
Differential Effect of Non-Steroidal Anti-Inflammatory Drugs Aspirin and Naproxen against TMPRSS2-ERG (Fusion)-Driven and Non-Fusion-Driven Prostate Cancer
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Animal Diets
2.3. Euthanasia and necropsy
2.4. Histopathological and Immunohistochemical Evaluation of Prostatic Tissues
2.5. Statistical and Microscopic Analyses
3. Results
3.1. Effects of Aspirin and Naproxen Intervention on Cancer Progression in Different Prostate Lobes of TMPRSS2-ERG Fusion-Driven and Non-Fusion-Driven Hi-Myc+/- PCa Models
3.2. Differential Effects of Aspirin and Naproxen Intervention on the Expression of Proliferative Markers and ETS Transcription Factor (ERG) in the Prostate of TMPRSS2-ERG Fusion-Driven and Non-Fusion-Driven Hi-Myc+/- PCa Models
3.2.1. C-Myc
3.2.2. ERG
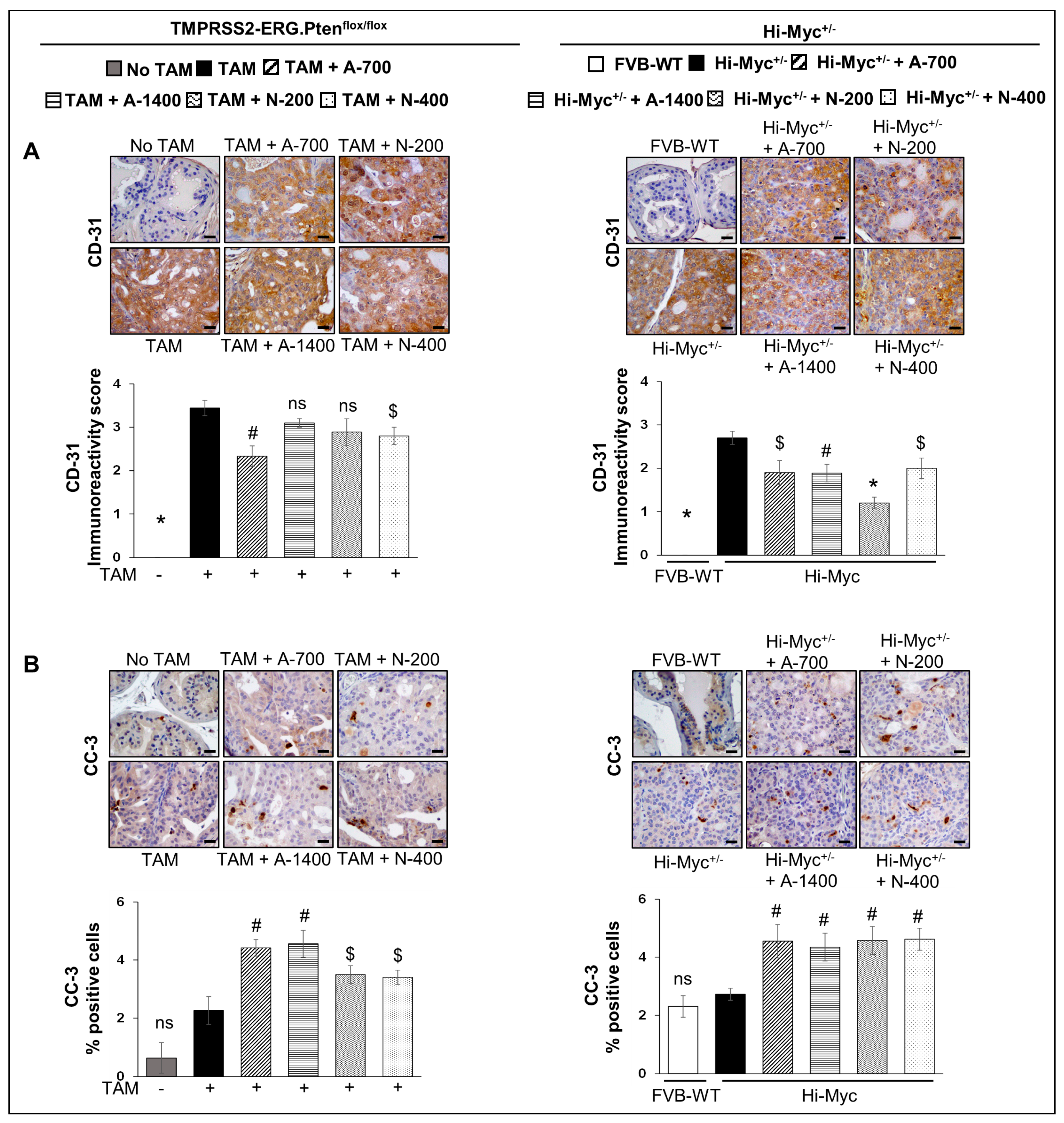
3.3. Differential Effects of Aspirin and Naproxen Intervention on Angiogenesis and Apoptosis Markers in the Prostate of TMPRSS2-ERG Fusion-Driven and Non-Fusion-Driven Hi-Myc+/− PCa Models
3.3.1. CD-31 (PECAM-1)
3.3.2. Cleaved Capsase-3
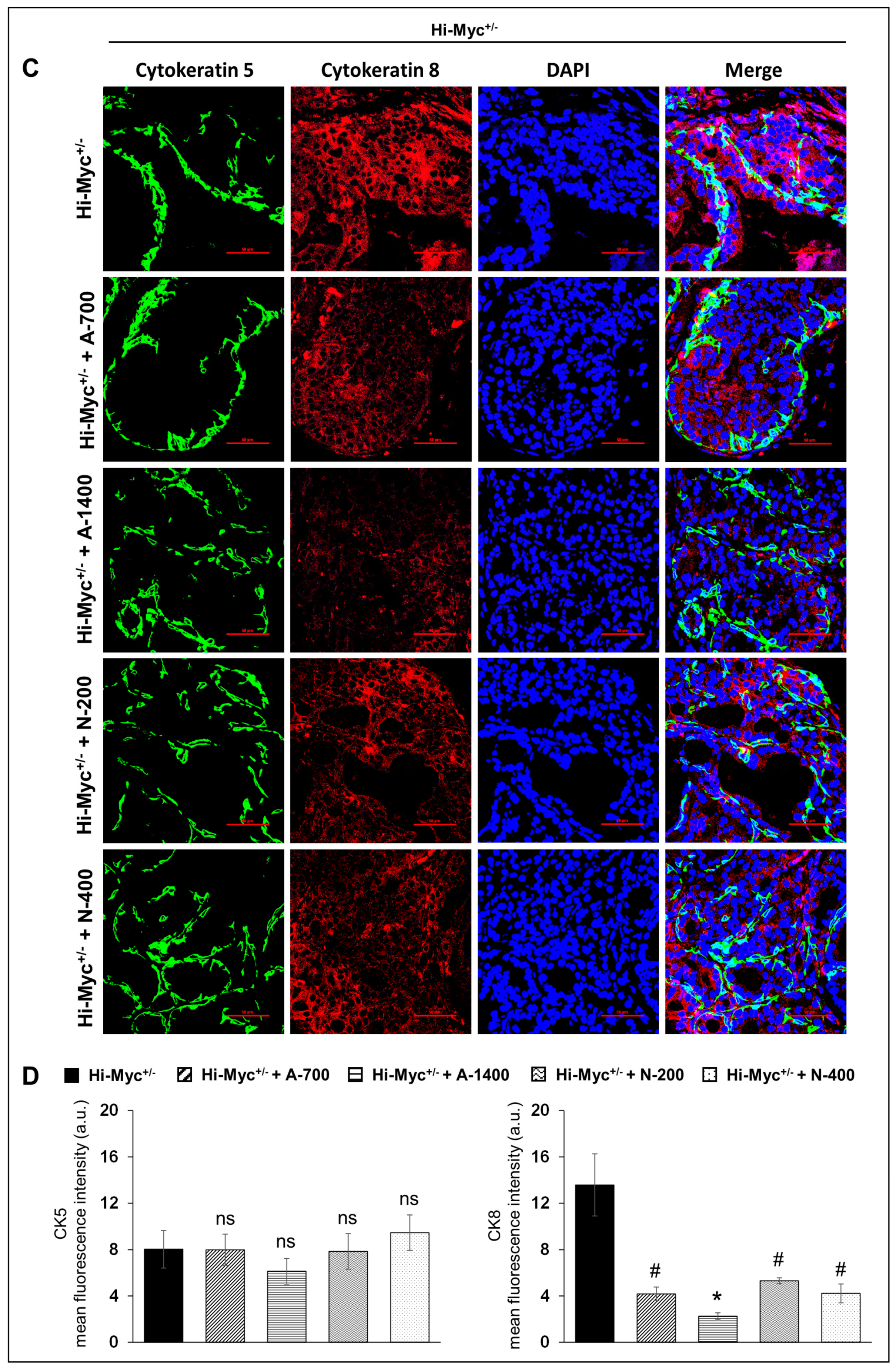
3.4. Differential Effects of Aspirin and Naproxen Intervention on the Cell Type Distribution (Basal, Luminal) in the Prostate of TMPRSS2-ERG Fusion-Driven and Non-Fusion-Driven Hi-Myc+/− PCa Models
(CK5/CK8) Basal/Luminal Epithelial Cell Markers
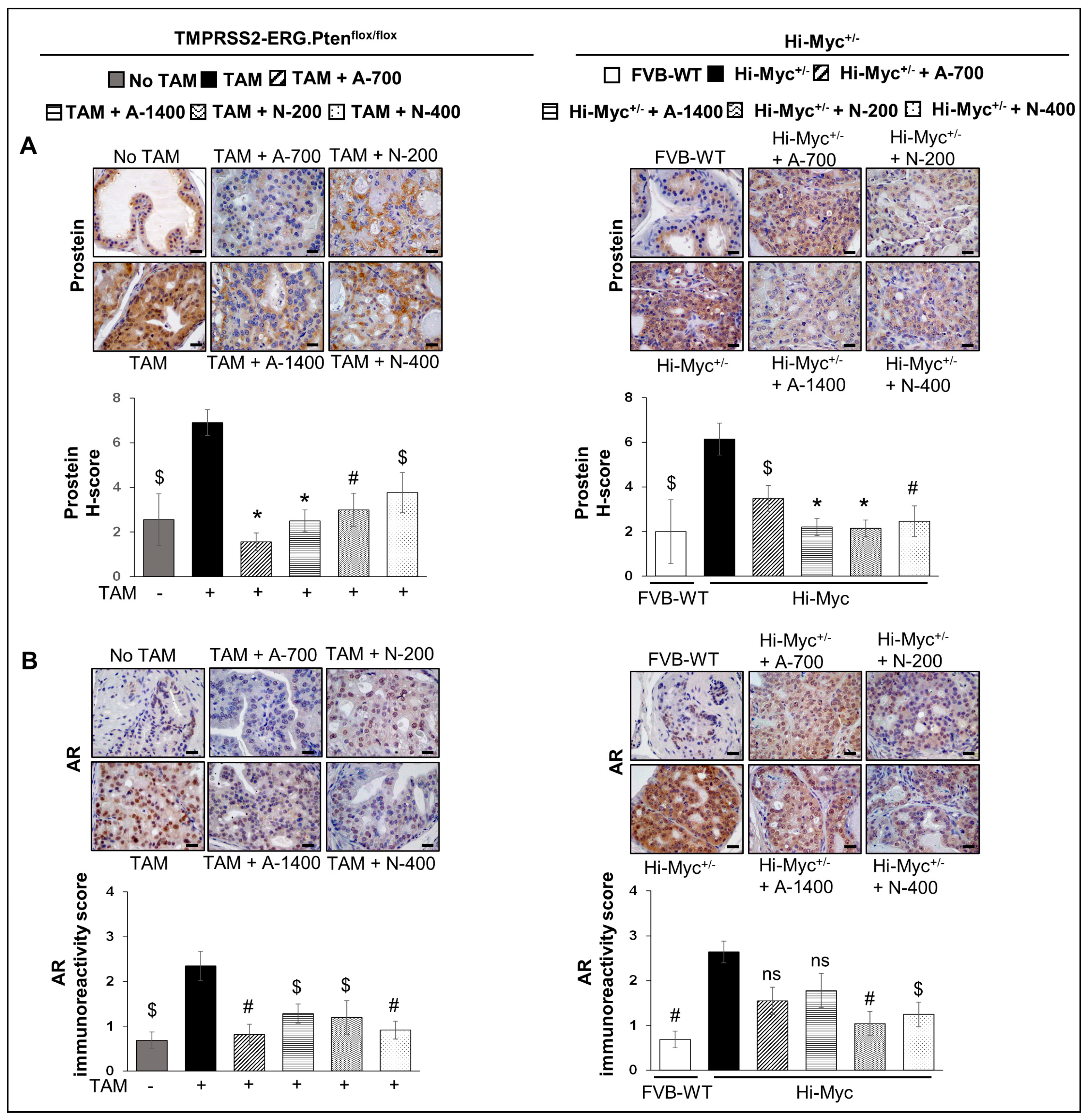
3.5. Differential Effects of Aspirin and Naproxen Intervention on the Expression of Prostate-Specific Solute Carrier Family Member (Prostein) and Androgen Receptor (AR) in the Prostate of TMPRSS2-ERG Fusion-Driven and Non-Fusion-Driven Hi-Myc+/− PCa Models
3.5.1. Prostein (SLC45A3)
3.5.2. AR
3.6. Differential Effects of Aspirin and Naproxen Interventions on the Expression of Inflammation-Related Markers in the Prostate of TMPRSS2-ERG Fusion-Driven and Non-Fusion-Driven Hi-Myc+/− PCa Models
3.6.1. NFκb (Total p65)
3.6.2. COX-1 and COX-2
