome. J. Biotechnol. 2009, 143, 139–144. [Google Scholar] [CrossRef]
- Krishna, T.S.; Kong, X.P.; Gary, S.; Burgers, P.M.; Kuriyan, J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 1994, 79, 1233–1243. [Google Scholar] [CrossRef]
- Williams, G.J.; Johnson, K.; Rudolf, J.; McMahon, S.A.; Carter, L.; Oke, M.; Liu, H.; Taylor, G.L.; White, M.F.; Naismith, J.H. Structure of the heterotrimeric PCNA from Sulfolobus solfataricus. Acta. Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 944–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirakawa, H.; Nagamune, T. Molecular assembly of P450 with ferredoxin and ferredoxin reductase by fusion to PCNA. Chembiochem 2010, 11, 1517–1520. [Google Scholar] [CrossRef]
- Tan, C.Y.; Hirakawa, H.; Nagamune, T. Supramolecular protein assembly supports immobilization of a cytochrome P450 monooxygenase system as water-insoluble gel. Sci. Rep. 2015, 5, 8648. [Google Scholar] [CrossRef] [Green Version]
- Reddington, S.C.; Howarth, M. Secrets of a covalent interaction for biomaterials and biotechnology: SpyTag and SpyCatcher. Curr. Opin. Chem. Biol. 2015, 29, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, A.R.; Alam, M.K.; Geyer, C.R. Post-translational Assembly of Protein Parts into Complex Devices by Using SpyTag/SpyCatcher Protein Ligase. Chembiochem 2019, 20, 319–328. [Google Scholar] [CrossRef]
- Hatlem, D.; Trunk, T.; Linke, D.; Leo, J.C. Catching a SPY: Using the SpyCatcher-SpyTag and Related Systems for Labeling and Localizing Bacterial Proteins. Int. J. Mol. Sci. 2019, 20, 2129. [Google Scholar] [CrossRef] [Green Version]
- Keeble, A.H.; Howarth, M. Power to the protein: Enhancing and combining activities using the Spy toolbox. Chem. Sci. 2020, 11, 7281–7291. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [Green Version]
- Keeble, A.H.; Banerjee, A.; Ferla, M.P.; Reddington, S.C.; Anuar, I.; Howarth, M. Evolving Accelerated Amidation by SpyTag/SpyCatcher to Analyze Membrane Dynamics. Angew. Chem. Int. Ed. Engl. 2017, 56, 16521–16525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeble, A.H.; Howarth, M. Insider information on successful covalent protein coupling with help from SpyBank. Methods Enzymol. 2019, 617, 443–461. [Google Scholar] [CrossRef] [PubMed]
- Veggiani, G.; Nakamura, T.; Brenner, M.D.; Gayet, R.V.; Yan, J.; Robinson, C.V.; Howarth, M. Programmable polyproteams built using twin peptide superglues. Proc. Natl. Acad. Sci. USA 2016, 113, 1202–1207. [Google Scholar] [CrossRef] [Green Version]
- Khairil Anuar, I.N.A.; Banerjee, A.; Keeble, A.H.; Carella, A.; Nikov, G.I.; Howarth, M. Spy&Go purification of SpyTag-proteins using pseudo-SpyCatcher to access an oligomerization toolbox. Nat. Commun. 2019, 10, 1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beesley, J.L.; Woolfson, D.N. The de novo design of α-helical peptides for supramolecular self-assembly. Curr. Opin. Biotechnol. 2019, 58, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Steen Redeker, E.; Ta, D.T.; Cortens, D.; Billen, B.; Guedens, W.; Adriaensens, P. Protein engineering for directed immobilization. Bioconjug. Chem. 2013, 24, 1761–1777. [Google Scholar] [CrossRef]
- Meldal, M.; Schoffelen, S. Recent advances in covalent, site-specific protein immobilization. F1000Research 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Dorr, B.M.; Ham, H.O.; An, C.; Chaikof, E.L.; Liu, D.R. Reprogramming the specificity of sortase enzymes. Proc. Natl. Acad. Sci. USA 2014, 111, 13343–13348. [Google Scholar] [CrossRef] [Green Version]
- Schmohl, L.; Schwarzer, D. Sortase-mediated ligations for the site-specific modification of proteins. Curr. Opin. Chem. Biol. 2014, 22, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Ritzefeld, M. Sortagging: A Robust and Efficient Chemoenzymatic Ligation Strategy. Chem. Eur. J. 2014, 20, 8516–8529. [Google Scholar] [CrossRef]
