
The AKT1E17K Allele Promotes Breast Cancer in Mice

 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation and Analysis of Transgenic Mice
- TRF, 5′-GGATCGACGGTATCGTAGAGTCGAGGCCG-3′
- L2R 5′-GCCAATGAAGGTGCCATCATTCTTGAGGAGGAAG-3′; primers for MMTV-Cre: MMTV-F, 5′-CTGATCTGAGCTCTGAGTG-3′
- MMTV-R 5′-GTGAAACAGCATTGCTGTCACTT-3′.
2.2. Western Blot and Antibodies
2.3. Quantitative Reverse Transcription Real-Time PCR (qRT-PCR)
2.4. Enzymatic Dissociation of Murine Mammary Glands
2.5. Histological Analysis and Immunohistochemistry
2.6. Gene Expression Profiling
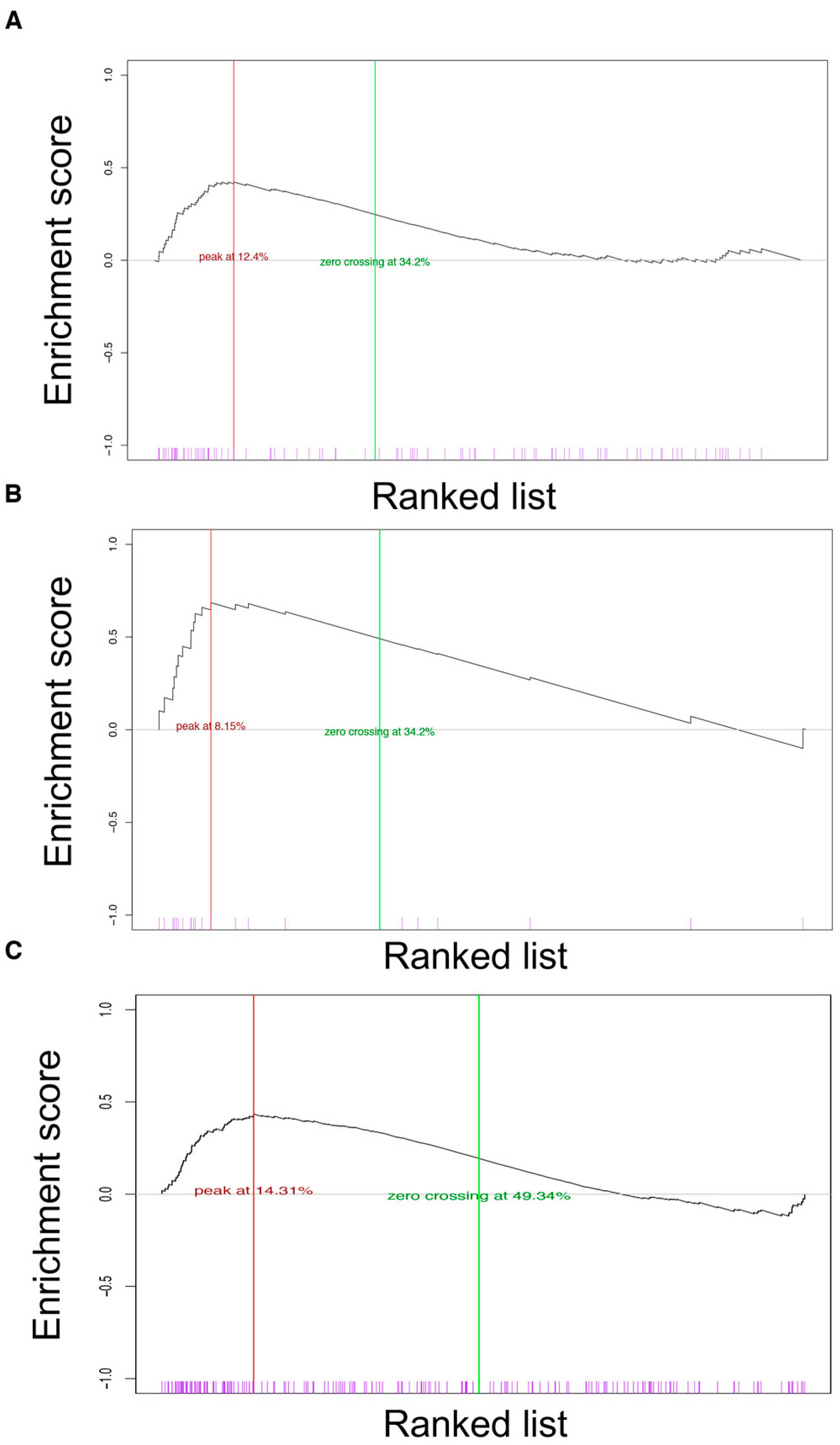
2.7. GSEA Analysis
2.8. Integrative Analysis of Murine Models of Breast Cancer
2.9. Validation of Murine Gene Expression Data in the Human Breast Cancer Dataset from TCGA
2.10. Statistical Analysis
3. Results
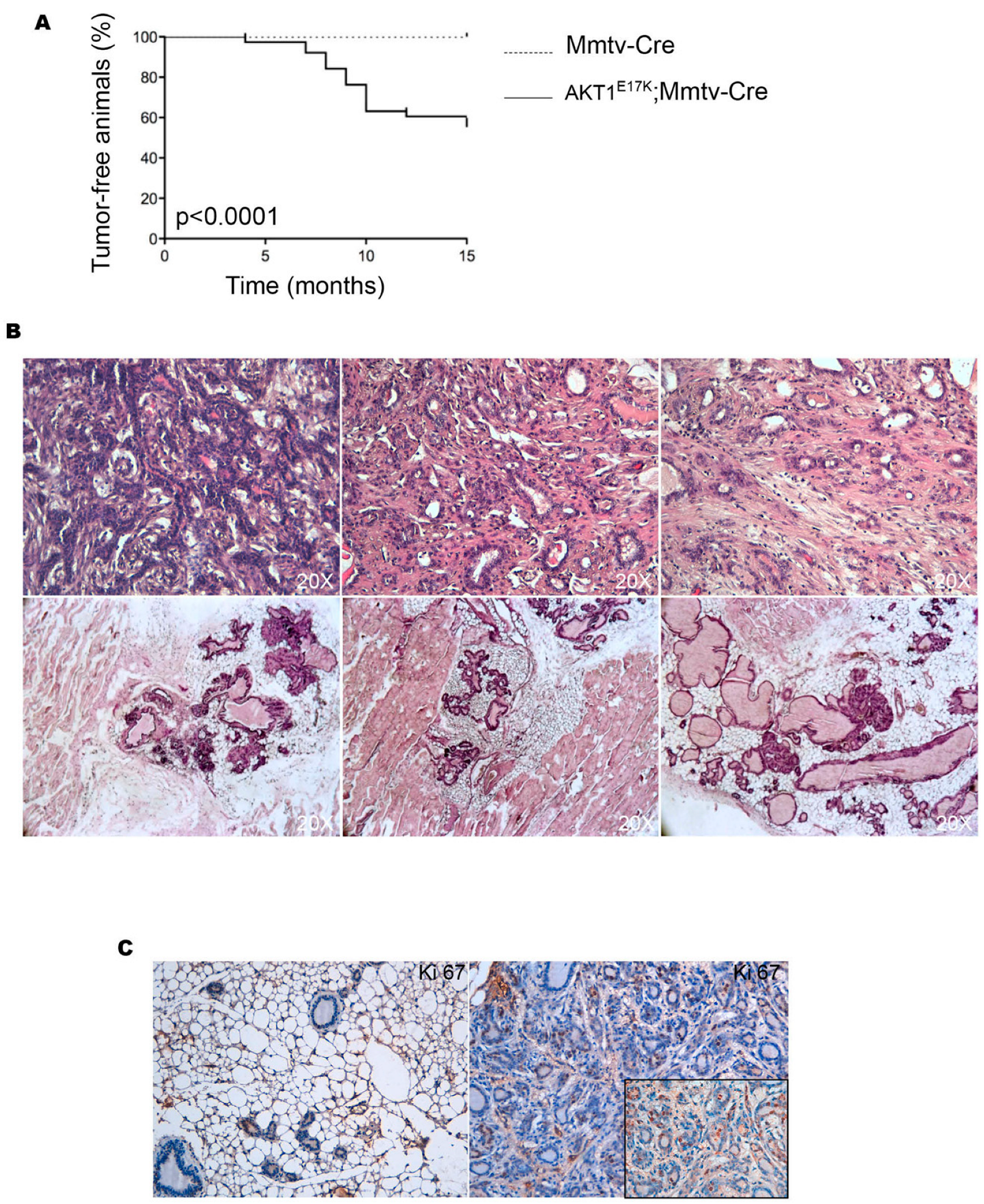
3.1. Mutant AKT1E17K Promotes Mammary Tumor Development
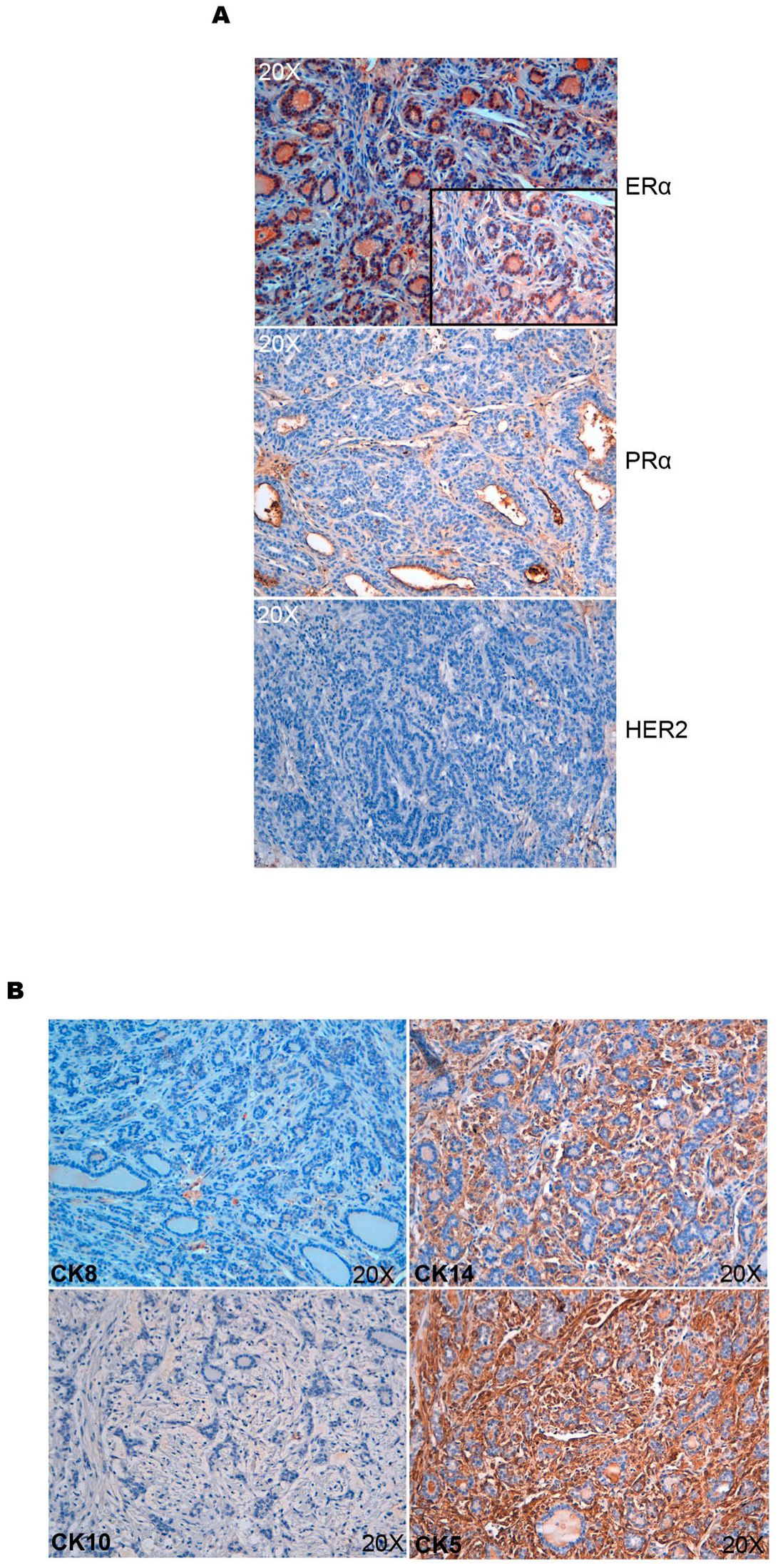
3.2. Characterization of Mammary Tumors Developed in R26AKT1E17K; MMTV-Cre Mice
3.3. Mutant AKT1E17K Promotes Activation of AKT-Pathway in Mouse Mammary Tumors
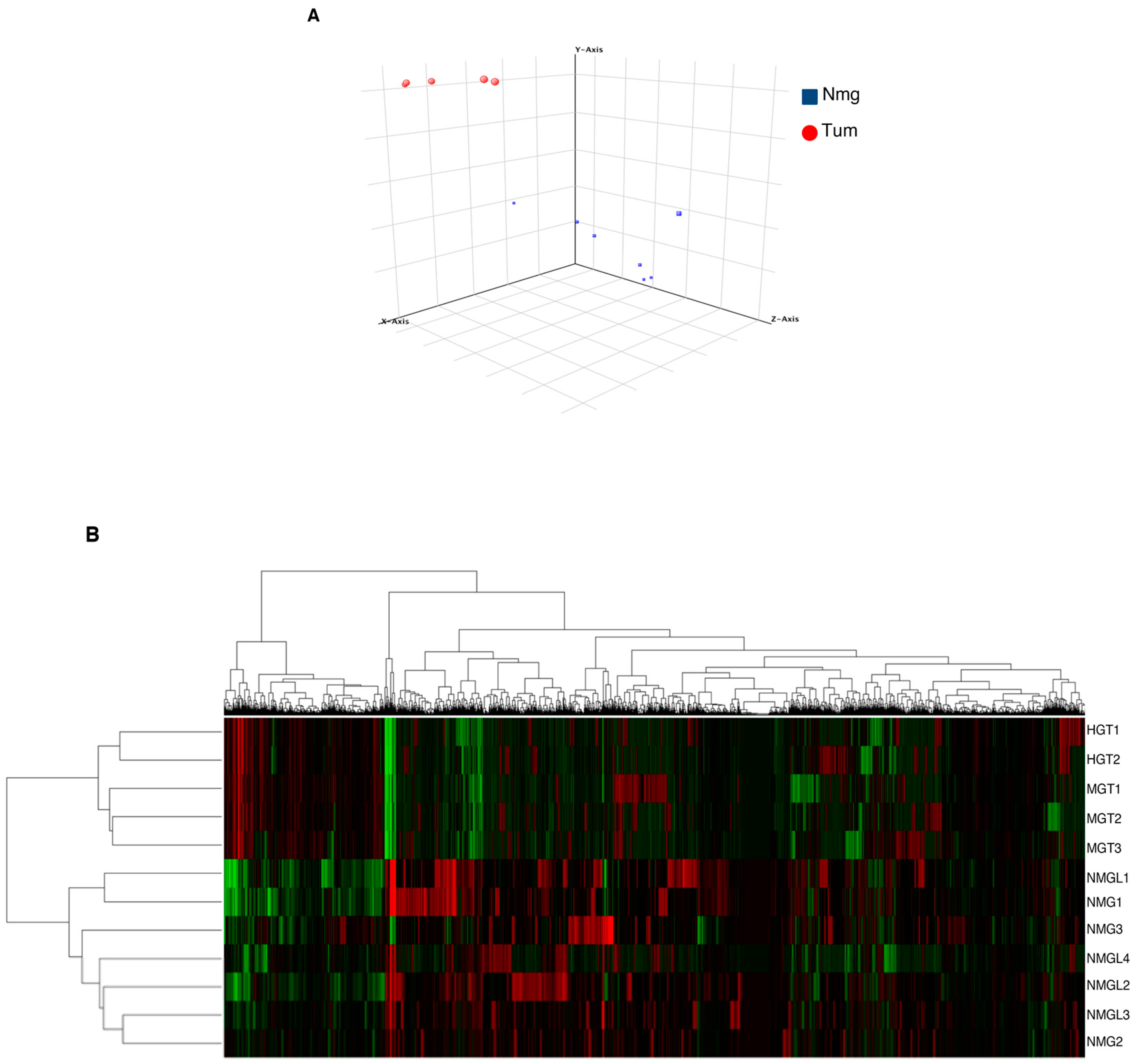
3.4. Gene Expression Analysis of Breast Tumors in R26AKT1E17K; MMTV-Cre Mice
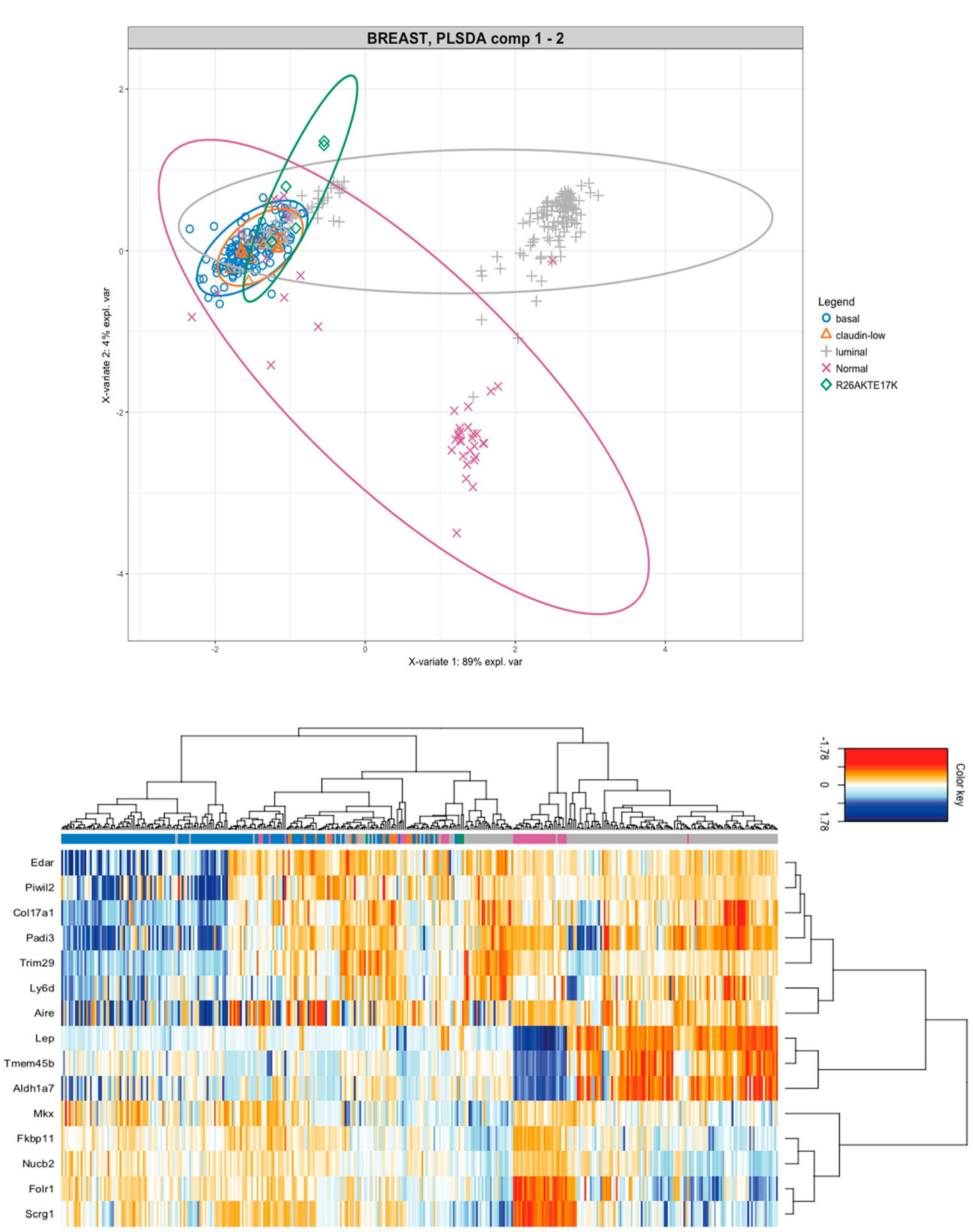
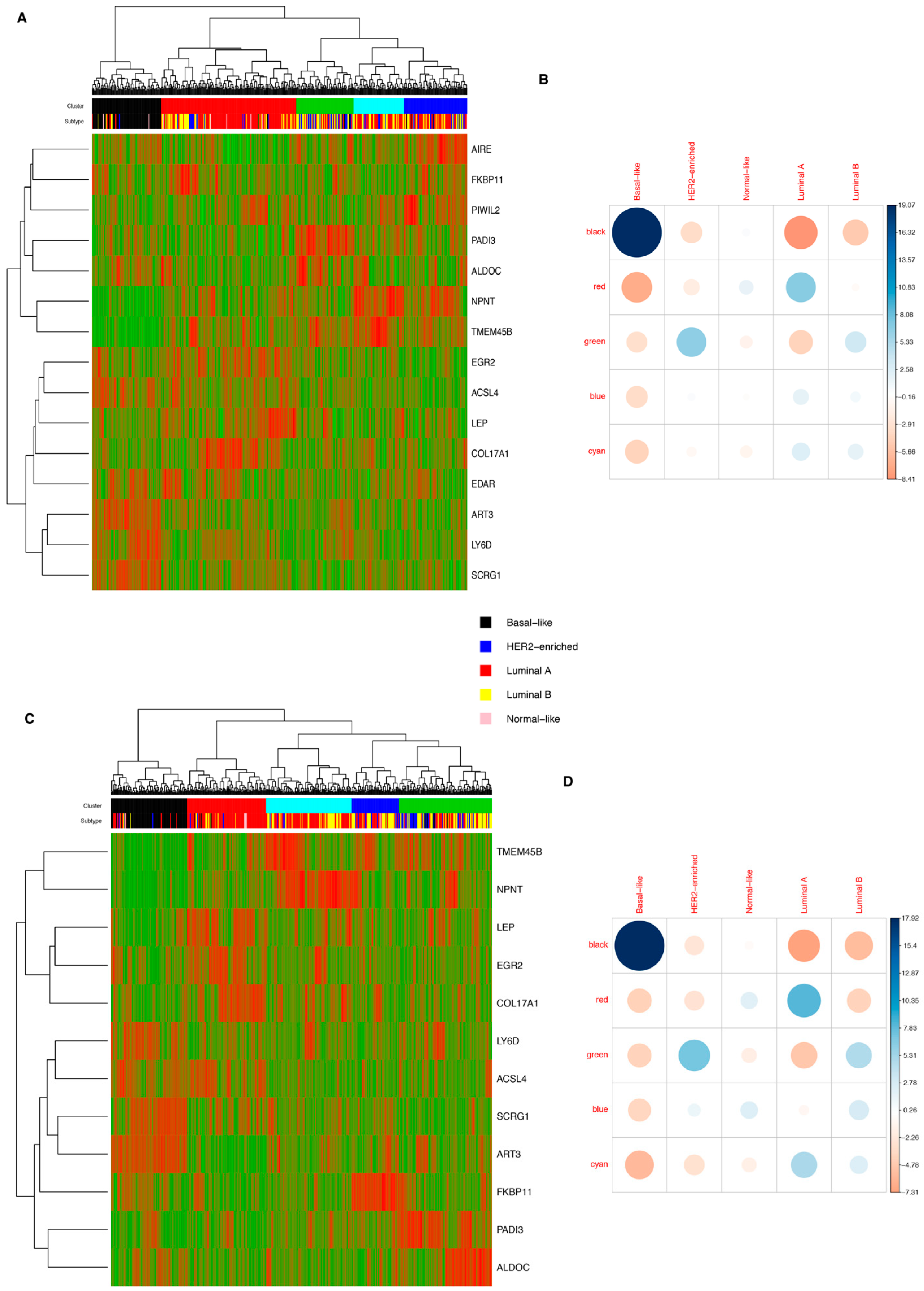
3.5. Molecular Classification of Tumors Induced by AKT1E17K in Mice
3.6. Comparison of the Expression Profile of Tumors Induced by AKT1E17K in Mice with the Profile of Human Breast Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist 2011, 16 (Suppl. S1), 12–19. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, M.; Anzeneder, T.; Schulz, A.; Beckmann, G.; Byrne, A.T.; Jeffers, M.; Pena, C.; Politz, O.; Kochert, K.; Vonk, R.; et al. AKT1 (E17K) mutation profiling in breast cancer: Prevalence, concurrent oncogenic alterations, and blood-based detection. BMC Cancer 2016, 16, 622. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.Y.; Di, M.Y.; Yuan, J.Q.; Shen, W.X.; Zheng, D.Y.; Chen, J.Z.; Mao, C.; Tang, J.L. The prognostic value of phosphorylated Akt in breast cancer: A systematic review. Sci. Rep. 2015, 5, 7758. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhao, Y.; Wang, C.; Ju, H.; Liu, W.; Zhang, X.; Miao, S.; Wang, L.; Sun, Q.; Song, W. Rhomboid domain-containing protein 1 promotes breast cancer progression by regulating the p-Akt and CDK2 levels. Cell Commun. Signal. 2018, 16, 65. [Google Scholar] [CrossRef] [Green Version]
- Dummler, B.; Hemmings, B.A. Physiological roles of PKB/Akt isoforms in development and disease. Biochem. Soc. Trans. 2007, 35, 231–235. [Google Scholar] [CrossRef]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef] [PubMed]
- Sakr, R.A.; Weigelt, B.; Chandarlapaty, S.; Andrade, V.P.; Guerini-Rocco, E.; Giri, D.; Ng, C.K.; Cowell, C.F.; Rosen, N.; Reis-Filho, J.S.; et al. PI3K pathway activation in high-grade ductal carcinoma in situ--implications for progression to invasive breast carcinoma. Clin. Cancer Res. 2014, 20, 2326–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troxell, M.L.; Levine, J.; Beadling, C.; Warrick, A.; Dunlap, J.; Presnell, A.; Patterson, J.; Shukla, A.; Olson, N.R.; Heinrich, M.C.; et al. High prevalence of PIK3CA/AKT pathway mutations in papillary neoplasms of the breast. Mod. Pathol. 2010, 23, 27–37. [Google Scholar] [CrossRef]