3.7. Differential Effects of Aspirin and Naproxen Intervention on the Expression of Epithelial to Mesenchymal Transition Markers in the Prostate of TMPRSS2-ERG Fusion-Driven and Non-Fusion-Driven Hi-Myc+/− PCa Models
E-Cadherin and Vimentin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PCa | prostate cancer |
| TMPRSS2 | transmembrane protease serine 2 |
| Ets | erythroblastosis virus E26 |
| ERG | Ets related gene |
| TAM | tamoxifen |
| LUT | lower urogenital tract |
| PIN | prostatic intraepithelial neoplasia |
| LGPIN | low-grade PIN |
| HGPIN | high-grade PIN |
| WD | well differentiated |
| MD | moderately differentiated |
| PD | poorly differentiated |
| PCNA | proliferation cell nuclear antigen |
| PECAM-1/CD-31 | platelet endothelial cell adhesion molecule-1 |
| DAB | 3,3′-diaminobenzidine |
| IHC | immunohistochemistry |
References
- Salinas, C.A.; Kwon, E.M.; FitzGerald, L.M.; Feng, Z.; Nelson, P.S.; Ostrander, E.A.; Peters, U.; Stanford, J.L. Use of aspirin and other nonsteroidal anti-inflammatory medications in relation to prostate cancer risk. Am. J. Epidemiol. 2010, 172, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.L.; Chery, L.; Holt, S.; Lin, D.W.; Luedeke, M.; Rinckleb, A.E.; Maier, C.; Stanford, J.L. Aspirin and NSAID use in association with molecular subtypes of prostate cancer defined by TMPRSS2:ERG fusion status. Prostate Cancer Prostatic. Dis. 2016, 19, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Doat, S.; Cenee, S.; Tretarre, B.; Rebillard, X.; Lamy, P.J.; Bringer, J.P.; Iborra, F.; Murez, T.; Sanchez, M.; Menegaux, F. Nonsteroidal anti-inflammatory drugs (NSAIDs) and prostate cancer risk: Results from the EPICAP study. Cancer Med. 2017, 6, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, L.; Ai, G.; Spitale, R.C.; Bhat, G.J. Molecular targets of aspirin and cancer prevention. Br. J. Cancer 2014, 111, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, H.; Kawahara, T. Nonsteroidal anti-inflammatory drugs and prostatic diseases. Biomed. Res. Int. 2014, 2014, 436123. [Google Scholar] [CrossRef]