- Broguiere, N.; Formica, F.; Barreto, G.; Zenobi-Wong, M. Sortase A as a cross-linking enzyme in tissue engineering. Acta Biomater. 2018, 77, 182–190. [Google Scholar] [CrossRef]
- Singh, S.; Gupta, K.; Shukla, S.; Sampathkumar, S.G.; Roy, R.P. Sortase-click strategy for defined protein conjugation on a heptavalent cyclodextrin scaffold. PLoS ONE 2019, 14, e0217369. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Tanaka, T.; Kondo, A. Sortase A-Catalyzed Site-Specific Coimmobilization on Microparticles via Streptavidin. Langmuir 2012, 28, 3553–3557. [Google Scholar] [CrossRef]
- McConnell, S.A.; Cannon, K.A.; Morgan, C.; McAllister, R.; Amer, B.R.; Clubb, R.T.; Yeates, T.O. Designed Protein Cages as Scaffolds for Building Multienzyme Materials. ACS Synth. Biol. 2020, 9, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Mateu, M.G. Assembly, Engineering and Applications of Virus-Based Protein Nanoparticles. Adv. Exp. Med. Biol. 2016, 940, 83–120. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, J.D.; Hagan, M.F. Mechanisms of virus assembly. Annu. Rev. Phys. Chem. 2015, 66, 217–239. [Google Scholar] [CrossRef] [Green Version]
- Raff, J.; Matys, S.; Suhr, M.; Vogel, M.; Gunther, T.; Pollmann, K. S-Layer-Based Nanocomposites for Industrial Applications. Adv. Exp. Med. Biol. 2016, 940, 245–279. [Google Scholar] [CrossRef]
- Sleytr, U.B.; Schuster, B.; Egelseer, E.M.; Pum, D. S-layers: Principles and applications. Fems Microbiol. Rev. 2014, 38, 823–864. [Google Scholar] [CrossRef] [PubMed]
- Demchuk, A.M.; Patel, T.R. The biomedical and bioengineering potential of protein nanocompartments. Biotechnol. Adv. 2020, 41, 107547. [Google Scholar] [CrossRef]
- Glasgow, J.E.; Capehart, S.L.; Francis, M.B.; Tullman-Ercek, D. Osmolyte-Mediated Encapsulation of Proteins inside MS2 Viral Capsids. ACS Nano 2012, 6, 8658–8664. [Google Scholar] [CrossRef]
- Jones, J.A.; Giessen, T.W. Advances in encapsulin nanocompartment biology and engineering. Biotechnol. Bioeng. 2021, 118, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Kerfeld, C.A.; Sutter, M. Engineered bacterial microcompartments: Apps for programming metabolism. Curr. Opin. Biotechnol. 2020, 65, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Palmer, D.J.; Warren, M.J. Biotechnological Advances in Bacterial Microcompartment Technology. Trends Biotechnol. 2019, 37, 325–336. [Google Scholar] [CrossRef]
- Planamente, S.; Frank, S. Bio-engineering of bacterial microcompartments: A mini review. Biochem. Soc. Trans. 2019, 47, 765–777. [Google Scholar] [CrossRef]
- Yeates, T.O. Geometric Principles for Designing Highly Symmetric Self-Assembling Protein Nanomaterials. Annu. Rev. Biophys. 2017, 46, 23–42. [Google Scholar] [CrossRef]
- Okesola, B.O.; Mata, A. Multicomponent self-assembly as a tool to harness new properties from peptides and proteins in material design. Chem. Soc. Rev. 2018, 47, 3721–3736. [Google Scholar] [CrossRef]
- Kuan, S.L.; Bergamini, F.R.G.; Weil, T. Functional protein nanostructures: A chemical toolbox. Chem. Soc. Rev. 2018, 47, 9069–9105. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, K. Synthetic approaches to construct viral capsid-like spherical nanomaterials. Chem. Commun. 2018, 54, 8944–8959. [Google Scholar] [CrossRef]
- Majerle, A.; Schmieden, D.T.; Jerala, R.; Meyer, A.S. Synthetic Biology for Multiscale Designed Biomimetic Assemblies: From Designed Self-Assembling Biopolymers to Bacterial Bioprinting. Biochemistry 2019, 58, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Hamley, I.W. Protein Assemblies: Nature-Inspired and Designed Nanostructures. Biomacromolecules 2019, 20, 1829–1848. [Google Scholar] [CrossRef] [Green Version]
