
Circulating Tumor DNA Profiling of a Diffuse Large B Cell Lymphoma Patient with Secondary Acute Myeloid Leukemia

 , ,
, , 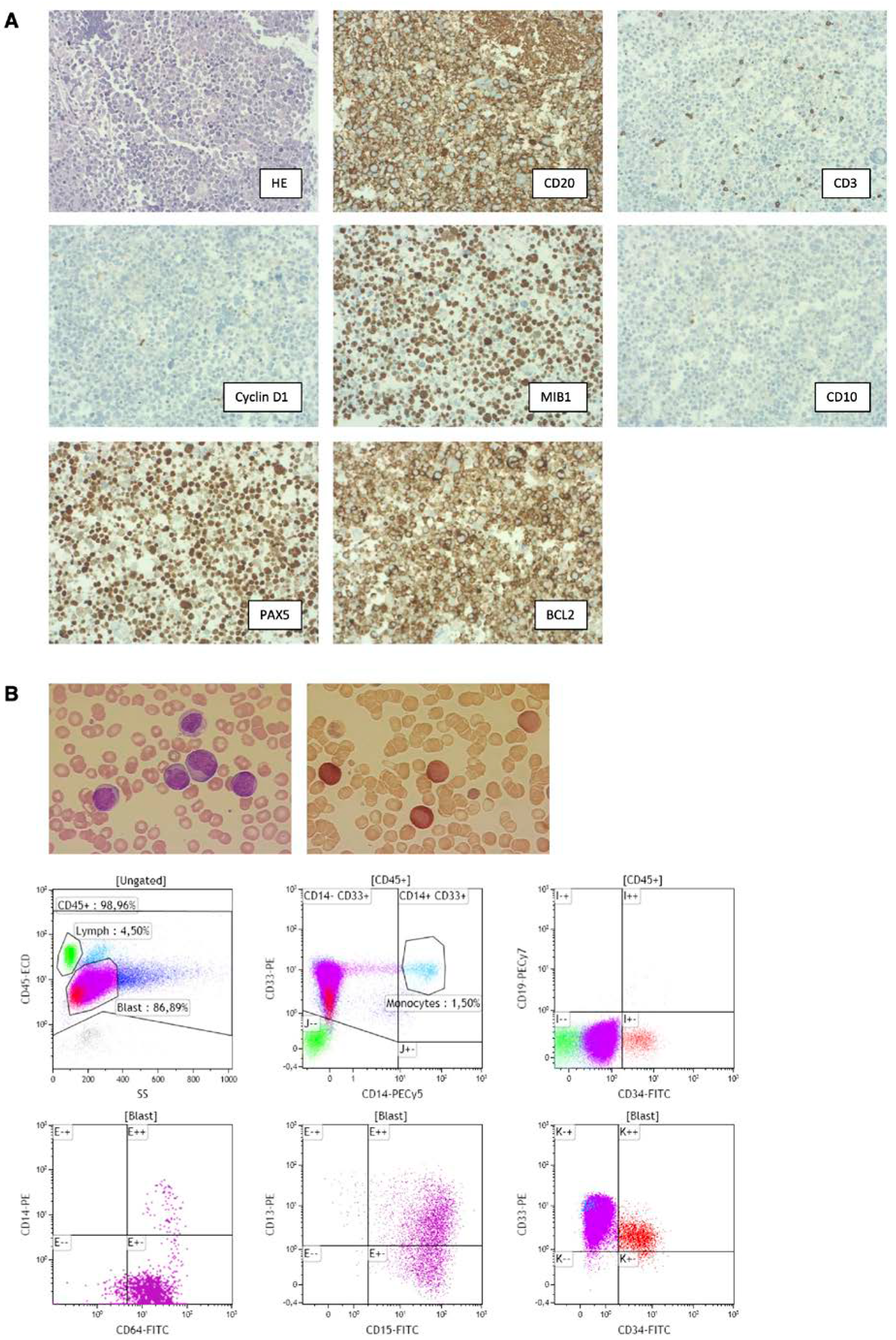{kind=link}
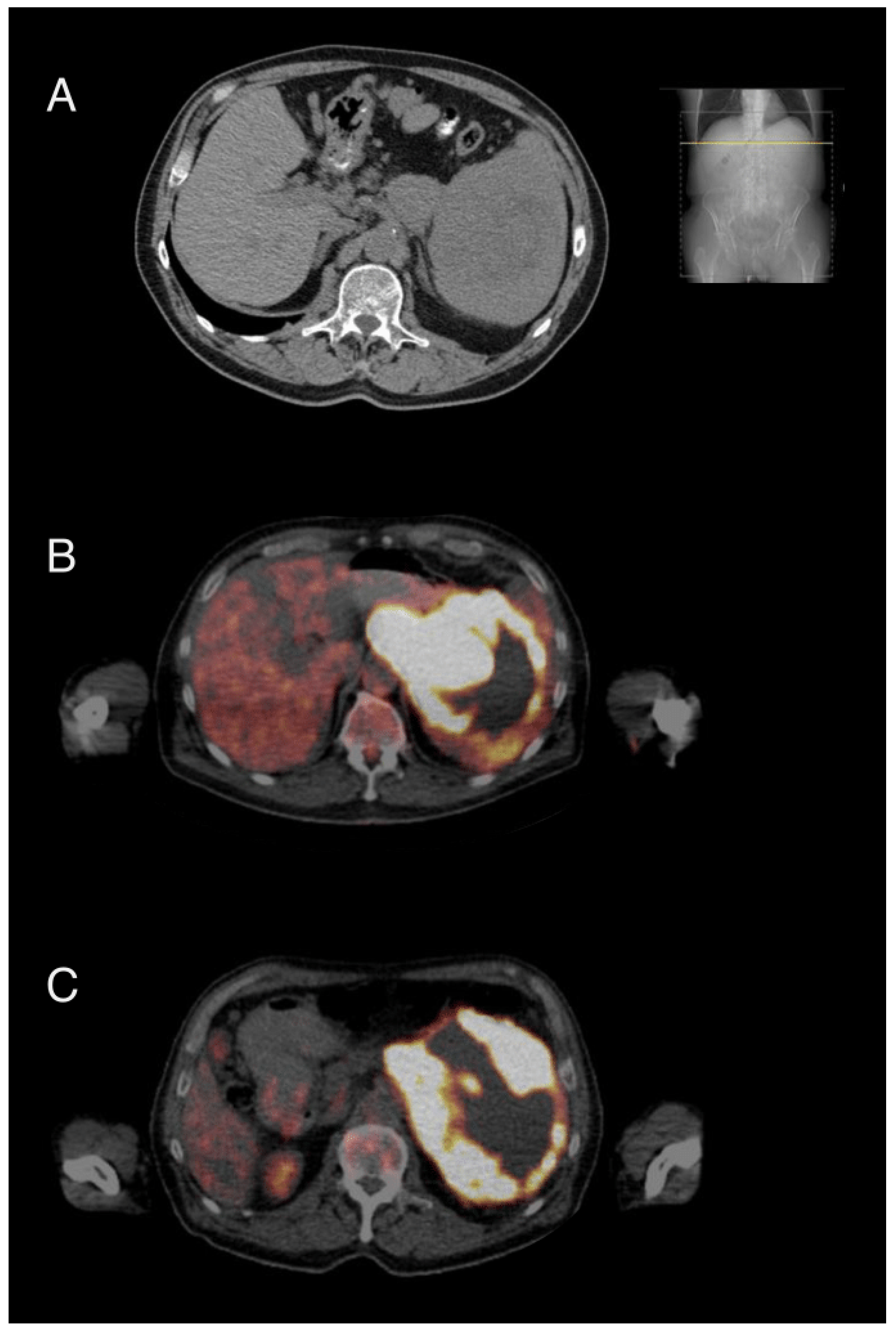{kind=link}
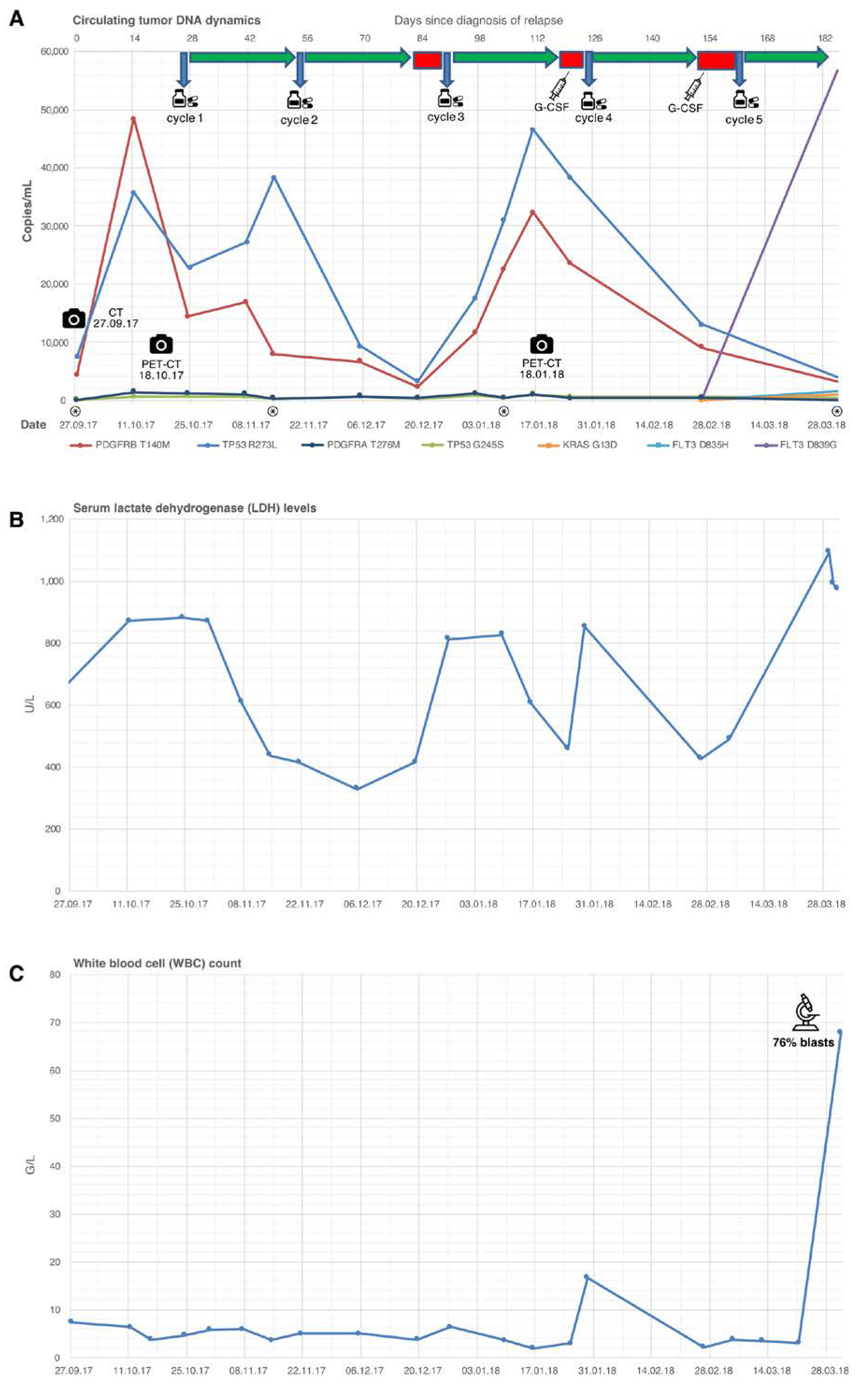{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient and Samples
2.2. Targeted NGS of cfDNA and Tumor-Specific ddPCR Assays
2.3. Droplet Digital PCR Data Analysis
3. Results and Discussion
3.1. Patient Course of Treatment
3.2. Mutational Profiling of Cell-Free Tumor DNA in Blood Plasma
3.3. Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coiffier, B. Diffuse large cell lymphoma. Curr. Opin. Oncol. 2001, 13, 325–334. [Google Scholar] [CrossRef]
- Groves, F.D.; Linet, M.S.; Travis, L.B.; Devesa, S.S. Cancer surveillance series: Non-Hodgkin’s lymphoma incidence by histologic subtype in the United States from 1978 through 1995. JNCI J. Natl. Cancer Inst. 2000, 92, 1240–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sant, M.; Allemani, C.; Tereanu, C.; De Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadié, M.; Simonetti, A.; Lutz, J.M.; et al. Incidence of hematologic malignancies in Europe by morphologic subtype: Results of the HAEMACARE project. Blood 2010, 116, 3724–3734. [Google Scholar] [CrossRef]
- Barrington, S.F.; Mikhaeel, N.G.; Kostakoglu, L.; Meignan, M.; Hutchings, M.; Müeller, S.P.; Schwartz, L.H.; Zucca, E.; Fisher, R.I.; Trotman, J.; et al. Role of Imaging in the Staging and Response Assessment of Lymphoma: Consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J. Clin. Oncol. 2014, 32, 3048–3058. [Google Scholar] [CrossRef]
- Tilly, H.; da Silva, M.G.; Vitolo, U.; Jack, A.; Meignan, M.; Lopez-Guillermo, A.; Walewski, J.; André, M.; Johnson, P.W.; Pfreundschuh, M.; et al. Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26 (Suppl. 5), v116–v125. [Google Scholar] [CrossRef]