- Shao, N.; Feng, N.; Wang, Y.; Mi, Y.; Li, T.; Hua, L. Systematic review and meta-analysis of COX-2 expression and polymorphisms in prostate cancer. Mol. Biol. Rep. 2012, 39, 10997–11004. [Google Scholar] [CrossRef]
- Kirschenbaum, A.; Liu, X.; Yao, S.; Levine, A.C. The role of cyclooxygenase-2 in prostate cancer. Urology 2001, 58, 127–131. [Google Scholar] [CrossRef]
- Taverna, G.; Pedretti, E.; Di Caro, G.; Borroni, E.M.; Marchesi, F.; Grizzi, F. Inflammation and prostate cancer: Friends or foe? Inflamm. Res. 2015, 64, 275–286. [Google Scholar] [CrossRef]
- McGettigan, P.; Henry, D. Cardiovascular risk with non-steroidal anti-inflammatory drugs: Systematic review of population-based controlled observational studies. PLoS Med. 2011, 8, e1001098. [Google Scholar] [CrossRef]
- King, J.C.; Xu, J.; Wongvipat, J.; Hieronymus, H.; Carver, B.S.; Leung, D.H.; Taylor, B.S.; Sander, C.; Cardiff, R.D.; Couto, S.S.; et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet. 2009, 41, 524–526. [Google Scholar] [CrossRef]
- Gao, D.; Zhan, Y.; Di, W.; Moore, A.R.; Sher, J.J.; Guan, Y.; Wang, S.; Zhang, Z.; Murphy, D.A.; Sawyers, C.L.; et al. A Tmprss2-CreERT2 Knock-In Mouse Model for Cancer Genetic Studies on Prostate and Colon. PLoS ONE 2016, 11, e0161084. [Google Scholar] [CrossRef]
- Raina, K.; Kant, R.; Prasad, R.R.; Kandhari, K.; Tomar, M.; Mishra, N.; Kumar, R.; Fox, J.T.; Sei, S.; Shoemaker, R.H.; et al. Characterization of stage-specific tumor progression in TMPRSS2-ERG (fusion)-driven and non-fusion-driven prostate cancer in GEM models. Mol. Carcinog. 2022, 61, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Akinyeke, T.; Matsumura, S.; Wang, X.; Wu, Y.; Schalfer, E.D.; Saxena, A.; Yan, W.; Logan, S.K.; Li, X. Metformin targets c-MYC oncogene to prevent prostate cancer. Carcinogenesis 2013, 34, 2823–2832. [Google Scholar] [CrossRef] [PubMed]