- Cannon, K.A.; Ochoa, J.M.; Yeates, T.O. High-symmetry protein assemblies: Patterns and emerging applications. Curr. Opin. Struct. Biol. 2019, 55, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Bale, J.B.; Gonen, S.; Liu, Y.; Sheffler, W.; Ellis, D.; Thomas, C.; Cascio, D.; Yeates, T.O.; Gonen, T.; King, N.P.; et al. Accurate design of megadalton-scale two-component icosahedral protein complexes. Science 2016, 353, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, N.P.; Sheffler, W.; Sawaya, M.R.; Vollmar, B.S.; Sumida, J.P.; Andre, I.; Gonen, T.; Yeates, T.O.; Baker, D. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science 2012, 336, 1171–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.T.; Cascio, D.; Yeates, T.O. Structure of a 16-nm cage designed by using protein oligomers. Science 2012, 336, 1129. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.E.; Colovos, C.; Yeates, T.O. Nanohedra: Using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 2217–2221. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sasson, A.J.; Watson, J.L.; Sheffler, W.; Johnson, M.C.; Bittleston, A.; Somasundaram, L.; Decarreau, J.; Jiao, F.; Chen, J.; Mela, I.; et al. Design of biologically active binary protein 2D materials. Nature 2021, 589, 468–473. [Google Scholar] [CrossRef]
- Worsdorfer, B.; Woycechowsky, K.J.; Hilvert, D. Directed evolution of a protein container. Science 2011, 331, 589–592. [Google Scholar] [CrossRef]
- Frey, R.; Mantri, S.; Rocca, M.; Hilvert, D. Bottom-up Construction of a Primordial Carboxysome Mimic. J. Am. Chem. Soc. 2016, 138, 10072–10075. [Google Scholar] [CrossRef]
- Sasaki, E.; Bohringer, D.; van de Waterbeemd, M.; Leibundgut, M.; Zschoche, R.; Heck, A.J.; Ban, N.; Hilvert, D. Structure and assembly of scalable porous protein cages. Nat. Commun. 2017, 8, 14663. [Google Scholar] [CrossRef] [Green Version]
- Terasaka, N.; Azuma, Y.; Hilvert, D. Laboratory evolution of virus-like nucleocapsids from nonviral protein cages. Proc. Natl. Acad. Sci. USA 2018, 115, 5432–5437. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Cardone, G.; Restrepo, D.; Zavattieri, P.D.; Baker, T.S.; Tezcan, F.A. Self-assembly of coherently dynamic, auxetic, two-dimensional protein crystals. Nature 2016, 533, 369–373. [Google Scholar] [CrossRef]
- Chen, Z.; Johnson, M.C.; Chen, J.; Bick, M.J.; Boyken, S.E.; Lin, B.; De Yoreo, J.J.; Kollman, J.M.; Baker, D.; DiMaio, F. Self-Assembling 2D Arrays with de Novo Protein Building Blocks. J. Am. Chem. Soc. 2019, 141, 8891–8895. [Google Scholar] [CrossRef] [PubMed]
- Brodin, J.D.; Carr, J.R.; Sontz, P.A.; Tezcan, F.A. Exceptionally stable, redox-active supramolecular protein assemblies with emergent properties. Proc. Natl. Acad. Sci. USA 2014, 111, 2897–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberstein, R.; Suzuki, Y.; Paesani, F.; Tezcan, F.A. Engineering the entropy-driven free-energy landscape of a dynamic nanoporous protein assembly. Nat. Chem. 2018, 10, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Golub, E.; Subramanian, R.H.; Esselborn, J.; Alberstein, R.G.; Bailey, J.B.; Chiong, J.A.; Yan, X.D.; Booth, T.; Baker, T.S.; Tezcan, F.A. Constructing protein polyhedra via orthogonal chemical interactions. Nature 2020, 578, 172–176. [Google Scholar] [CrossRef]
- Kakkis, A.; Gagnon, D.; Esselborn, J.; Britt, R.D.; Tezcan, F.A. Metal-Templated Design of Chemically Switchable Protein Assemblies with High-Affinity Coordination Sites. Angew. Chem. Int. Ed. Engl. 2020, 59, 21940–21944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Alberstein, R.G.; De Yoreo, J.J.; Tezcan, F.A. Assembly of a patchy protein into variable 2D lattices via tunable multiscale interactions. Nat. Commun. 2020, 11, 3770. [Google Scholar] [CrossRef]
