Supported Tris-Triazole Ligands for Batch and Continuous-Flow Copper-Catalyzed Huisgen 1,3-Dipolar Cycloaddition Reactions


 , , ,
, , ,  ,
,
Abstract
: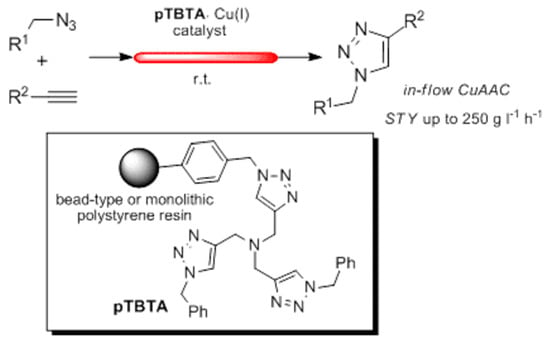
1. Introduction
2. Results
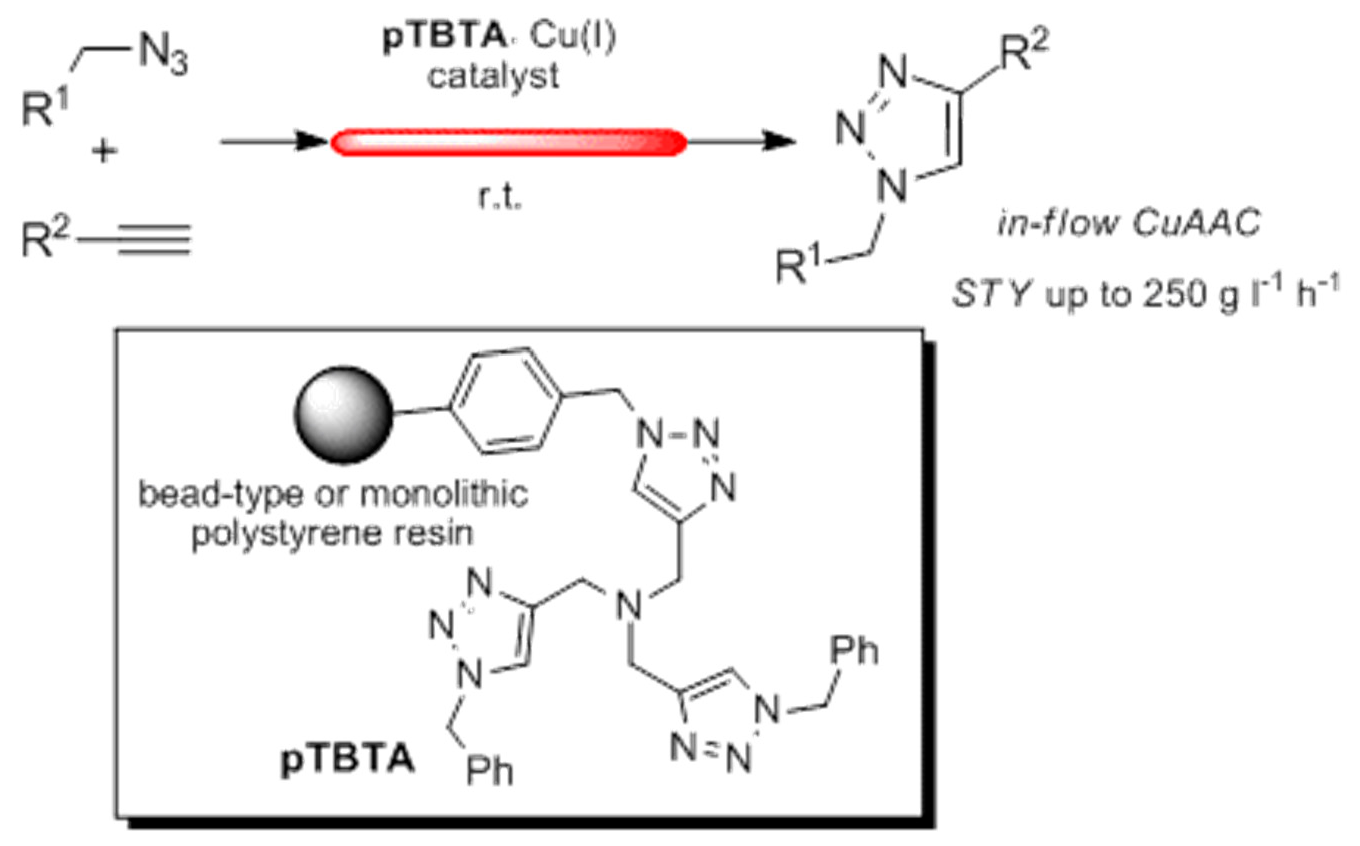
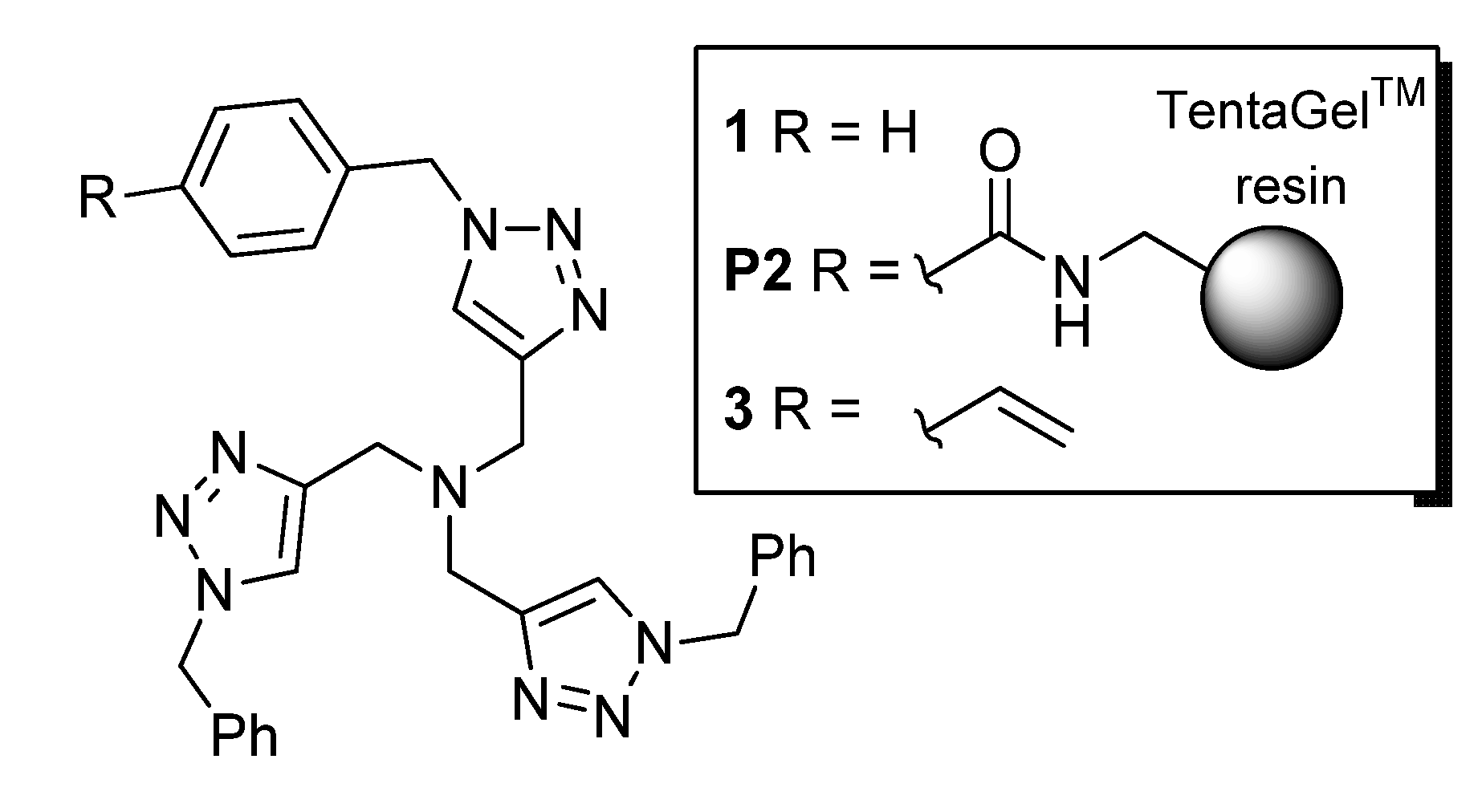
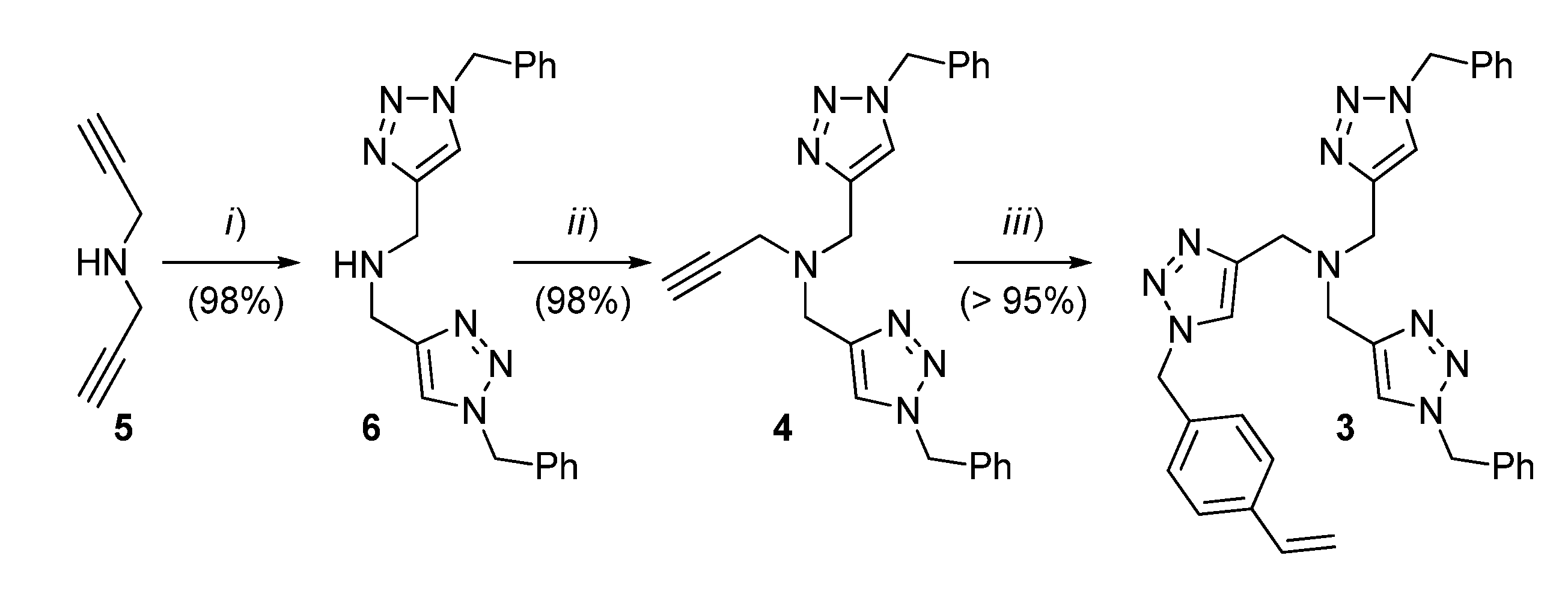
2.1. Preparation of the Functional Monomer 3
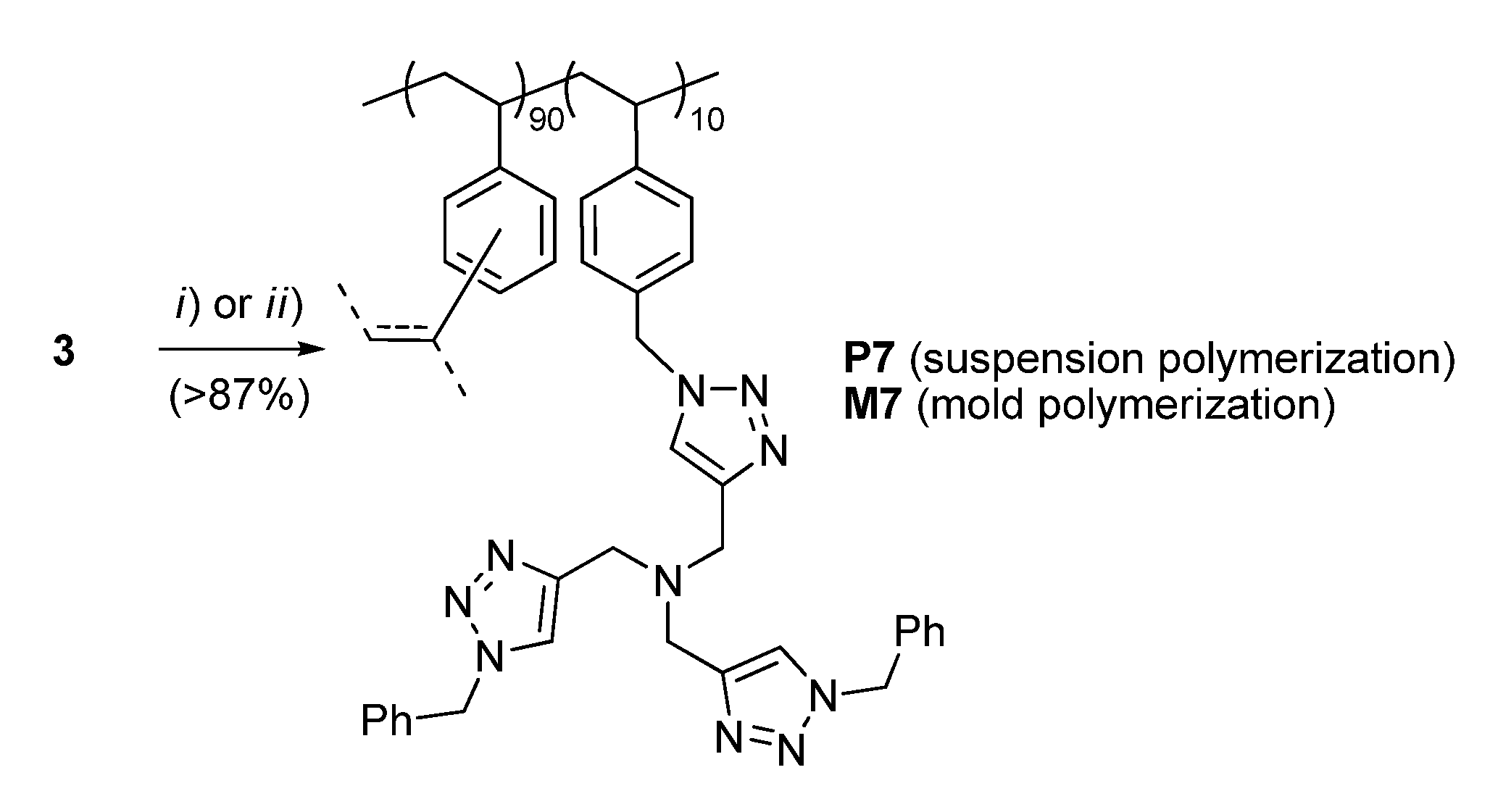
2.2. Preparation of the Bead-Type Supported Catalysts
2.3. Preparation of the Monolithic Reactors
2.4. CuAAC Reactions in Batch
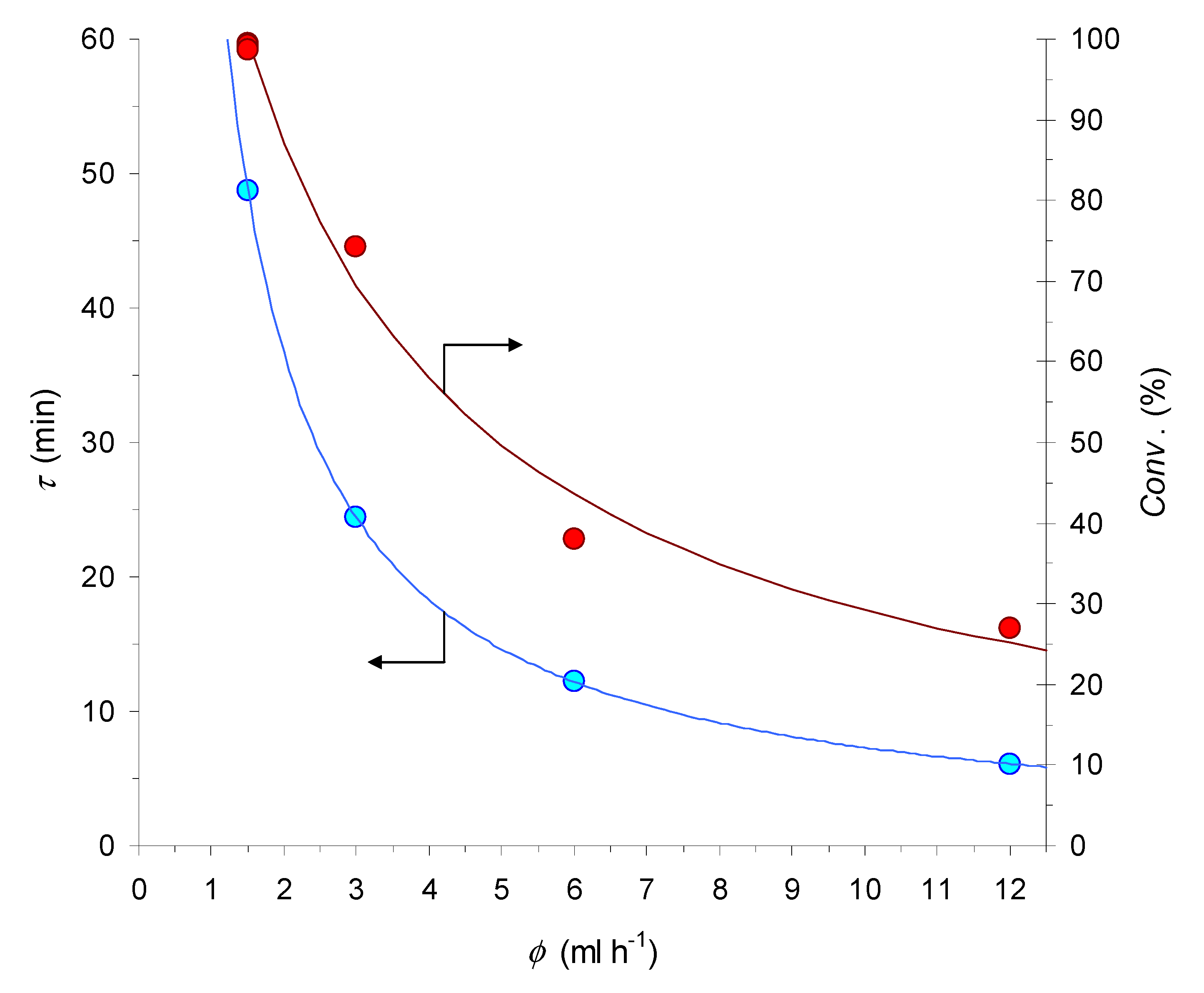
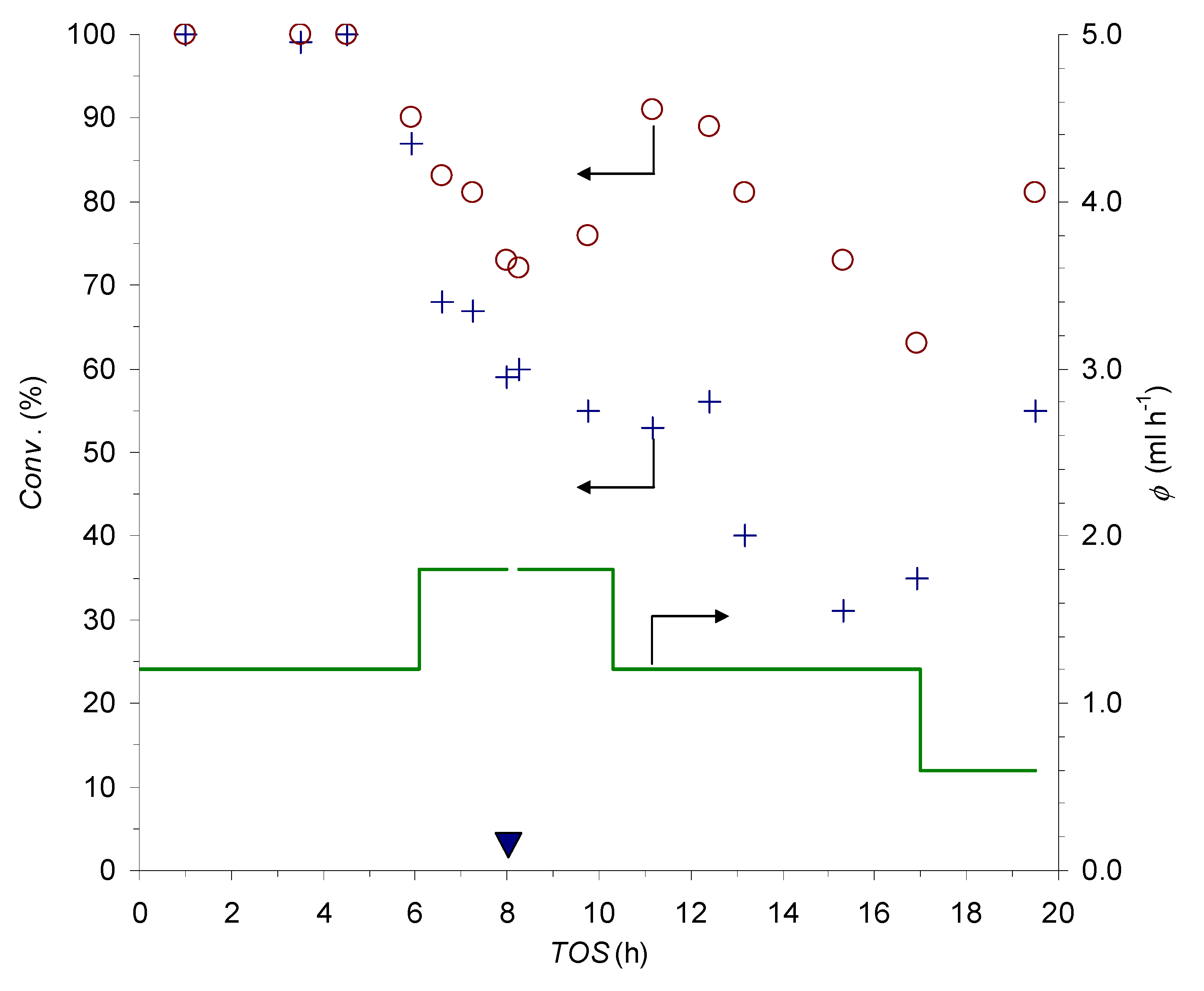
2.5. CuAAC Reactions in Flow
3. Discussion
3.1. Preparation of the TBTA Monomer and Supported Catalysts
3.2. Catalysis in Batch and in Flow
4. Materials and Methods
4.1. CuAAC in Batch with P7-Supported Catalysts: General Procedure
4.2. CuAAC in Flow: General Procedure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Gil, M.V.; Arevalo, M.J.; Lopez, O. Click chemistry—What‘s in a name? Triazole synthesis and beyond. Synthesis 2007, 2007, 1589–1620. [Google Scholar] [CrossRef]
- Wang, C.; Ikhlef, D.; Kahlal, S.; Saillard, J.-Y.; Astruc, D. Metal-catalyzed azide-alkyne “click” reactions: Mechanistic overview and recent trends. Coord. Chem. Rev. 2016, 316, 1–20. [Google Scholar] [CrossRef]
- Tabacaru, A.; Furdui, B.; Ghinea, I.O.; Carac, G.; Dinica, R.M. Recent advances in click chemistry reactions mediated by transition metal based systems. Inorg. Chim. Acta 2017, 455, 329–349. [Google Scholar] [CrossRef]
- Huo, J.; Hu, H.; Zhang, M.; Hu, X.; Chen, M.; Chen, D.; Liu, J.; Xiao, G.; Wang, Y.; Wen, Z. A mini review of the synthesis of poly-1,2,3-triazole-based functional materials. RSC Adv. 2017, 7, 2281–2287. [Google Scholar] [CrossRef]
- Pickens, C.J.; Johnson, S.N.; Pressnall, M.M.; Leon, M.A.; Berkland, C.J. Practical considerations, challenges, and limitations of bioconjugation via azide-alkyne cycloaddition. Bioconjugate Chem. 2018, 29, 686–701. [Google Scholar] [CrossRef]
- Haldon, E.; Nicasio, M.C.; Perez, P.J. Copper-catalysed azide-alkyne cycloadditions (CuAAC): An update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as Copper(I)-stabilizing ligands in catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef]
