
Analysis of Circulating Tumour Cells in Early-Stage Uveal Melanoma: Evaluation of Tumour Marker Expression to Increase Capture

 , , , and
, , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Sample Collection
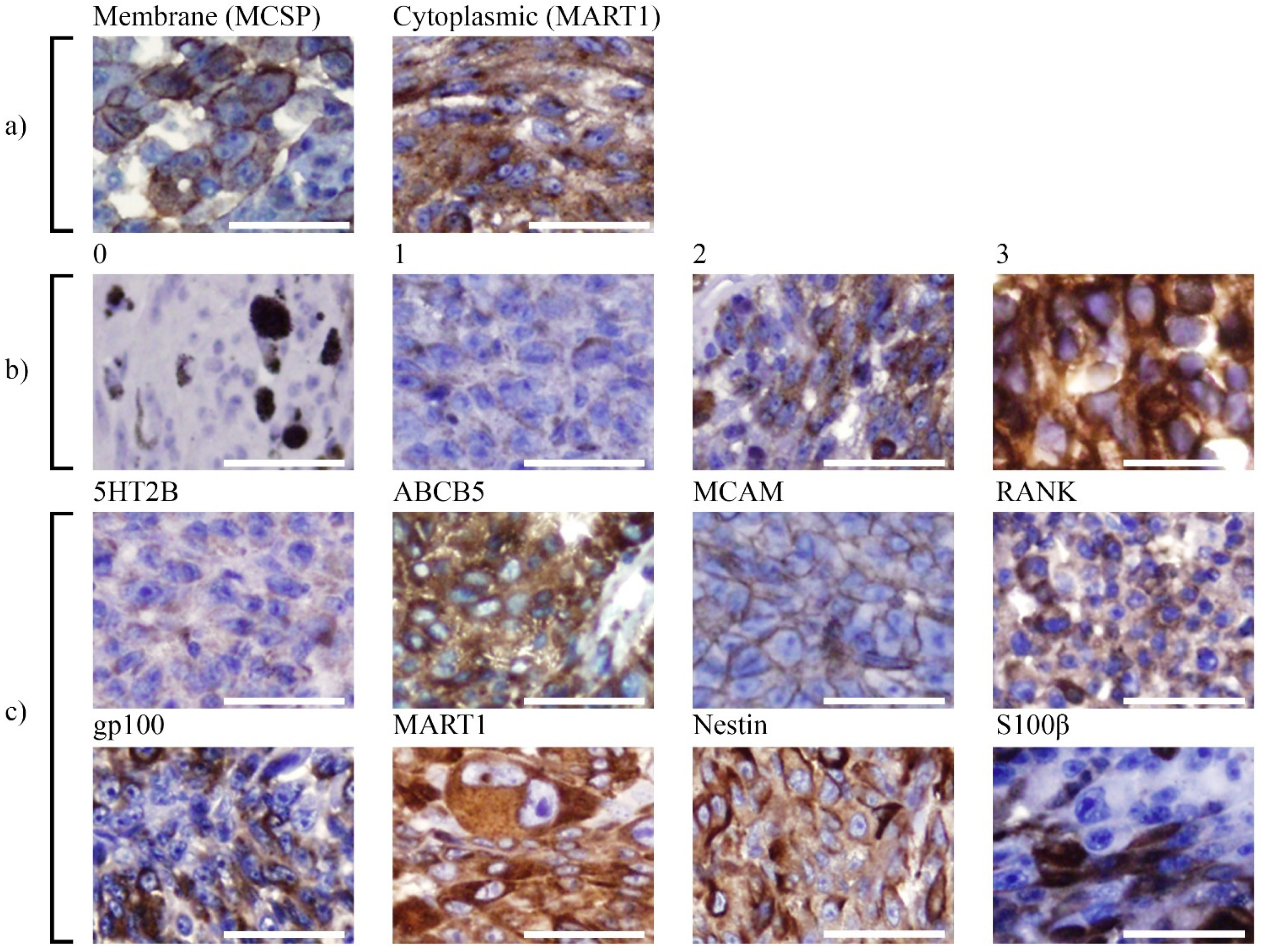
2.2. Immunohistochemistry
2.3. Cell Lines
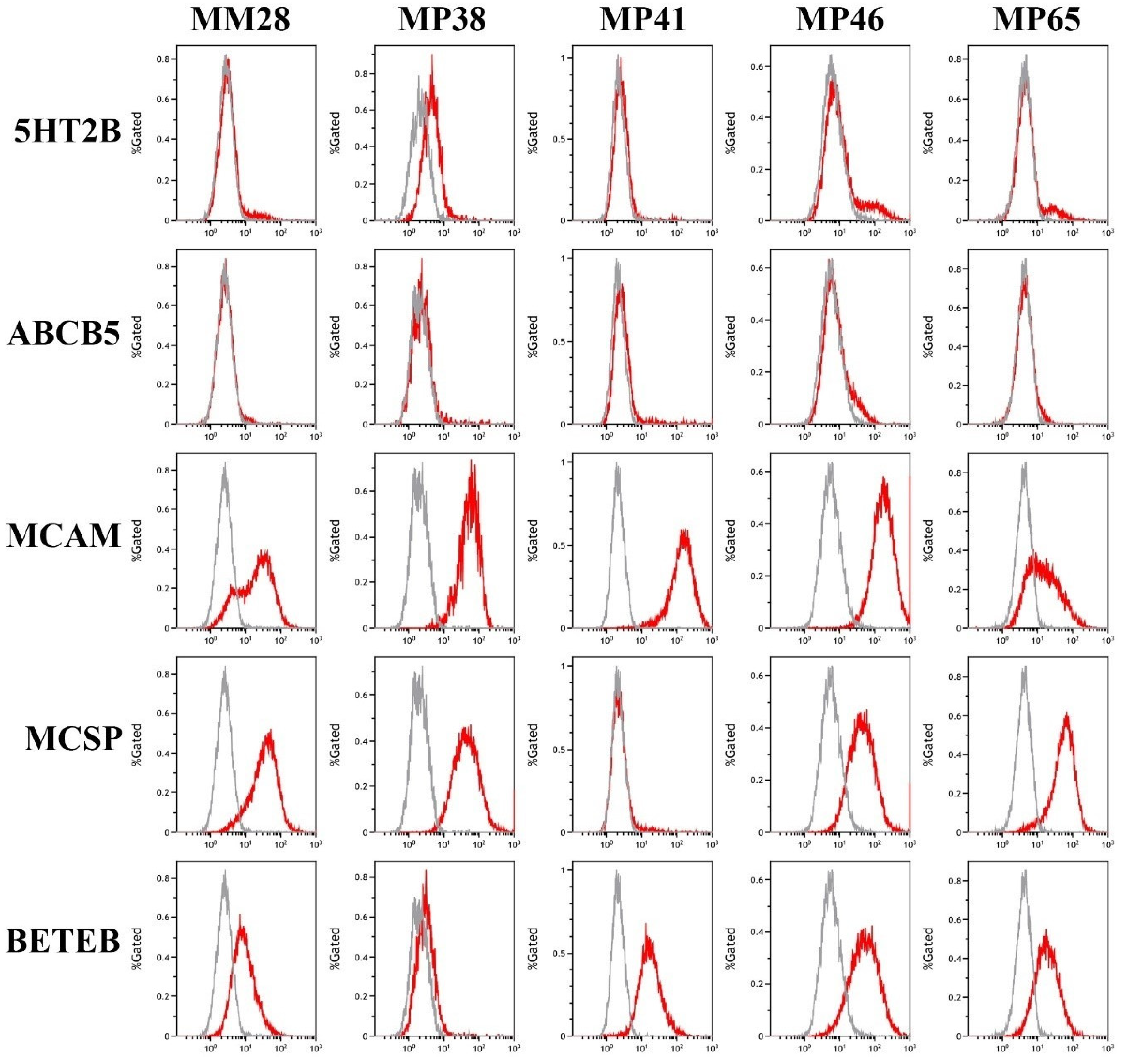
2.4. Flow Cytometry
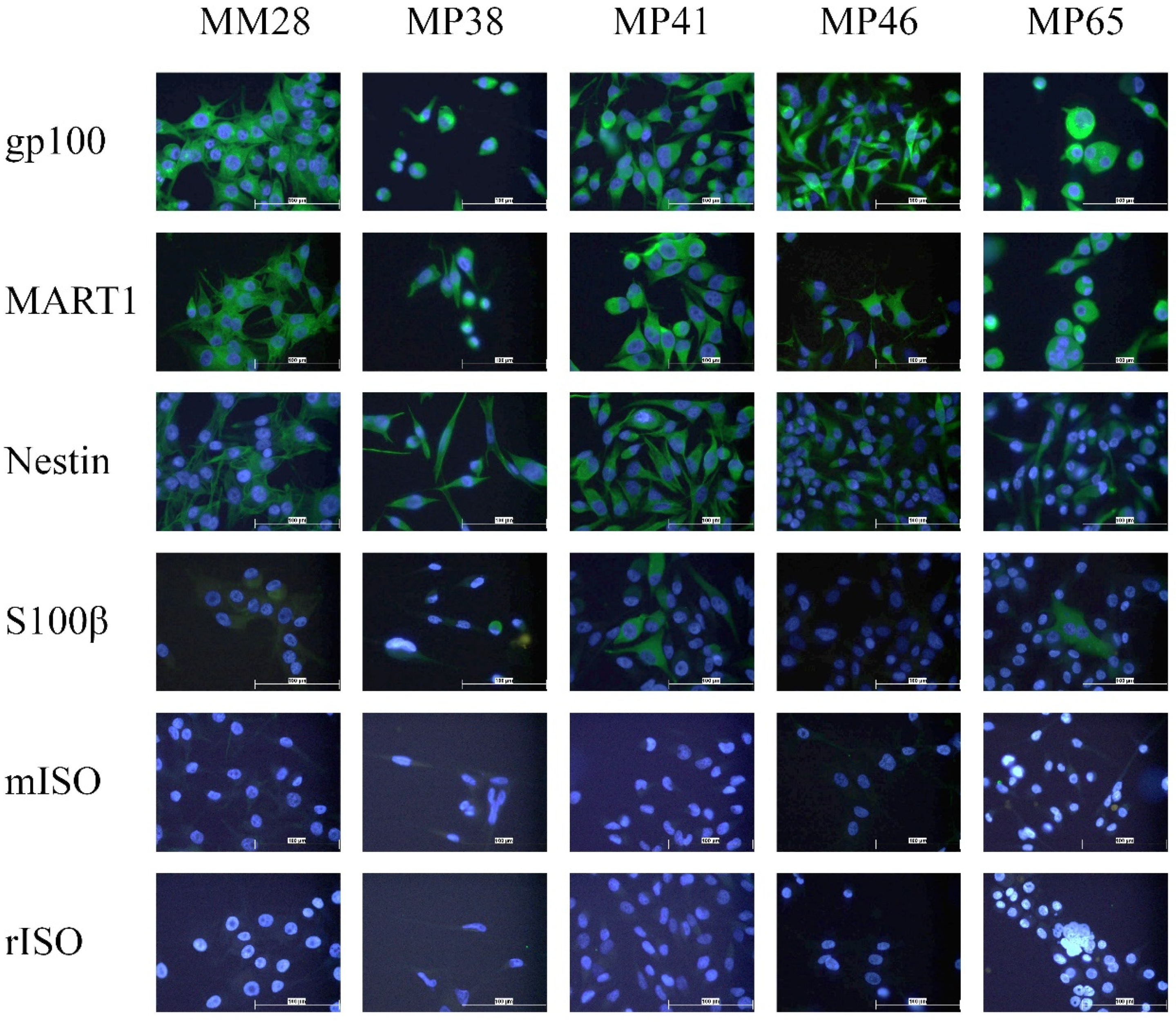
2.5. Immunocytochemistry
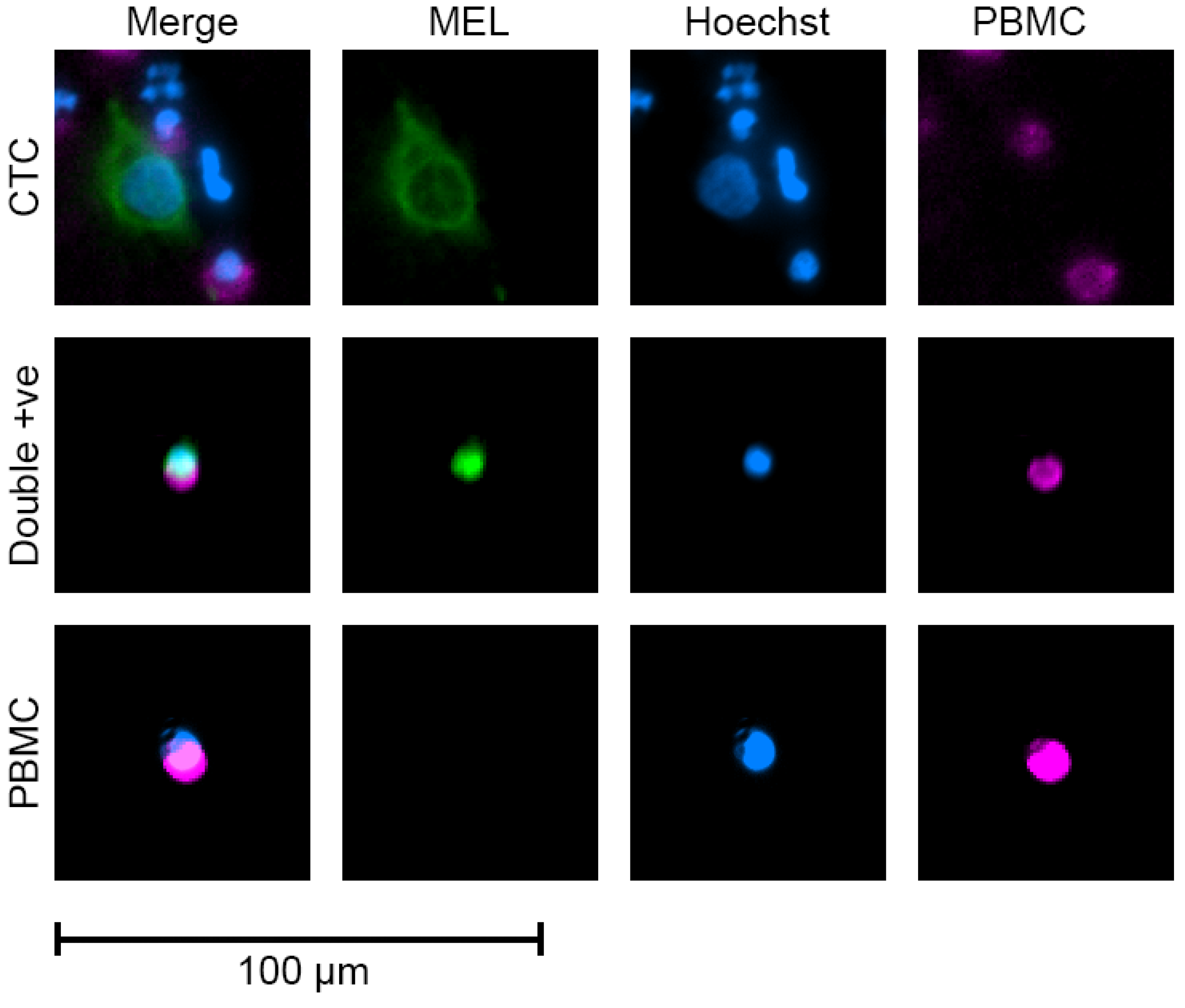
2.6. Circulating Tumour Cell Capture and Quantification
2.7. Statistical Analysis
3. Results
3.1. Analysis of Marker Expression in Primary UM Tumours
3.2. Analysis of Marker Expression in Primary and Metastatic UM Cell Lines
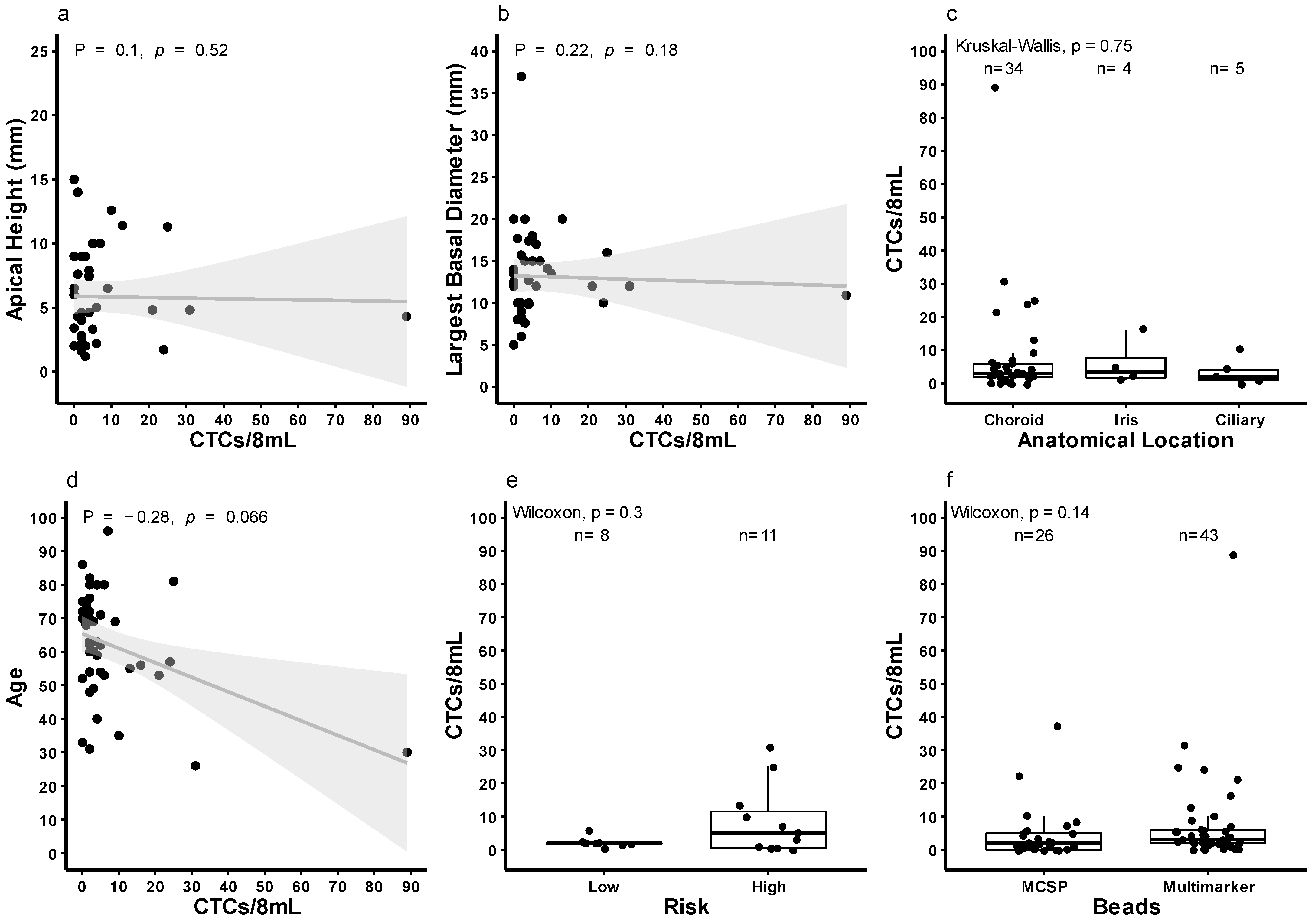
3.3. Quantification of CTCs in Patients with Localised Disease Using Multi-Marker Immunomagnetic Beads