- Manea, F.; Garda, V.G.; Rad, B.; Ajo-Franklin, C.M. Programmable assembly of 2D crystalline protein arrays into covalently stacked 3D bionanomaterials. Biotechnol. Bioeng. 2020, 117, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Quin, M.B.; Sanders, M.A.; Johnson, E.T.; Schmidt-Dannert, C. Engineered protein nano-compartments for targeted enzyme localization. PLoS ONE 2012, 7, e33342. [Google Scholar] [CrossRef] [Green Version]
- Held, M.; Quin, M.B.; Schmidt-Dannert, C. Eut bacterial microcompartments: Insights into their function, structure, and bioengineering applications. J. Mol. Microbiol. Biotechnol. 2013, 23, 308–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quin, M.B.; Perdue, S.A.; Hsu, S.Y.; Schmidt-Dannert, C. Encapsulation of multiple cargo proteins within recombinant Eut nanocompartments. Appl. Microbiol. Biotechnol. 2016, 100, 9187–9200. [Google Scholar] [CrossRef]
- Schmidt-Dannert, S.; Zhang, G.; Johnston, T.; Quin, M.B.; Schmidt-Dannert, C. Building a toolbox of protein scaffolds for future immobilization of biocatalysts. Appl. Microbiol. Biotechnol. 2018, 102, 8373–8388. [Google Scholar] [CrossRef]
- Zhang, G.; Schmidt-Dannert, S.; Quin, M.B.; Schmidt-Dannert, C. Protein-based scaffolds for enzyme immobilization. Methods Enzymol. 2019, 617, 323–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Johnston, T.; Quin, M.B.; Schmidt-Dannert, C. Developing a Protein Scaffolding System for Rapid Enzyme Immobilization and Optimization of Enzyme Functions for Biocatalysis. ACS Synth. Biol. 2019, 8, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Schoonen, L.; van Hest, J.C. Compartmentalization Approaches in Soft Matter Science: From Nanoreactor Development to Organelle Mimics. Adv. Mater. 2016, 28, 1109–1128. [Google Scholar] [CrossRef]
- Quin, M.B.; Wallin, K.K.; Zhang, G.; Schmidt-Dannert, C. Spatial organization of multi-enzyme biocatalytic cascades. Org. Biomol. Chem. 2017, 15, 4260–4271. [Google Scholar] [CrossRef]
- Schwarz, B.; Uchida, M.; Douglas, T. Biomedical and Catalytic Opportunities of Virus-Like Particles in Nanotechnology. Adv. Virus Res. 2017, 97, 1–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupka, I.; Heddle, J.G. Artificial protein cages—inspiration, construction, and observation. Curr. Opin. Struct. Biol. 2020, 64, 66–73. [Google Scholar] [CrossRef]
- Lv, C.; Zhang, X.; Liu, Y.; Zhang, T.; Chen, H.; Zang, J.; Zheng, B.; Zhao, G. Redesign of protein nanocages: The way from 0D, 1D, 2D to 3D assembly. Chem. Soc. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.M.; Stewart, K.L.; Yeates, T.O.; Bobik, T.A. Advances in the World of Bacterial Microcompartments. Trends Biochem. Sci. 2021. [Google Scholar] [CrossRef]
- Sutter, M.; McGuire, S.; Ferlez, B.; Kerfeld, C.A. Structural Characterization of a Synthetic Tandem-Domain Bacterial Microcompartment Shell Protein Capable of Forming Icosahedral Shell Assemblies. ACS Synth. Biol. 2019, 8, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Hagen, A.R.; Plegaria, J.S.; Sloan, N.; Ferlez, B.; Aussignargues, C.; Burton, R.; Kerfeld, C.A. In Vitro Assembly of Diverse Bacterial Microcompartment Shell Architectures. Nano Lett. 2018, 18, 7030–7037. [Google Scholar] [CrossRef]
- Lončar, N.; Rozeboom, H.J.; Franken, L.E.; Stuart, M.C.A.; Fraaije, M.W. Structure of a robust bacterial protein cage and its application as a versatile biocatalytic platform through enzyme encapsulation. Biochem. Biophys. Res. Commun. 2020, 529, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.M.; Jung, S.M.; Coffman, J.L.; Lutz, S. Pore Engineering for Enhanced Mass Transport in Encapsulin Nanocompartments. ACS Synth. Biol. 2018, 7, 2514–2517. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Edwardson, T.G.W.; Hilvert, D. Tailoring lumazine synthase assemblies for bionanotechnology. Chem. Soc. Rev. 2018, 47, 3543–3557. [Google Scholar] [CrossRef]