- Rodionov, V.O.; Presolski, S.I.; Gardinier, S.; Lim, Y.-H.; Finn, M.G. Benzimidazole and related ligands for Cu-catalyzed azide−alkyne cycloaddition. J. Am. Chem. Soc. 2007, 129, 12696–12704. [Google Scholar] [CrossRef]
- Wang, C.; Wang, D.; Yu, S.; Cornilleau, T.; Ruiz, J.; Salmon, L.; Astruc, D. Design and applications of an efficient amphiphilic “click” Cu(I) catalyst in water. ACS Catal. 2016, 6, 5424–5431. [Google Scholar] [CrossRef]
- Hong, V.; Presolski, S.I.; Ma, C.; Finn, M.G. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew. Chem. Int. Ed. 2009, 48, 9879–9883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dervaux, B.; Du Prez, F.E. Heterogeneous azide-alkyne click chemistry: Towards metal-free end products. Chem. Sci. 2012, 3, 959–966. [Google Scholar] [CrossRef]
- Monguchi, Y.; Sawama, Y.; Sajiki, H. Synthesis of triazole, indole, and five or six-membered saturated heterocyclic compounds. Heterocycles 2015, 91, 239–264. [Google Scholar]
- Chassaing, S.; Beneteau, V.; Pale, P. When CuAAC ’click chemistry’ goes heterogeneous. Catal. Sci. Technol. 2016, 6, 923–957. [Google Scholar] [CrossRef]
- Mandoli, A. Recent advances in recoverable systems for the copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC). Molecules 2016, 21, 1174. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.R.; Fokin, V.V. Polymer-supported copper(I) catalysts for the experimentally simplified azide–alkyne cycloaddition. QSAR Comb. Sci. 2007, 26, 1274–1279. [Google Scholar] [CrossRef]
- Sigma-Aldrich Product Cat. 696773. Available online: https://www.sigmaaldrich.com/catalog/product/aldrich/696773 (accessed on 28 February 2020).
- De Bo, G.; Kuschel, S.; Leigh, D.A.; Lewandowski, B.; Papmeyer, M.; Ward, J.W. Efficient assembly of threaded molecular machines for sequence-specific synthesis. J. Am. Chem. Soc. 2014, 136, 5811–5814. [Google Scholar] [CrossRef]
- Lammens, M.; Skey, J.; Wallyn, S.; O’Reilly, R.; Du Prez, F. Polymeric ligands as homogeneous, reusable catalyst systems for copper assisted click chemistry. Chem. Commun. 2010, 46, 8719–8721. [Google Scholar] [CrossRef]