- Malanga, D.; Scrima, M.; De Marco, C.; Fabiani, F.; De Rosa, N.; De Gisi, S.; Malara, N.; Savino, R.; Rocco, G.; Chiappetta, G.; et al. Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung. Cell Cycle 2008, 7, 665–669. [Google Scholar] [CrossRef] [Green Version]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, F.E.; Felicioni, L.; Buttitta, F.; Lamba, S.; Cardone, L.; Rodolfo, M.; Scarpa, A.; Leenstra, S.; Frattini, M.; Barbareschi, M.; et al. AKT1E17K in human solid tumours. Oncogene 2008, 27, 5648–5650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoji, K.; Oda, K.; Nakagawa, S.; Hosokawa, S.; Nagae, G.; Uehara, Y.; Sone, K.; Miyamoto, Y.; Hiraike, H.; Hiraike-Wada, O.; et al. The oncogenic mutation in the pleckstrin homology domain of AKT1 in endometrial carcinomas. Br. J. Cancer 2009, 101, 145–148. [Google Scholar] [CrossRef] [PubMed]
- De Marco, C.; Malanga, D.; Rinaldo, N.; De Vita, F.; Scrima, M.; Lovisa, S.; Fabris, L.; Carriero, M.V.; Franco, R.; Rizzuto, A.; et al. Mutant AKT1-E17K is oncogenic in lung epithelial cells. Oncotarget 2015, 6, 39634–39650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgraf, K.E.; Pilling, C.; Falke, J.J. Molecular mechanism of an oncogenic mutation that alters membrane targeting: Glu17Lys modifies the PIP lipid specificity of the AKT1 PH domain. Biochemistry 2008, 47, 12260–12269. [Google Scholar] [CrossRef] [Green Version]
- Malanga, D.; Belmonte, S.; Colelli, F.; Scarfo, M.; De Marco, C.; Oliveira, D.M.; Mirante, T.; Camastra, C.; Gagliardi, M.; Rizzuto, A.; et al. AKT1E17K Is Oncogenic in Mouse Lung and Cooperates with Chemical Carcinogens in Inducing Lung Cancer. PLoS ONE 2016, 11, e0147334. [Google Scholar] [CrossRef] [Green Version]