- Uchida, M.; McCoy, K.; Fukuto, M.; Yang, L.; Yoshimura, H.; Miettinen, H.M.; LaFrance, B.; Patterson, D.P.; Schwarz, B.; Karty, J.A.; et al. Modular Self-Assembly of Protein Cage Lattices for Multistep Catalysis. ACS Nano 2018, 12, 942–953. [Google Scholar] [CrossRef]
- Giessen, T.W.; Silver, P.A. A Catalytic Nanoreactor Based on in Vivo Encapsulation of Multiple Enzymes in an Engineered Protein Nanocompartment. Chembiochem 2016, 17, 1931–1935. [Google Scholar] [CrossRef]
- Lizatovic, R.; Assent, M.; Barendregt, A.; Dahlin, J.; Bille, A.; Satzinger, K.; Tupina, D.; Heck, A.J.R.; Wennmalm, S.; Andre, I. A Protein-Based Encapsulation System with Calcium-Controlled Cargo Loading and Detachment. Angew. Chem. Int. Ed. Engl. 2018, 57, 11334–11338. [Google Scholar] [CrossRef]
- Fernandez-Trillo, F.; Grover, L.M.; Stephenson-Brown, A.; Harrison, P.; Mendes, P.M. Vesicles in Nature and the Laboratory: Elucidation of Their Biological Properties and Synthesis of Increasingly Complex Synthetic Vesicles. Angew. Chem. Int. Ed. Engl. 2017, 56, 3142–3160. [Google Scholar] [CrossRef] [Green Version]
- Rideau, E.; Dimova, R.; Schwille, P.; Wurm, F.R.; Landfester, K. Liposomes and polymersomes: A comparative review towards cell mimicking. Chem. Soc. Rev. 2018, 47, 8572–8610. [Google Scholar] [CrossRef] [Green Version]
- Blackman, L.D.; Varlas, S.; Arno, M.C.; Fayter, A.; Gibson, M.I.; O’Reilly, R.K. Permeable Protein-Loaded Polymersome Cascade Nanoreactors by Polymerization-Induced Self-Assembly. ACS Macro Lett. 2017, 6, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Gaitzsch, J.; Appelhans, D.; Wang, L.; Battaglia, G.; Voit, B. Synthetic Bio-nanoreactor: Mechanical and Chemical Control of Polymersome Membrane Permeability. Angew. Chem. Int. Ed. Engl. 2012, 51, 4448–4451. [Google Scholar] [CrossRef]
- De Hoog, H.-P.M.; Arends, I.W.C.E.; Rowan, A.E.; Cornelissen, J.J.L.M.; Nolte, R.J.M. A hydrogel-based enzyme-loaded polymersome reactor. Nanoscale 2010, 2, 709–716. [Google Scholar] [CrossRef] [Green Version]
- Alves, N.J.; Moore, M.; Johnson, B.J.; Dean, S.N.; Turner, K.B.; Medintz, I.L.; Walper, S.A. Environmental Decontamination of a Chemical Warfare Simulant Utilizing a Membrane Vesicle-Encapsulated Phosphotriesterase. ACS Appl. Mater. Interfaces 2018, 10, 15712–15719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Gallego, F.; Schmidt-Dannert, C. Multi-enzymatic synthesis. Curr. Opin. Chem. Biol. 2010, 14, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Wegst, U.G.K.; Bai, H.; Saiz, E.; Tomsia, A.P.; Ritchie, R.O. Bioinspired structural materials. Nat. Mater. 2015, 14, 23–36. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Examples of enzyme cascades co-localized on nucleic acid-based scaffolds. (a) Tethering of xylose reductase (XR) and xylitol dehydrogenase (XDH) on a DNA scaffold for the production of xylulose from xylose [37]. (b) Co-localization of a co-factor self-sufficient three enzyme system via Watson–Crick pairing of DNA tethers. Malate dehydrogenase (MDH) catalyzes malic acid oxidation to oxaloacetate which is converted to pyruvate by oxaloacetate decarboxylase (OAD). Lactate dehydrogenase (LDH) reduces pyruvate and completes the reduction–oxidation cycle of NAD+ and NADH of MDH and LDH [38]. (c) Co-assembly of MDH, glucose-6-phosphate dehydrogenase (G6pDH), and lactate LDH on DNA tiles with nanometric control over their spatial arrangement [74].