- Wallyn, S.; Lammens, M.; O’Reilly, R.K.; Du Prez, F. Highly active, thermo-responsive polymeric catalytic system for reuse in aqueous and organic CuAAC reactions. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 2878–2885. [Google Scholar] [CrossRef] [Green Version]
- Movahedi, A.; Moth-Poulsen, K.; Ekloef, J.; Nyden, M.; Kann, N. One-pot synthesis of TBTA-functionalized coordinating polymers. React. Funct. Polym. 2014, 82, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, A.E.; Ye, Q.; Collard, L.; Le Duff, C.; d’Haese, C.; Deumer, G.; Haufroid, V.; Nysten, B.; Riant, O.; Jonas, A.M. Effects of thickness and grafting density on the activity of polymer-brush-immobilized tris(triazolyl) Copper(I) catalysts. ChemCatChem 2015, 7, 856–864. [Google Scholar] [CrossRef]
- Yamada Yoichi, M.A.; Ohno, A.; Sato, T.; Uozumi, Y. Instantaneous click chemistry by a copper-containing polymeric-membrane-installed microflow catalytic reactor. Chem. Eur. J. 2015, 21, 17269–17273. [Google Scholar] [CrossRef] [PubMed]
- Mandity, I.M.; Oetvoes, S.B.; Szolosi, G.; Fueloep, F. Harnessing the versatility of continuous-flow processes: Selective and efficient reactions. Chem. Rec. 2016, 16, 1018–1033. [Google Scholar] [CrossRef]
- Pan, S.; Yan, S.; Osako, T.; Uozumi, Y. Batch and continuous-flow Huisgen 1,3-dipolar cycloadditions with an amphiphilic resin-supported triazine-based polyethyleneamine dendrimer copper catalyst. ACS Sustain. Chem. Eng. 2017, 5, 10722–10734. [Google Scholar] [CrossRef]
- Manzano, J.S.; Weinstein, Z.B.; Sadow, A.D.; Slowing, I.I. Direct 3D printing of catalytically active structures. ACS Catal. 2017, 7, 7567–7577. [Google Scholar] [CrossRef] [Green Version]
- Hatit, M.Z.C.; Reichenbach, L.F.; Tobin, J.M.; Vilela, F.; Burley, G.A.; Watson, A.J.B. A flow platform for degradation-free CuAAC bioconjugation. Nat. Commun. 2018, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Wu, K.; Yang, D.; Tian, J.; Huang, Z.; Filatov, A.S.; Lei, A.; Lin, X.-M. Low-pressure flow chemistry of CuAAC click reaction catalyzed by nanoporous AuCu membrane. ACS Appl. Mater. Interfaces 2018, 10, 25930–25935. [Google Scholar] [CrossRef]
- Mandoli, A. Catalyst recycling in continuous flow reactors. In Catalyst Immobilization. Methods and Applications; Benaglia, M., Puglisi, A., Eds.; Wiley-VCH: Weinheim, Germany, 2019; pp. 257–306. [Google Scholar]