- Laird, P.W.; Zijderveld, A.; Linders, K.; Rudnicki, M.A.; Jaenisch, R.; Berns, A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991, 19, 4293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Dai, X.; Chen, A.; Bai, Z. Integrative investigation on breast cancer in ER, PR and HER2-defined subgroups using mRNA and miRNA expression profiling. Sci. Rep. 2014, 4, 6566. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Jaccard, P. Etude compartive de la distribution florale dans une portion des alpes et du Jura. Bullettin Del La Soc. Vaud. Des Sci. Nat. 1901, 37, 547–579. [Google Scholar]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Pfefferle, A.D.; Herschkowitz, J.I.; Usary, J.; Harrell, J.C.; Spike, B.T.; Adams, J.R.; Torres-Arzayus, M.I.; Brown, M.; Egan, S.E.; Wahl, G.M.; et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 2013, 14, R125. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, R.A.; Ooi, S.L.; Wu, Z.; Boeke, J.D. Use of mixture models in a microarray-based screening procedure for detecting differentially represented yeast mutants. Stat. Appl. Genet. Mol. Biol. 2003, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Wold, H. Estimation of Principal Components and Related Models by Iterative Least Squares; Academic Press: New York, NY, USA, 1966. [Google Scholar]
- Nguyen, D.V.; Rocke, D.M. Tumor classification by partial least squares using microarray gene expression data. Bioinformatics 2002, 18, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Cao, K.A.; Boitard, S.; Besse, P. Sparse PLS discriminant analysis: Biologically relevant feature selection and graphical displays for multiclass problems. BMC Bioinform. 2011, 12, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. Ser. B Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Risso, D.; Schwartz, K.; Sherlock, G.; Dudoit, S. GC-content normalization for RNA-Seq data. BMC Bioinform. 