- Godley, L.A.; Larson, R.A. Therapy-Related Myeloid Leukemia. Semin. Oncol. 2008, 35, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Clarke, C.A.; Rosenberg, A.S.; Advani, R.H.; Jonas, B.A.; Flowers, C.R.; Keegan, T.H.M. Subsequent primary malignancies after diffuse large B-cell lymphoma in the modern treatment era. Br. J. Haematol. 2017, 178, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Morton, L.M.; Curtis, R.E.; Linet, M.S.; Bluhm, E.C.; Tucker, M.A.; Caporaso, N.; Ries, L.A.; Fraumeni, J.F., Jr. Second malignancy risks after non-Hodgkin’s lymphoma and chronic lymphocytic leukemia: Differences by lymphoma subtype. J. Clin. Oncol. 2010, 28, 4935–4944. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Sun, P.; Chen, C.; Xia, Y.; Wang, Y.; Liu, P.P.; Bi, X.W.; Shao, Y.W.; Ou, Q.X.; Wu, X.; Yang, H.; et al. Mutation Profiling of Malignant Lymphoma by Next-Generation Sequencing of Circulating Cell-Free DNA. J. Cancer 2019, 10, 323–331. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Jardin, F.; Tilly, H. The value of liquid biopsy in diagnosis and monitoring of diffuse large b-cell lymphoma: Recent developments and future potential. Expert Rev. Mol. Diagn. 2017, 17, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Hohaus, S.; Giachelia, M.; Massini, G.; Mansueto, G.; Vannata, B.; Bozzoli, V.; Criscuolo, M.; D’Alò, F.; Martini, M.; Larocca, L.M.; et al. Cell-free circulating DNA in Hodgkin’s and non-Hodgkin’s lymphomas. Ann. Oncol. 2009, 20, 1408–1413. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Green, M.R.; Bratman, S.V.; Scherer, F.; Liu, C.L.; Kunder, C.A.; Takahashi, K.; Glover, C.; Keane, C.; Kihira, S.; et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood 2015, 125, 3679–3687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2017, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Horak, P.; Griffith, M.; Danos, A.M.; Pitel, B.A.; Madhavan, S.; Liu, X.; Chow, C.; Williams, H.; Carmody, L.; Barrow-Laing, L.; et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genet. Med. 2022. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Jiang, C.; Tse, P.; Achacoso, N.; Alexeeff, S.; Solorzano, A.V.; Chung, E.; Hu, W.; Truong, T.G.; Arora, A.; et al. TP53 Gain-of-Function and Non-Gain-of-Function Mutations Are Differentially Associated With Sidedness-Dependent Prognosis in Metastatic Colorectal Cancer. J. Clin. Oncol. 2021, 40, 171–179. [Google Scholar] [CrossRef]
- Yokouchi, H.; Nishihara, H.; Harada, T.; Yamazaki, S.; Kikuchi, H.; Oizumi, S.; Uramoto, H.; Tanaka, F.; Harada, M.; Akie, K.; et al. Detection of somatic TP53 mutation in surgically resected small-cell lung cancer by targeted exome sequencing: Association with longer relapse-free survival. Heliyon 2020, 6, e04439. [Google Scholar] [CrossRef]
- Yoo, K.H.; Kim, N.K.; Kwon, W.I.; Lee, C.; Kim, S.Y.; Jang, J.; Ahn, J.; Kang, M.; Jang, H.; Kim, S.T.; et al. Genomic Alterations in Biliary Tract Cancer Using Targeted Sequencing. Transl. Oncol. 2016, 9, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Rushton, C.K.; Arthur, S.E.; Alcaide, M.; Cheung, M.; Jiang, A.; Coyle, K.M.; Cleary, K.L.S.; Thomas, N.; Hilton, L.K.; Michaud, N.; et al. Genetic and evolutionary patterns of treatment resistance in relapsed B-cell lymphoma. Blood Adv. 2020, 4, 2886–2898. [Google Scholar] [CrossRef]