- Santos de Sa, D.; Bustamante, R.d.A.; Rocha, C.E.R.; Diniz da Silva, V.; Rodrigues, E.J.d.R.; Muller, C.D.B.; Ghavami, K.; Massi, A.; Pandoli, O.G. Fabrication of lignocellulose-based microreactors: Copper-functionalized bamboo for continuous-flow CuAAC click reactions. ACS Sustain. Chem. Eng. 2019, 7, 3267–3273. [Google Scholar] [CrossRef]
- Zhang, C.; Feng, X.; Wang, B.; Mao, Z.; Xu, H.; Zhong, Y.; Zhang, L.; Chen, X.; Sui, X. Nanocellulose sponges as efficient continuous flow reactors. Carbohydr. Polym. 2019, 224, 115184. [Google Scholar] [CrossRef]
- Orlandi, S.; Mandoli, A.; Pini, D.; Salvadori, P. An insoluble polymer-bound bis-oxazoline copper(II) complex: A highly efficient heterogeneous catalyst for the enantioselective Mukaiyama aldol reaction. Angew. Chem. Int. Ed. 2001, 40, 2519–2521. [Google Scholar] [CrossRef]
- Mandoli, A.; Orlandi, S.; Pini, D.; Salvadori, P. Insoluble polystyrene-bound bis(oxazoline): Batch and continuous-flow heterogeneous enantioselective glyoxylate-ene reaction. Tetrahedron Asymmetry 2004, 15, 3233–3244. [Google Scholar] [CrossRef]
- Mandoli, A.; Lessi, M.; Pini, D.; Evangelisti, C.; Salvadori, P. Remarkable efficiency improvement in the preparation of insoluble polymer-bound (IPB) enantioselective catalytic systems by the use of silicone chemistry. Adv. Synth. Catal. 2008, 350, 375–379. [Google Scholar] [CrossRef]
- Mandoli, A.; Garzelli, R.; Orlandi, S.; Pini, D.; Lessi, M.; Salvadori, P. Some factors affecting the catalytic efficiency in the enantioselective cyclopropanation of olefins by the use of insoluble polystyrene-bound bisoxazoline-copper(I) complex. Catal. Today 2009, 140, 51–57. [Google Scholar] [CrossRef]
- Jumde, R.P.; Evangelisti, C.; Mandoli, A.; Scotti, N.; Psaro, R. Aminopropyl-silica-supported Cu nanoparticles: An efficient catalyst for continuous-flow Huisgen azide-alkyne cycloaddition (CuAAC). J. Catal. 2015, 324, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Balerna, A.; Evangelisti, C.; Tiozzo, C. XAFS structural characterization of cu vapour derived catalysts supported on poly-4-vinylpyridine and carbon. X-ray Spectrom. 2017, 46, 82–87. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, P.; Zhu, L. Structural determinants of alkyne reactivity in copper-catalyzed azide-alkyne cycloadditions. Molecules 2016, 21, 1697. [Google Scholar] [CrossRef] [Green Version]