2011, 12, 480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.H., Jr. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.A. mixOmics: An R package for omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [Green Version]
- Ponzo, M.G.; Lesurf, R.; Petkiewicz, S.; O’Malley, F.P.; Pinnaduwage, D.; Andrulis, I.L.; Bull, S.B.; Chughtai, N.; Zuo, D.; Souleimanova, M.; et al. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 12903–12908. [Google Scholar] [CrossRef] [Green Version]
- Usary, J.; Darr, D.B.; Pfefferle, A.D.; Perou, C.M. Overview of Genetically Engineered Mouse Models of Distinct Breast Cancer Subtypes. Curr. Protoc. Pharmacol. 2016, 72, 14.38.1–14.38.11. [Google Scholar] [CrossRef] [Green Version]
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; Rasmussen, K.E.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S.; et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007, 8, R76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, D.S.; Brinkhaus, H.; Muller, U.; Muller, M.; Cardiff, R.D.; Bentires-Alj, M. Luminal expression of PIK3CA mutant H1047R in the mammary gland induces heterogeneous tumors. Cancer Res. 2011, 71, 4344–4351. [Google Scholar] [CrossRef] [Green Version]
- Reis-Filho, J.S.; Pusztai, L. Gene expression profiling in breast cancer: Classification, prognostication, and prediction. Lancet 2011, 378, 1812–1823. [Google Scholar] [CrossRef]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, K.H.; Lauring, J. Recurrent AKT mutations in human cancers: Functional consequences and effects on drug sensitivity. Oncotarget 2016, 7, 