Figure 1.
Examples of enzyme cascades co-localized on nucleic acid-based scaffolds. (a) Tethering of xylose reductase (XR) and xylitol dehydrogenase (XDH) on a DNA scaffold for the production of xylulose from xylose [37]. (b) Co-localization of a co-factor self-sufficient three enzyme system via Watson–Crick pairing of DNA tethers. Malate dehydrogenase (MDH) catalyzes malic acid oxidation to oxaloacetate which is converted to pyruvate by oxaloacetate decarboxylase (OAD). Lactate dehydrogenase (LDH) reduces pyruvate and completes the reduction–oxidation cycle of NAD+ and NADH of MDH and LDH [38]. (c) Co-assembly of MDH, glucose-6-phosphate dehydrogenase (G6pDH), and lactate LDH on DNA tiles with nanometric control over their spatial arrangement [74].

Figure 2.
Assembly of enzymes with high-affinity protein and peptide-interaction domains. (a) Proliferating cellular nuclear antigen (PCNA)-based heterotrimeric complex formation of a cytochrome P450s (CYP450) with its redox partners (PdX, and PdR) to a ring-shaped electron transfer scaffold (PUPPET) [97]. (b) Co-localization of enzyme assemblies driven by the interaction of RIAD and RIDD peptide pairs that result in disulfide bond formation [39]. (c) Scaffolding of enzymes on a designer cellulosome via specific interactions of orthogonal dockerin and cohesin pairs. A cellulose binding domain (CBM) can be fused to the cellulosome protein scaffold allows for attachment to regenerated amorphous cellulose (RAC).
Figure 2.
Assembly of enzymes with high-affinity protein and peptide-interaction domains. (a) Proliferating cellular nuclear antigen (PCNA)-based heterotrimeric complex formation of a cytochrome P450s (CYP450) with its redox partners (PdX, and PdR) to a ring-shaped electron transfer scaffold (PUPPET) [97]. (b) Co-localization of enzyme assemblies driven by the interaction of RIAD and RIDD peptide pairs that result in disulfide bond formation [39]. (c) Scaffolding of enzymes on a designer cellulosome via specific interactions of orthogonal dockerin and cohesin pairs. A cellulose binding domain (CBM) can be fused to the cellulosome protein scaffold allows for attachment to regenerated amorphous cellulose (RAC).

Figure 3.
CRISPR/dCas9 directed nanometric assembly of enzymes. Violacein biosynthetic enzymes are covalently ligated to dCas9 via the SpyTag/SpyCatcher mediated isopeptide bond formation. Complex formation of the enzyme conjugated dCas9′s with guide RNAs (sgRNA) allows for the programmable arrangement of five violacein biosynthetic enzymes (VioA-E) on a DNA scaffold with sgRNA specific target sequences [50].
Figure 3.
CRISPR/dCas9 directed nanometric assembly of enzymes. Violacein biosynthetic enzymes are covalently ligated to dCas9 via the SpyTag/SpyCatcher mediated isopeptide bond formation. Complex formation of the enzyme conjugated dCas9′s with guide RNAs (sgRNA) allows for the programmable arrangement of five violacein biosynthetic enzymes (VioA-E) on a DNA scaffold with sgRNA specific target sequences [50].

Figure 4.
Self-assembling protein scaffolds. (a) Arrays are assembled from the bacterial S-layer protein SbsB. Genetic engineering S-layer proteins for Spy/SnoopTag display facilitates the stacking of layers with bifunctional Spy/SnoopCatcher domain proteins [154]. (b) Scaffolding system for multi-enzyme biocatalysis. The bacterial microcompartment shell protein EutM assembles into hexameric arrays to which biocatalysts can be attached via N- or C-terminal genetic fusion of interacting polypeptide tags like the SpyTag/SpyCatcher pair. The spatial organization of enzyme cascades is optimized by varying the molar ratios of catalysts and scaffold building blocks. A scaffold toolbox created from EutM homologs provides access to arrays with different assembly and surface properties for enzyme cascade optimization [46].
Figure 4.