- Svec, F.; Frechet, J.M.J. Kinetic control of pore formation in macroporous polymers. Formation of “molded” porous materials with high flow characteristics for separations or catalysis. Chem. Mater. 1995, 7, 707–715. [Google Scholar] [CrossRef]
- Viklund, C.; Svec, F.; Fréchet, J.M.J.; Irgum, K. Monolithic, “molded”, porous materials with high flow characteristics for separations, catalysis, or solid-phase chemistry: Control of porous properties during polymerization. Chem. Mater. 1996, 8, 744–750. [Google Scholar] [CrossRef]
- Peters, E.C.; Svec, F.; Fréchet, J.M.J. Rigid macroporous polymer monoliths. Adv. Mater. (Weinh. Ger.) 1999, 11, 1169–1181. [Google Scholar] [CrossRef]
- Altava, B.; Burguete, M.I.; Fraile, J.M.; García, J.I.; Luis, S.V.; Mayoral, J.A.; Vicent, M.J. How important is the inert matrix of supported enantiomeric catalysts? Reversal of topicity with two polystyrene backbones. Angew. Chem. Int. Ed. 2000, 39, 1503–1506. [Google Scholar] [CrossRef]
- Sherrington, D.C. Preparation, structure and morphology of polymer supports. Chem. Commun. 1998, 2275–2286. [Google Scholar] [CrossRef]
- Hu, H.; Ohno, A.; Sato, T.; Mase, T.; Uozumi, Y.; Yamada, Y.M.A. Self-assembled polymeric pyridine copper catalysts for Huisgen cycloaddition with alkynes and acetylene gas: Application in synthesis of tazobactam. Org. Process Rev. Dev. 2019, 23, 493–498. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Z.; Huang, X.; Zhang, Q. Determination, correlation, and application of sodium l-ascorbate solubility in nine pure solvents and two binary solvents at temperatures from 278.15 to 323.15 k. J. Chem. Eng. Data 2018, 63, 233–245. [Google Scholar] [CrossRef]
- El Kadib, A.; Chimenton, R.; Sachse, A.; Fajula, F.; Galarneau, A.; Coq, B. Functionalized inorganic monolithic microreactors for high productivity in fine chemicals catalytic synthesis. Angew. Chem. Int. Ed. 2009, 48, 4969–4972. [Google Scholar] [CrossRef]