4241–4251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.C.; Voisin, V.; Wang, S.; Wang, D.Y.; Jones, R.A.; Datti, A.; Uehling, D.; Al-awar, R.; Egan, S.E.; Bader, G.D.; et al. Combined deletion of Pten and p53 in mammary epithelium accelerates triple-negative breast cancer with dependency on eEF2K. EMBO Mol. Med. 2014, 6, 1542–1560. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Kim, J.; Elshimali, Y.; Sarkissyan, M.; Vadgama, J.V. Activation of Akt1 accelerates carcinogen-induced tumorigenesis in mammary gland of virgin and post-lactating transgenic mice. BMC Cancer 2014, 14, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, M.L.; Lien, E.C.; Toker, A. Oncogenic AKT1E17K mutation induces mammary hyperplasia but prevents HER2-driven tumorigenesis. Oncotarget 2016, 7, 17301–17313. [Google Scholar] [CrossRef] [Green Version]
- Schwertfeger, K.L.; Richert, M.M.; Anderson, S.M. Mammary gland involution is delayed by activated Akt in transgenic mice. Mol. Endocrinol. 2001, 15, 867–881. [Google Scholar] [CrossRef]
- Adams, J.R.; Xu, K.; Liu, J.C.; Agamez, N.M.; Loch, A.J.; Wong, R.G.; Wang, W.; Wright, K.L.; Lane, T.F.; Zacksenhaus, E.; et al. Cooperation between Pik3ca and p53 mutations in mouse mammary tumor formation. Cancer Res. 2011, 71, 2706–2717. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Aparicio, C.; Canamero, M.; Cecilia, Y.; Pequeno, B.; Renner, O.; Ferrer, I.; Carnero, A. Exploring the gain of function contribution of AKT to mammary tumorigenesis in mouse models. PLoS ONE 2010, 5, e9305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Aparicio, C.; Perez-Gallego, L.; Pequeno, B.; Leal, J.F.; Renner, O.; Carnero, A. Mice expressing myrAKT1 in the mammary gland develop carcinogen-induced ER-positive mammary tumors that mimic