- Ellwood-Yen, K.; Graeber, T.G.; Wongvipat, J.; Iruela-Arispe, M.L.; Zhang, J.; Matusik, R.; Thomas, G.V.; Sawyers, C.L. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003, 4, 223–238. [Google Scholar] [CrossRef]
- Saha, A.; Blando, J.; Tremmel, L.; DiGiovanni, J. Effect of Metformin, Rapamycin, and Their Combination on Growth and Progression of Prostate Tumors in HiMyc Mice. Cancer Prev. Res. 2015, 8, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Madka, V.; Zhang, Y.; Singh, A.; Biddick, L.; Li, Q.; Lightfoot, S.; Steele, V.E.; Lubet, R.A.; et al. Intermittent Dosing Regimens of Aspirin and Naproxen Inhibit Azoxymethane-Induced Colon Adenoma Progression to Adenocarcinoma and Invasive Carcinoma. Cancer Prev. Res. 2019, 12, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Raina, K.; Blouin, M.J.; Singh, R.P.; Majeed, N.; Deep, G.; Varghese, L.; Glode, L.M.; Greenberg, N.M.; Hwang, D.; Cohen, P.; et al. Dietary feeding of silibinin inhibits prostate tumor growth and progression in transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2007, 67, 11083–11091. [Google Scholar] [CrossRef]
- Raina, K.; Rajamanickam, S.; Singh, R.P.; Deep, G.; Chittezhath, M.; Agarwal, R. Stage-specific inhibitory effects and associated mechanisms of silibinin on tumor progression and metastasis in transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2008, 68, 6822–6830. [Google Scholar] [CrossRef]
- Shappell, S.B.; Thomas, G.V.; Roberts, R.L.; Herbert, R.; Ittmann, M.M.; Rubin, M.A.; Humphrey, P.A.; Sundberg, J.P.; Rozengurt, N.; Barrios, R.; et al. Prostate pathology of genetically engineered mice: Definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004, 64, 2270–2305. [Google Scholar] [CrossRef]
- Raina, K.; Ravichandran, K.; Rajamanickam, S.; Huber, K.M.; Serkova, N.J.; Agarwal, R. Inositol hexaphosphate inhibits tumor growth, vascularity, and metabolism in TRAMP mice: A multiparametric magnetic resonance study. Cancer Prev. Res. 2013, 6, 40–50. [Google Scholar] [CrossRef]