- Megia-Fernandez, A.; Ortega-Muñoz, M.; Lopez-Jaramillo, J.; Hernandez-Mateo, F.; Santoyo-Gonzalez, F. Non-magnetic and magnetic supported Copper(I) chelating adsorbents as efficient heterogeneous catalysts and copper scavengers for click chemistry. Adv. Synth. Catal. 2010, 352, 3306–3320. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Campbell-Verduyn, L.; Elsinga, P.H.; Mirfeizi, L.; Dierckx, R.A.; Feringa, B.L. Copper-free ‘click’: 1,3-dipolar cycloaddition of azides and arynes. Org. Biomol. Chem. 2008, 6, 3461–3463. [Google Scholar] [CrossRef]
- Presolski, S.I.; Hong, V.; Cho, S.-H.; Finn, M.G. Tailored ligand acceleration of the Cu-catalyzed azide−alkyne cycloaddition reaction: Practical and mechanistic implications. J. Am. Chem. Soc. 2010, 132, 14570–14576. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Díez-González, S.; Correa, A.; Cavallo, L.; Nolan, S.P. (nhc)Copper(I)-catalyzed [3+2] cycloaddition of azides and mono- or disubstituted alkynes. Chem. Eur. J. 2006, 12, 7558–7564. [Google Scholar] [CrossRef]
- Sarkar, A.; Mukherjee, T.; Kapoor, S. PVP-stabilized copper nanoparticles: A reusable catalyst for “click” reaction between terminal alkynes and azides in nonaqueous solvents. J. Phys. Chem. C 2008, 112, 3334–3340. [Google Scholar] [CrossRef]
- Sharghi, H.; Khalifeh, R.; Doroodmand, M.M. Copper nanoparticles on charcoal for multicomponent catalytic synthesis of 1,2,3-triazole derivatives from benzyl halides or alkyl halides, terminal alkynes and sodium azide in water as a “green” solvent. Adv. Synth. Catal. 2009, 351, 207–218. [Google Scholar] [CrossRef]
- Borgati, T.F.; Alves, R.B.; Teixeira, R.R.; de Freitas, R.P.; Perdigao, T.G.; da Silva, S.F.; dos Santos, A.A.; de Jesus, O.; Bastidas, A. Synthesis and phytotoxic activity of 1,2,3-triazole derivatives. J. Braz. Chem. Soc. 2013, 24, 953–961. [Google Scholar] [CrossRef]
- Chahdoura, F.; Pradel, C.; Gómez, M. Copper(I) oxide nanoparticles in glycerol: A convenient catalyst for cross-coupling and azide–alkyne cycloaddition processes. ChemCatChem 2014, 6, 2929–2936. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry a | CuX1-2 | Reducing | R1, R2 | Solvent | Conv. |
|---|---|---|---|---|---|
| (S/C) b | agent c | (volume ratio) | (%) d | ||
| 1 | CuCl (10) | - | Ph, CH2OH | CH2Cl2 | 71/>99 |
| 2 | CuCl (20) e | - | Ph, CH2OH | CH2Cl2 | 54/>99 |
| 3 | CuCl (50) e | - | Ph, CH2OH | CH2Cl2 | 33/84 |
| 4 | CuCl (50) e | - | Ph, CH2OH | CH2Cl2 | 31/83 |