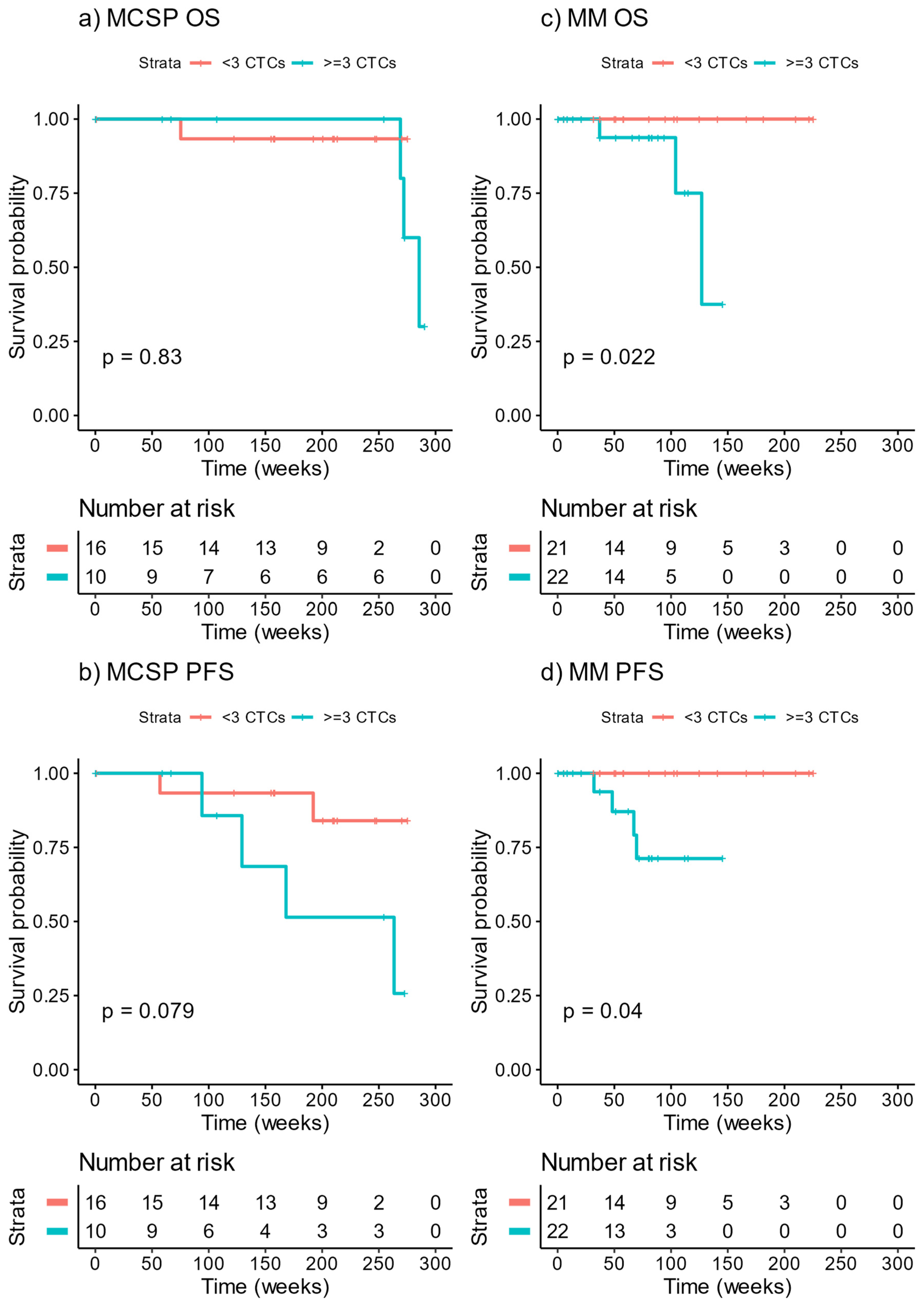
3.4. Survival of Patients Stratified by CTC Count at Baseline
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McLaughlin, C.C.; Wu, X.C.; Jemal, A.; Martin, H.J.; Roche, L.M.; Chen, V.W. Incidence of noncutaneous melanomas in the U.S. Cancer 2005, 103, 1000–1007. [Google Scholar] [CrossRef]
- Beasley, A.B.; Preen, D.B.; McLenachan, S.; Gray, E.S.; Chen, F.K. Incidence and Mortality of Uveal Melanoma in Australia (1982 to 2014). Br. J. Ophthalmol. 2021, in press. [Google Scholar]
- Blum, E.S.; Yang, J.; Komatsubara, K.M.; Carvajal, R.D. Clinical Management of Uveal and Conjunctival Melanoma. Oncology 2016, 30, 29–32. [Google Scholar]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [Green Version]