- Roschewski, M.; Dunleavy, K.; Pittaluga, S.; Moorhead, M.; Pepin, F.; Kong, K.; Shovlin, M.; Jaffe, E.S.; Staudt, L.M.; Lai, C.; et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: A correlative biomarker study. Lancet Oncol. 2015, 16, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Scherer, F.; Kurtz, D.M.; Newman, A.M.; Stehr, H.; Craig, A.F.; Esfahani, M.S.; Lovejoy, A.F.; Chabon, J.J.; Klass, D.M.; Liu, C.L.; et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transl. Med. 2016, 8, 364ra155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.L.; Arch, R.; Smith, L.L.; Bainton, N.; Neat, M.; Taylor, C.; Bonnet, D.; Cavenagh, J.D.; Andrew Lister, T.; Fitzgibbon, J. Development of a human acute myeloid leukaemia screening panel and consequent identification of novel gene mutation in FLT3 and CCND3. Br. J. Haematol. 2005, 128, 318–323. [Google Scholar] [CrossRef]
- Zou, X.L.; Zeng, K.; Xie, L.P.; Wang, L.; Chen, M.; Liu, T.; Niu, T. Acute promyelocytic leukemia with Flt3-TKD and WT1 mutations relapsing in a testicle and followed by systemic relapse. Acta Haematol. 2013, 130, 223–229. [Google Scholar] [CrossRef]
- Zhang, H.; Savage, S.; Schultz, A.R.; Bottomly, D.; White, L.; Segerdell, E.; Wilmot, B.; McWeeney, S.K.; Eide, C.A.; Nechiporuk, T.; et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat. Commun. 2019, 10, 244. [Google Scholar] [CrossRef]
- Daver, N.; Cortes, J.; Ravandi, F.; Patel, K.P.; Burger, J.A.; Konopleva, M.; Kantarjian, H. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood 2015, 125, 3236–3245. [Google Scholar] [CrossRef]
- Colović, M.; Suvajdžić, N.; Janković, G.; Tomin, D.; Colović, N.; Fekete, M.D.; Palibrk, V. Therapy-related myelodysplastic syndrome and acute myeloid leukemia in patients with chronic lymphocytic leukemia treated with fludarabine and cyclophosphamide. Biomed. Pharmacother. 2011, 65, 319–321. [Google Scholar] [CrossRef]