| 5 | CuCl (50) e | - | 4-Br-C6H4, CH2OH | CH2Cl2 | 30/83 |
| 6 | Cu(OTf)2 (10) | PH | Ph, CH2OH | CH2Cl2 | <5/21 |
| 7 | Cu(OTf)2 (20) e | - | Ph, CH2OH | CH2Cl2 | 19/40 |
| 8 | Cu(OTf)2 (20) | HQ | Ph, CH2OH | THF | <5/<5 |
| 9 | Cu(OTf)2 (20) e | ASC | Ph, CH2OH | MeOH/THF (4:1) | 78/>99 |
| 10 | Cu(OTf)2 (20) e | - | Ph, CH2OH | MeOH/THF (4:1) | 89/>99 |
| 11 | Cu(OTf)2 (100) e | - | Ph, CH2OH | MeOH/THF (4:1) | 50/99 |
| 12 | Cu(OTf)2 (100) e | ASC f | Ph, CH2OH | MeOH/THF (4:1) | 24/>99 |
| 13 | CuSO4 (167) | ASC | Ph, CH2OH | MeOH/THF (20:1) | 52/>99 |
| 14 | CuSO4 (167) e | - | Ph, n-Bu | MeOH/THF (20:1) | <5/57 g |
| 15 | CuSO4 (167) h | ASC | Ph, CH2OH | MeOH/THF (20:1) | >99/- |
| 16 | CuSO4 (167) h | ASC | Ph, n-Bu | MeOH/THF (20:1) | 15/93 |

| Entry a | Device Type | Reducing | Solvent | S/Cc | T | ф | τ | Conv. | STY |
|---|---|---|---|---|---|---|---|---|---|
| (length/cm) | agent b | (volume ratio) | (°C) | (mL h−1) d | (min) e | (%) f | (g L−1 h−1) g | ||
| 1 | MCFR (15) | HQ | THF | 5.0 | 24 | 0.6 | 190 | <5 | - |
| 2 | MCFR (15) h | PH | THF | 5.0 | 24 | 0.4 | 280 | 8 | <2 |
| 3 | MCFR (10) | ASC | MeOH/THF (4:1) | 67 | 24 | 1.5 | 48 | >99 | 34 |
| 4 | MCFR (10) h | (ASC) | MeOH/THF (4:1) | 16 | 24 | 12.0 | 6 | 27 | 74 |
| 5 | MCFR (10) h | (ASC) | MeOH/THF (4:1) | 16 | 24 | 6.0 | 12 | 38 | 52 |
| 6 | MCFR (10) h | (ASC) | MeOH/THF (4:1) | 47 | 24 | 3.0 | 24 | 74 | 51 |
| 7 | MCFR (10) h | (ASC) | MeOH/THF (4:1) | 77 | 24 | 1.5 | 48 | 99 | 34 |
| 8 | PCFR (15) | HQ | THF | 5.3 | 24 | 0.6 | 205 | <5 | - |
| 9 | PCFR (15) h | PH | THF | 5.3 | 24 | 0.4 | 310 | 47 | 4 |
| 10 | PCFR (15) h | ASC | MeOH/THF (4:1) | 7.2 | 24 | 6.0 | 19 | 57 | 70 |
| 11 | PCFR (15) h | (ASC) | MeOH/THF (4:1) | 6.5 | 60 | 3.0 | 39 | >99 | 60 |
| 12 | PCFR (15) h | (ASC) | MeOH/THF (4:1) | 6.5 | 60 | 3.0 | 39 | 89 | 53 |
| 13 | PCFR (15) h | (ASC) | MeOH/THF (4:1) | 6.5 | 60 | 3.0 | 39 | 97 | 58 |
| 14 | PCFR (15) h | (ASC) | MeOH/THF (4:1) | 6.5 | 60 | 3.0 | 39 | 91 | 55 |
| 15 | PCFR (15) h | (ASC) | MeOH/THF (4:1) | 6.5 | 60 | 3.0 | 39 | 85 | 51 |

| Entry a | Device | R1, R2 | S/Cb | Solvent | [Azide] | ф | τ | Yield | STY | Cu |
|---|---|---|---|---|---|---|---|---|---|---|
| (v/v) | (M) | (mL h−1) c | (min) d | (%) e | (g L−1 h−1) f | (ppm) g | ||||