- Vichitvejpaisal, P.; Dalvin, L.A.; Mazloumi, M.; Ewens, K.G.; Ganguly, A.; Shields, C.L. Genetic Analysis of Uveal Melanoma in 658 Patients Using the Cancer Genome Atlas Classification of Uveal Melanoma as A, B, C, and D. Ophthalmology 2019, 126, 1445–1453. [Google Scholar] [CrossRef]
- Harbour, J.W.; Roberson, E.D.O.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef]
- Pereira, P.R.; Odashiro, A.N.; Lim, L.A.; Miyamoto, C.; Blanco, P.L.; Odashiro, M.; Maloney, S.; De Souza, D.F.; Burnier, M.N., Jr. Current and emerging treatment options for uveal melanoma. J. Clin. Ophthalmol. 2013, 7, 1669–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suesskind, D.; Ulmer, A.; Schiebel, U.; Fierlbeck, G.; Spitzer, B.; Spitzer, M.S.; Bartz-Schmidt, K.U.; Grisanti, S. Circulating melanoma cells in peripheral blood of patients with uveal melanoma before and after different therapies and association with prognostic parameters: A pilot study. Acta Ophthalmol. 2011, 89, 17–24. [Google Scholar] [CrossRef]
- Ulmer, A.; Beutel, J.; Susskind, D.; Hilgers, R.D.; Ziemssen, F.; Luke, M.; Rocken, M.; Rohrbach, M.; Fierlbeck, G.; Bartz-Schmidt, K.U.; et al. Visualization of circulating melanoma cells in peripheral blood of patients with primary uveal melanoma. Clin. Cancer Res. 2008, 14, 4469–4474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beasley, A.; Isaacs, T.; Khattak, M.A.; Freeman, J.B.; Allcock, R.; Chen, F.K.; Pereira, M.R.; Yau, K.; Bentel, J.; Vermeulen, T.; et al. Clinical Application of Circulating Tumor Cells and Circulating Tumor DNA in Uveal Melanoma. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, C.; Cani, A.K.; Larios, J.M.; Hovelson, D.H.; Aung, K.; Darga, E.P.; Cannell, E.M.; Baratta, P.J.; Liu, C.