- Smith, R.E. Risk for the development of treatment-related acute myelocytic leukemia and myelodysplastic syndrome among patients with breast cancer: Review of the literature and the National Surgical Adjuvant Breast and Bowel Project experience. Clin. Breast Cancer 2003, 4, 273–279. [Google Scholar] [CrossRef]
- Lesesve, J.F.; Schneider, P.; Dolgopolov, I.; Bastard, C.; Lenormand, B.; Cambon-Michot, E.; Callat, M.P.; Cavelier, B.; Tron, P.H.; Vannier, J.P. Therapy-related acute myeloid leukemia with t(8;21) in a child with previous Ewing’s sarcoma. Med. Pediatric Oncol. 1997, 29, 132–134. [Google Scholar] [CrossRef]
- Morton, L.M.; Dores, G.M.; Schonfeld, S.J.; Linet, M.S.; Sigel, B.S.; Lam, C.J.K.; Tucker, M.A.; Curtis, R.E. Association of Chemotherapy for Solid Tumors with Development of Therapy-Related Myelodysplastic Syndrome or Acute Myeloid Leukemia in the Modern Era. JAMA Oncol. 2018, 5, 318–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calip, G.S.; Moran, K.M.; Sweiss, K.I.; Patel, P.R.; Wu, Z.; Adimadhyam, S.; Lee, T.A.; Ko, N.Y.; Quigley, J.G.; Chiu, B.C. Myelodysplastic syndrome and acute myeloid leukemia after receipt of granulocyte colony-stimulating factors in older patients with non-Hodgkin lymphoma. Cancer 2018, 125, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Calip, G.S.; Malmgren, J.A.; Lee, W.J.; Schwartz, S.M.; Kaplan, H.G. Myelodysplastic syndrome and acute myeloid leukemia following adjuvant chemotherapy with and without granulocyte colony-stimulating factors for breast cancer. Breast Cancer Res. Treat. 2015, 154, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Sakamaki, S.; Matsunaga, T.; Hirayama, Y.; Kuga, T.; Niitsu, Y. Haematological study of healthy volunteers 5 years after G-CSF. Lancet 1995, 346, 1432–1433. [Google Scholar] [CrossRef]