- Chen, Y.; Chi, P.; Rockowitz, S.; Iaquinta, P.J.; Shamu, T.; Shukla, S.; Gao, D.; Sirota, I.; Carver, B.S.; Wongvipat, J.; et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat. Med. 2013, 19, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Jeon, H.G.; Jeong, B.C.; Seo, S.I.; Jeon, S.S.; Lee, H.M.; Choi, H.Y.; Kang, S.Y.; Choi, Y.L.; Kwon, G.Y. Correlation of ERG immunohistochemistry with molecular detection of TMPRSS2-ERG gene fusion. J. Clin. Pathol. 2016, 69, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Llodra, S.; Juanpere, N.; de Muga, S.; Lorenzo, M.; Gil, J.; Font-Tello, A.; Agell, L.; Albero-Gonzalez, R.; Segales, L.; Merino, J.; et al. ERG overexpression plus SLC45A3 (prostein) and PTEN expression loss: Strong association of the triple hit phenotype with an aggressive pathway of prostate cancer progression. Oncotarget 2017, 8, 74106–74118. [Google Scholar] [CrossRef] [PubMed]
- Perner, S.; Rupp, N.J.; Braun, M.; Rubin, M.A.; Moch, H.; Dietel, M.; Wernert, N.; Jung, K.; Stephan, C.; Kristiansen, G. Loss of SLC45A3 protein (prostein) expression in prostate cancer is associated with SLC45A3-ERG gene rearrangement and an unfavorable clinical course. Int. J. Cancer 2013, 132, 807–812. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Fujita, K.; Nonomura, N. Influence of Diet and Nutrition on Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 1447. [Google Scholar] [CrossRef]
- Reed, D.; Raina, K.; Agarwal, R. Nutraceuticals in prostate cancer therapeutic strategies and their neo-adjuvant use in diverse populations. NPJ Precis. Oncol. 2018, 2, 15. [Google Scholar] [CrossRef]
- Hamilton, Z.; Parsons, J.K. Prostate Cancer Prevention: Concepts and Clinical Trials. Curr. Urol. Rep. 2016, 17, 35. [Google Scholar] [CrossRef]
- Singh, R.P.; Agarwal, R. Mechanisms of action of novel agents for prostate cancer chemoprevention. Endocr. Relat. Cancer 2006, 13, 751–778. [Google Scholar] [CrossRef]
- Brasky, T.M.; Velicer, C.M.; Kristal, A.R.; Peters, U.; Potter, J.D.; White, E. Nonsteroidal anti-inflammatory drugs and prostate cancer risk in the VITamins And Lifestyle (VITAL) cohort. Cancer Epidemiol. Biomark. Prev. 2010, 19, 3185–3188. [Google Scholar] [CrossRef]