-J.; Chu, D.; et al. Comprehensive Mutation and Copy Number Profiling in Archived Circulating Breast Cancer Tumor Cells Documents Heterogeneous Resistance Mechanisms. Cancer Res. 2018, 78, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Mishima, Y.; Paiva, B.; Shi, J.; Park, J.; Manier, S.; Takagi, S.; Massoud, M.; Perilla-Glen, A.; Aljawai, Y.; Huynh, D.; et al. The Mutational Landscape of Circulating Tumor Cells in Multiple Myeloma. Cell Rep. 2017, 19, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidard, F.C.; Madic, J.; Mariani, P.; Piperno-Neumann, S.; Rampanou, A.; Servois, V.; Cassoux, N.; Desjardins, L.; Milder, M.; Vaucher, I.; et al. Detection rate and prognostic value of circulating tumor cells and circulating tumor DNA in metastatic uveal melanoma. Int. J. Cancer 2014, 134, 1207–1213. [Google Scholar] [CrossRef]
- Tura, A.; Luke, J.; Merz, H.; Reinsberg, M.; Luke, M.; Jager, M.J.; Grisanti, S. Identification of circulating melanoma cells in uveal melanoma patients by dual-marker immunoenrichment. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4395–4404. [Google Scholar] [CrossRef] [Green Version]
- Freeman, J.B.; Gray, E.S.; Millward, M.; Pearce, R.; Ziman, M. Evaluation of a multi-marker immunomagnetic enrichment assay for the quantification of circulating melanoma cells. J. Transl. Med. 2012, 10, 192. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Brunner, G.; Celebi, J.T. A Melanoma Subtype: Uveal Melanoma. J. Am. Acad. Dermatol. 2011, 64, 1185–1186. [Google Scholar] [CrossRef] [Green Version]
- Amirouchene-Angelozzi, N.; Nemati, F.; Gentien, D.; Nicolas, A.; Dumont, A.; Carita, G.; Camonis, J.; Desjardins, L.; Cassoux, N.; Piperno-Neumann, S.; et al. Establishment of novel cell lines recapitulating the genetic landscape of uveal melanoma and preclinical validation of mTOR as a therapeutic target. Mol. Oncol. 2014, 8, 1508–1520. [Google Scholar] [CrossRef]
- Therneau, T.M. A Package for Survival Analysis in R. Available online: https://CRAN.R-project.org/package=survival (accessed on 13 September 2021).
- Therneau, T.M.; Grambsch, P.M.; Therneau, T.M.; Grambsch, P.M. The cox model. In Modeling Survival Data: Extending the Cox Model; Springer: Cham, Switzerland, 2000; pp. 39–77. [Google Scholar]
- Kassambara, A.; Kosinski, M.; Biecek, P. survminer: Drawing Survival Curves Using ’ggplot2’. Available online: https://CRAN.R-project.org/package=survminer (accessed on 13 September 2021).
- Dardis, C. survMisc: Miscellaneous Functions for Survival Data. R Package Version 0.5.5. Available online: https://CRAN.R-project.org/package=survMisc (accessed on 13 September 2021).