- Hölig, K. G-CSF in Healthy Allogeneic Stem Cell Donors. Transfus. Med. Hemother. 2013, 40, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Tigue, C.C.; McKoy, J.M.; Evens, A.M.; Trifilio, S.M.; Tallman, M.S.; Bennett, C.L. Granulocyte-colony stimulating factor administration to healthy individuals and persons with chronic neutropenia or cancer: An overview of safety considerations from the Research on Adverse Drug Events and Reports project. Bone Marrow Transplant. 2007, 40, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Baroni, T.E.; Wang, T.; Qian, H.; Dearth, L.R.; Truong, L.N.; Zeng, J.; Denes, A.E.; Chen, S.W.; Brachmann, R.K. A global suppressor motif for p53 cancer mutants. Proc. Natl. Acad. Sci. USA 2004, 101, 4930–4935. [Google Scholar] [CrossRef] [Green Version]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M. T-cell Responses to TP53 “Hotspot” Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin. Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerle, I.A.; Jägerhuber, L.; Secci, R.; Pfarr, N.; Blüm, P.; Roesch, R.; Götze, K.S.; Weichert, W.; Bassermann, F.; Ruland, J.; et al. Circulating Tumor DNA Profiling of a Diffuse Large B Cell Lymphoma Patient with Secondary Acute Myeloid Leukemia. Cancers 2022, 14, 1371. https://doi.org/10.3390/cancers14061371
Kerle IA, Jägerhuber L, Secci R, Pfarr N, Blüm P, Roesch R, Götze KS, Weichert W, Bassermann F, Ruland J, et al. Circulating Tumor DNA Profiling of a Diffuse Large B Cell Lymphoma Patient with Secondary Acute Myeloid Leukemia. Cancers. 2022; 14(6):1371. https://doi.org/10.3390/cancers14061371
Chicago/Turabian StyleKerle, Irina A., Ludwig Jägerhuber, Ramona Secci, Nicole Pfarr, Philipp Blüm, Romina Roesch, Katharina S. Götze, Wilko Weichert, Florian Bassermann, Jürgen Ruland, and et al. 2022. "Circulating Tumor DNA Profiling of a Diffuse Large B Cell Lymphoma Patient with Secondary Acute Myeloid Leukemia" Cancers 14, no. 6: 1371. https://doi.org/10.3390/cancers14061371