Self-assembling protein scaffolds. (a) Arrays are assembled from the bacterial S-layer protein SbsB. Genetic engineering S-layer proteins for Spy/SnoopTag display facilitates the stacking of layers with bifunctional Spy/SnoopCatcher domain proteins [154]. (b) Scaffolding system for multi-enzyme biocatalysis. The bacterial microcompartment shell protein EutM assembles into hexameric arrays to which biocatalysts can be attached via N- or C-terminal genetic fusion of interacting polypeptide tags like the SpyTag/SpyCatcher pair. The spatial organization of enzyme cascades is optimized by varying the molar ratios of catalysts and scaffold building blocks. A scaffold toolbox created from EutM homologs provides access to arrays with different assembly and surface properties for enzyme cascade optimization [46].

Figure 5.
Compartmentalization strategies for multi-enzyme catalysis. (a) Representative examples of protein-based compartments and their different sizes. Major vault particle from Rattus norvegicus, bacterial microcompartment from Haliangium ochraceum, encapsulin from Quasibacillus thermotolerans, lumazine synthase from Aquifex aeolicus and bacteriophage P22 virion capsid. (b) Encapsulation of multiple enzymes into porous protein shells via engineered targeting mechanisms (top) and enzyme encapsulation into synthetic polymersomes equipped with membrane channels and domains for enzyme cargo attachment. BMC: bacterial microcompartment shell. MVP, major vault protein; VLP, virus-like particle; BMC, bacterial microcompartment; PDB, protein data bank.
Figure 5.
Compartmentalization strategies for multi-enzyme catalysis. (a) Representative examples of protein-based compartments and their different sizes. Major vault particle from Rattus norvegicus, bacterial microcompartment from Haliangium ochraceum, encapsulin from Quasibacillus thermotolerans, lumazine synthase from Aquifex aeolicus and bacteriophage P22 virion capsid. (b) Encapsulation of multiple enzymes into porous protein shells via engineered targeting mechanisms (top) and enzyme encapsulation into synthetic polymersomes equipped with membrane channels and domains for enzyme cargo attachment. BMC: bacterial microcompartment shell. MVP, major vault protein; VLP, virus-like particle; BMC, bacterial microcompartment; PDB, protein data bank.

Figure 6.
Future platform for the fabrication of autonomously self-organizing biocatalytic materials from genetically programmable protein components. Biocatalysts and material building blocks can be produced in predefined ratios by cell-factories or cell free systems. Produced proteins self-assembly into hierarchical materials with preconfigured properties that are also genetically encoded. 3D-assembly is driven by the geometric assembly of engineered building blocks and their different ratios. Designed surface properties and placement of genetic tags for cargo attachment, mineralization, and surface attachment give rise to multifunctional, robust materials suitable for cascade operation in batch or flow-through modes.
Figure 6.
Future platform for the fabrication of autonomously self-organizing biocatalytic materials from genetically programmable protein components. Biocatalysts and material building blocks can be produced in predefined ratios by cell-factories or cell free systems. Produced proteins self-assembly into hierarchical materials with preconfigured properties that are also genetically encoded. 3D-assembly is driven by the geometric assembly of engineered building blocks and their different ratios. Designed surface properties and placement of genetic tags for cargo attachment, mineralization, and surface attachment give rise to multifunctional, robust materials suitable for cascade operation in batch or flow-through modes.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Representative examples of enzyme systems co-localized by the different approaches discussed in this contribution.
Table 1.
Representative examples of enzyme systems co-localized by the different approaches discussed in this contribution.