| 1 h | R1 | Ph, CH2OH | 347 | MeOH/THF (4/1) | 0.20 | 0.6–1.8 | 11–32 | 88 i | 63 | - |
| 2 | R1l | Ph, CH2OH | 178 | MeOH/THF (3/2) | 0.20 | 0.6 | 32 | 49 | 64 | - |
| 3 h | R2 | Ph, CH2OH | 349 | MeOH/H2O (4/1) | 0.21 | 0.6–1.8 | 11–32 | 68 i | 63 | - |
| 4 | R2j | Ph, n-Bu | 174 | MeOH/THF (4/1) | 0.20 | 0.6 | 32 | 39 | 71 | 138 |
| 5 | R3 | Ph, CH2OH | 71 | MeOH/THF (4/1) | 0.20 | 0.6 | 32 | 91 | 64 | 175 |
| 6 | R3j | Ph, CH2CH2OH | 70 | MeOH/THF (4/1) | 0.20 | 0.6 | 32 | 92 | 68 | - |
| 7 | R3j | Ph, Ph | 71 | MeOH/THF (4/1) | 0.21 | 0.6 | 32 | 78 k | 80 | - |
| 8 | R3j | Ph, Ph | 73 | MeOH/THF (1/1) | 0.21 | 0.6 | 32 | 96 | 82 | 148 |
| 9 | R4 | Bn, CH(OH)CH3 | 126 | MeOH/THF (4/1) | 0.18 | 0.6 | 32 | 83 | 65 | - |
| 10 | R4j | Ph, (CH2)4C≡CH | 147 | MeOH/THF (4/1) | 0.21 | 0.6 | 32 | 38 l | 131 | - |
| 11 | R4j | Ph, CH2NHCH2C≡CH | 291 | MeOH/THF (1/1) | 0.42 | 0.6 | 32 | 95 | 250 | - |
| 12 | R5 | Bn, C(OH)(CH3)2 | 125 | MeOH/THF (4/1) | 0.18 | 0.6 | 32 | 90 | 69 | - |
| 13 | R5j | Ph, CH2NHCH2C≡CH | 141 | MeOH/THF (4/1) | 0.21 | 0.6 | 32 | 97 | 125 | 853 |
| 14 | R5j | Ph, CH2NHCH2C≡CH | 276 | MeOH/THF (1/1) | 0.42 | 0.6 | 32 | 60 m | 249 | 257 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pucci, A.; Albano, G.; Pollastrini, M.; Lucci, A.; Colalillo, M.; Oliva, F.; Evangelisti, C.; Marelli, M.; Santalucia, D.; Mandoli, A. Supported Tris-Triazole Ligands for Batch and Continuous-Flow Copper-Catalyzed Huisgen 1,3-Dipolar Cycloaddition Reactions. Catalysts 2020, 10, 434. https://doi.org/10.3390/catal10040434
Pucci A, Albano G, Pollastrini M, Lucci A, Colalillo M, Oliva F, Evangelisti C, Marelli M, Santalucia D, Mandoli A. Supported Tris-Triazole Ligands for Batch and Continuous-Flow Copper-Catalyzed Huisgen 1,3-Dipolar Cycloaddition Reactions. Catalysts. 2020; 10(4):434. https://doi.org/10.3390/catal10040434
Chicago/Turabian StylePucci, Alessandra, Gianluigi Albano, Matteo Pollastrini, Antonio Lucci, Marialuigia Colalillo, Fabrizio Oliva, Claudio Evangelisti, Marcello Marelli, Delio Santalucia, and Alessandro Mandoli. 2020. "Supported Tris-Triazole Ligands for Batch and Continuous-Flow Copper-Catalyzed Huisgen 1,3-Dipolar Cycloaddition Reactions" Catalysts 10, no. 4: 434. https://doi.org/10.3390/catal10040434