- Jacobs, E.J.; Thun, M.J.; Bain, E.B.; Rodriguez, C.; Henley, S.J.; Calle, E.E. A large cohort study of long-term daily use of adult-strength aspirin and cancer incidence. J. Natl. Cancer Inst. 2007, 99, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, J.Q.; Xie, L.; Wang, J.; Li, T.; He, Y.; Gao, Y.; Qin, X.; Li, S. Effect of aspirin and other non-steroidal anti-inflammatory drugs on prostate cancer incidence and mortality: A systematic review and meta-analysis. BMC Med. 2014, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.M.; Franco, E.L.; Turner, D.; Platt, R.W.; Beck, P.; Skarsgard, D.; Tonita, J.; Sharpe, C.; Aprikian, A.G. Use of non-steroidal anti-inflammatory drugs and prostate cancer risk: A population-based nested case-control study. PLoS ONE 2011, 6, e16412. [Google Scholar] [CrossRef]
- Skriver, C.; Dehlendorff, C.; Borre, M.; Brasso, K.; Sorensen, H.T.; Hallas, J.; Larsen, S.B.; Tjonneland, A.; Friis, S. Low-dose aspirin or other nonsteroidal anti-inflammatory drug use and prostate cancer risk: A nationwide study. Cancer Causes Control 2016, 27, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Veitonmaki, T.; Murtola, T.J.; Maattanen, L.; Taari, K.; Stenman, U.H.; Tammela, T.L.; Auvinen, A. Prostate cancer risk and nonsteroidal antiinflammatory drug use in the Finnish prostate cancer screening trial. Br. J. Cancer 2014, 111, 1421–1431. [Google Scholar] [CrossRef]
- St John, J.; Powell, K.; Conley-Lacomb, M.K.; Chinni, S.R. TMPRSS2-ERG Fusion Gene Expression in Prostate Tumor Cells and Its Clinical and Biological Significance in Prostate Cancer Progression. J. Cancer Sci. Ther. 2012, 4, 94–101. [Google Scholar] [CrossRef]
- Kumar-Sinha, C.; Tomlins, S.A.; Chinnaiyan, A.M. Recurrent gene fusions in prostate cancer. Nat. Rev. Cancer 2008, 8, 497–511. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Wang, X.; Yan, H.; Cui, B.; Wang, Q.; Wu, J.; Cui, X.; Li, J.; Ou, T.; Yang, K. Intake of Non-steroidal Anti-inflammatory Drugs and the Risk of Prostate Cancer: A Meta-Analysis. Front. Oncol. 2018, 8, 437. [Google Scholar] [CrossRef]