| Biocatalysts | Methods | Substrate | Product | Co-Factor | Ref. |
|---|---|---|---|---|---|
| XR, XDH | DNA origami | Xylose | Xylulose | NAD+, NADH | [37] |
| MDH, OAD, LDH | DNA origami | Malic acid | Lactate | NAD+, NADH | [38] |
| MenF, D, H | RIAD, RIDD | Chorismate | MKH2 | — | [39] |
| ACO, CAD | PDZ, SH3 | Glucose | Itaconic acid | — | [40,41] |
| LeuDH, FDH | PDZ | Trimethlypyruvate (TMP) | l-tert-Leucine | NAD+, NADH | [42] |
| ACAT, HMGS, HMGR | PDZ, SH3, GBD | Acetyl-CoA | Mevalonate | — | [43] |
| Xyn, Agu, β-Xyl, XylB | Cohesin–dockerin | Glucuromoxylan | d-Xylonic acid | — | [44] |
| MTS, MTH | Spy toolbox | Trehalose | Latopentaose | — | [45] |
| ADH, AmDH | Spy toolbox, EutM | (S)-2-Hexanol | (R)-2-Aminohexane | NAD+, NADH | [46] |
| ADH, Gre2P, GDH | SBP, Spy, Halo-tags | 5-Nitrononane-2,8-dione (NDK) | Meso anti-NDK | NADPH, NADP | [47] |
| LeuDH, FDH | Spy toolbox | TMP | l-tert-Leucine | NAD+, NADH | [48] |
| ACAT, HMGS, HMGR | Spy, Snoop toolbox | Acetyl-CoA | Mevalonate | — | [49] |
| VioA, B, C, D, E | Spy toolbox, DNA origami | l-Tryptophan | Violacein | — | [50] |
| CelB, GALK, GLUK | P22 VLPs | Lactose | G-1-P, G-6-P | ATP, ADP, AMP | [51] |
| TnaA, FMO | Liposome | l-Tryptophan | Indigo | — | [52] |
| CalB, GOx, HRP | Polymersome | Glucose acetate | Gluconolactone | — | [53] |
| PAMO, CalB, ADH | Polymersome | 7-((4-Oxopentyl)oxy)-3H-phenoxazin-3-one | Resorufin | NAD+, NADPH | [54] |
| AGE, NAL, CSS | Polymersome | GlgNAc | CMP-Neu5Ac | — | [55] |
Abbreviations of enzyme names: XR, xylose reductase; XDH, xylitol dehydrogenase; MDH, malate dehydrogenase; OAD, oxaloacetate decarboxylase; LDH, Lactate dehydrogenase; MenF, menaquinone isochorismate synthase F; MenD, menaquinone isochorismate synthase D; MenH, menaquinone isochorismate synthase H; ACO, aconitase; CAD, cis-aconitate decarboxylase; LeuDH, leucine dehydrogenase; FDH, formate dehydrogenase; ACAT, acetoacetyl-CoA thiolase; HMGS, hydroxy-methylglutatryl-CoA synthase; HMGR, hydroxy-methylglutatryl-CoA reductase; Xyn, xylanase; Agu, α-glucuronidase; β-Xyl, β-xylosidase; XylB, xylose dehydrogenase; MTS, maltooligosyl trehalose synthase; MTH, maltooligosyl trehalose trehalohydrolase; ADH, alcohol dehydrogenase; AmDH, amine dehydrogenase; Gre2P, methylglyoxal reductase; GDH, glucose dehydrogenase; VioA, violacein synthase A; VioB, violacein synthase B; VioC, violacein synthase C; VioD, violacein synthase D; VioE, violacein synthase E; CelB, β-glucosidase; GALK, galactokinase; GLUK, glucokinase; TnaA, pyridoxal phosphate-dependent tryptophanase; FMO, flavin monooxygenase; CalB, Candida antarctica lipase B; GOx, glucose oxidase; HRP, horseradish peroxidase; PAMO, phenylacetone monooxygenase; AGE, N-acyl-d-glucosamine 2-epimerase; NAL, N-acetylneuraminate lyase; CSS, CMP-sialic acid synthetase; RIAD, RI anchoring disruptor; RIDD, RI binding domain; PDZ, PSD95/DlgA/Zo-1 domain from the adaptor protein syntrophin; SH3, SRC homology 3 domain; GBD, GTPase binding domain; EutM, ethanolamine utilization bacterial microcompartment shell protein; SBP, streptavidin binding peptide; VLPs, virus-like particles.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Seo, M.-J.; Schmidt-Dannert, C. Organizing Multi-Enzyme Systems into Programmable Materials for Biocatalysis. Catalysts 2021, 11, 409. https://doi.org/10.3390/catal11040409
AMA Style
Seo M-J, Schmidt-Dannert C. Organizing Multi-Enzyme Systems into Programmable Materials for Biocatalysis. Catalysts. 2021; 11(4):409. https://doi.org/10.3390/catal11040409
Chicago/Turabian StyleSeo, Min-Ju, and Claudia Schmidt-Dannert. 2021. "Organizing Multi-Enzyme Systems into Programmable Materials for Biocatalysis" Catalysts 11, no. 4: 409. https://doi.org/10.3390/catal11040409
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.