human breast cancer. Carcinogenesis 2007, 28, 584–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, P.K.; Grisanzio, C.; O’Connell, F.; Barry, M.; Brito, J.M.; Xu, Q.; Guney, I.; Berger, R.; Herman, P.; Bikoff, R.; et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell 2008, 14, 146–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauring, J.; Cosgrove, D.P.; Fontana, S.; Gustin, J.P.; Konishi, H.; Abukhdeir, A.M.; Garay, J.P.; Mohseni, M.; Wang, G.M.; Higgins, M.J.; et al. Knock in of the AKT1E17K mutation in human breast epithelial cells does not recapitulate oncogenic PIK3CA mutations. Oncogene 2010, 29, 2337–2345. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.S.; West, K.; Streicher, S.; Dennis, P.A. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol. Cancer Ther. 2002, 1, 707–717. [Google Scholar]
- Campbell, R.A.; Bhat-Nakshatri, P.; Patel, N.M.; Constantinidou, D.; Ali, S.; Nakshatri, H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: A new model for anti-estrogen resistance. J. Biol. Chem. 2001, 276, 9817–9824. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Lim, E.; Wu, D.; Pal, B.; Bouras, T.; Asselin-Labat, M.L.; Vaillant, F.; Yagita, H.; Lindeman, G.J.; Smyth, G.K.; Visvader, J.E. Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast Cancer Res. 2010, 12, R21. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R26AKT1E17K; MMTV-Cre | Human BC “Basal-Like” | |||||
|---|---|---|---|---|---|---|
| Hallmark Gene Set | es | p-Value | NES | es | p-Value | NES |
| E2F TARGETS | 0.64 | 0.00 | 2.64 | 0.76 | 0.00 | 2.53 |
| G2M CHECKPOINT | 0.59 | 0.00 | 2.42 | 0.73 | 0.00 | 2.42 |
| MYC TARGETS V1 | 0.57 | 0.00 | 2.30 | 0.67 | 0.00 | 2.20 |