- Srinivas, S.; Feldman, D. A phase II trial of calcitriol and naproxen in recurrent prostate cancer. Anticancer Res. 2009, 29, 3605–3610. [Google Scholar]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.G.; Ohgi, K.; Zhang, J.; Rose, D.W.; Fu, X.D.; Glass, C.K.; et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009, 139, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.S.; Amin, M.A.; Li, X.; Kalyana-Sundaram, S.; Veeneman, B.A.; Wang, L.; Ghosh, A.; Aslam, A.; Ramanand, S.G.; Rabquer, B.J.; et al. Inflammation-Induced Oxidative Stress Mediates Gene Fusion Formation in Prostate Cancer. Cell Rep. 2016, 17, 2620–2631. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raina, K.; Kandhari, K.; Kant, R.; Prasad, R.R.; Mishra, N.; Maurya, A.K.; Fox, J.T.; Sei, S.; Shoemaker, R.H.; Bosland, M.C.; et al. Differential Effect of Non-Steroidal Anti-Inflammatory Drugs Aspirin and Naproxen against TMPRSS2-ERG (Fusion)-Driven and Non-Fusion-Driven Prostate Cancer. Cancers 2023, 15, 5054. https://doi.org/10.3390/cancers15205054
Raina K, Kandhari K, Kant R, Prasad RR, Mishra N, Maurya AK, Fox JT, Sei S, Shoemaker RH, Bosland MC, et al. Differential Effect of Non-Steroidal Anti-Inflammatory Drugs Aspirin and Naproxen against TMPRSS2-ERG (Fusion)-Driven and Non-Fusion-Driven Prostate Cancer. Cancers. 2023; 15(20):5054. https://doi.org/10.3390/cancers15205054
Chicago/Turabian StyleRaina, Komal, Kushal Kandhari, Rama Kant, Ram Raj Prasad, Neha Mishra, Akhilendra K. Maurya, Jennifer T. Fox, Shizuko Sei, Robert H. Shoemaker, Maarten C. Bosland, and et al. 2023. "Differential Effect of Non-Steroidal Anti-Inflammatory Drugs Aspirin and Naproxen against TMPRSS2-ERG (Fusion)-Driven and Non-Fusion-Driven Prostate Cancer" Cancers 15, no. 20: 5054. https://doi.org/10.3390/cancers15205054