| MITOTIC SPINDLE | 0.51 | 0.00 | 2.06 | 0.53 | 0.00 | 1.78 |
| MTORC1 SIGNALING | 0.32 | 0.02 | 1.30 | 0.48 | 0.00 | 1.59 |
| WNT BETA CATENIN SIGNALING | 0.52 | 0.00 | 1.67 | 0.52 | 0.03 | 1.40 |
| TNFA SIGNALING VIA NFKB | 0.39 | 0.00 | 1.60 | 0.38 | 0.03 | 1.24 |
| INFLAMMATORY RESPONSE | 0.33 | 0.01 | 1.35 | 0.41 | 0.01 | 1.35 |
| MYC TARGETS V2 | 0.44 | 0.01 | 1.48 | 0.68 | 0.00 | 1.97 |
| GLYCOLYSIS | 0.36 | 0.00 | 1.46 | 0.39 | 0.02 | 1.28 |
| UNFOLDED PROTEIN RESPONSE | 0.47 | 0.00 | 1.79 | 0.42 | 0.02 | 1.33 |
| PEROXISOME | 0.41 | 0.00 | −1.57 | −0.44 | 0.02 | −1.38 |
| FATTY ACID METABOLISM | 0.53 | 0.00 | −2.11 | −0.44 | 0.01 | −1.42 |
| BILE ACID METABOLISM | 0.43 | 0.00 | −1.65 | −0.50 | 0.00 | −1.54 |
| ESTROGEN RESPONSE LATE | 0.43 | 0.00 | 1.79 | −0.56 | 0.00 | −1.85 |
| ESTROGEN RESPONSE EARLY | 0.36 | 0.00 | 1.48 | −0.69 | 0.00 | −2.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malanga, D.; Laudanna, C.; Mirante, T.; Colelli, F.; Migliozzi, S.; Zoppoli, P.; Santamaria, G.; Roberto, L.; De Marco, C.; Scarfò, M.; et al. The AKT1E17K Allele Promotes Breast Cancer in Mice. Cancers 2022, 14, 2645. https://doi.org/10.3390/cancers14112645
Malanga D, Laudanna C, Mirante T, Colelli F, Migliozzi S, Zoppoli P, Santamaria G, Roberto L, De Marco C, Scarfò M, et al. The AKT1E17K Allele Promotes Breast Cancer in Mice. Cancers. 2022; 14(11):2645. https://doi.org/10.3390/cancers14112645
Chicago/Turabian StyleMalanga, Donatella, Carmelo Laudanna, Teresa Mirante, Fabiana Colelli, Simona Migliozzi, Pietro Zoppoli, Gianluca Santamaria, Luca Roberto, Carmela De Marco, Marzia Scarfò, and et al. 2022. "The AKT1E17K Allele Promotes Breast Cancer in Mice" Cancers 14, no. 11: 2645. https://doi.org/10.3390/cancers14112645