- Thill, M.; Berna, M.J.; Grierson, R.; Reinhart, I.; Voelkel, T.; Piechaczek, C.; Galambos, P.; Jager, M.J.; Richard, G.; Lange, C.; et al. Expression of CD133 and other putative stem cell markers in uveal melanoma. Melanoma Res. 2011, 21, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Chen, R. The DecisionDx-UM Gene Expression Profile Test Provides Risk Stratification and Individualized Patient Care in Uveal Melanoma. PLoS Curr. 2013, 5, ecurrents.eogt.af8ba80fc776c778f771ce778f775dc485d774a618. [Google Scholar] [CrossRef]
- Bande, M.F.; Santiago, M.; Muinelo-Romay, L.; Blanco, M.J.; Mera, P.; Capeans, C.; Pardo, M.; Pineiro, A. Detection of circulating melanoma cells in choroidal melanocytic lesions. BMC Res. Notes 2015, 8, 452. [Google Scholar] [CrossRef] [Green Version]
- Frank, N.Y.; Margaryan, A.; Huang, Y.; Schatton, T.; Waaga-Gasser, A.M.; Gasser, M.; Sayegh, M.H.; Sadee, W.; Frank, M.H. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005, 65, 4320–4333. [Google Scholar] [CrossRef] [Green Version]
- Hoashi, T.; Watabe, H.; Muller, J.; Yamaguchi, Y.; Vieira, W.D.; Hearing, V.J. MART-1 is required for the function of the melanosomal matrix protein PMEL17/GP100 and the maturation of melanosomes. J. Biol. Chem. 2005, 280, 14006–14016. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Xu, Q.; Zhang, N.; Du, X.; Xu, G.; Yan, X. CD146, from a melanoma cell adhesion molecule to a signaling receptor. Signal Transduct. Target. Ther. 2020, 5, 148. [Google Scholar] [CrossRef]
- Eisenstein, A.; Gonzalez, E.C.; Raghunathan, R.; Xu, X.; Wu, M.; McLean, E.O.; McGee, J.; Ryu, B.; Alani, R.M. Emerging Biomarkers in Cutaneous Melanoma. Mol. Diagn. Ther. 2018, 22, 203–218. [Google Scholar] [CrossRef]
- Lutz, N.W.; Banerjee, P.; Wilson, B.J.; Ma, J.; Cozzone, P.J.; Frank, M.H. Expression of Cell-Surface Marker ABCB5 Causes Characteristic Modifications of Glucose, Amino Acid and Phospholipid Metabolism in the G3361 Melanoma-Initiating Cell Line. PLoS ONE 2016, 11, e0161803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cools-Lartigue, J.J.; McCauley, C.S.; Marshall, J.-C.A.; Di Cesare, S.; Gregoire, F.; Antecka, E.; Logan, P.; Burnier, M.N. Immunomagnetic isolation and in vitro expansion of human uveal melanoma cell lines. Mol. Vis. 2008, 14, 50–55. [Google Scholar] [PubMed]
- Li, Y.; Madigan, M.C.; Lai, K.; Conway, R.M.; Billson, F.A.; Crouch, R.; Allen, B.J. Human uveal melanoma expresses NG2 immunoreactivity. Br. J. Ophthalmol. 2003, 87, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Campoli, M.R.; Chang, C.C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): A melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit. Rev. Immunol. 2004, 24, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Chung, J.Y.; Ylaya, K.; Williams, R.L.; Guerrero, N.; Nakatsuka, N.; Badie, C.; Hewitt, S.M. Factors influencing the degradation of archival formalin-fixed paraffin-embedded tissue sections. J. Histochem. Cytochem. 2011, 59, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Vara, J.A.; Webster, J.D.; DuSold, D.; Miller, M.A. Immunohistochemical evaluation of the effects of paraffin section storage on biomarker stability. Vet. Pathol. 2014, 51, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, T.J.; Trancikova, D.; Ruiter, D.J.; van Muijen, G.N. High expression of immunotherapy candidate proteins gp100, MART-1, tyrosinase and TRP-1 in uveal melanoma. Br. J. Cancer 1998, 78, 1156–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beutel, J.; Wegner, J.; Wegner, R.; Ziemssen, F.; Nassar, K.; Rohrbach, J.M.; Hilgers, R.D.; Luke, M.; Grisanti, S. Possible implications of MCAM expression in metastasis and non-metastatic of primary uveal melanoma patients. Curr. Eye Res. 2009, 34, 1004–1009. [Google Scholar] [CrossRef]
- Gray, E.S.; Reid, A.L.; Bowyer, S.; Calapre, L.; Siew, K.; Pearce, R.; Cowell, L.; Frank, M.H.; Millward, M.; Ziman, M. Circulating Melanoma Cell Subpopulations: Their Heterogeneity and Differential Responses to Treatment. J. Investig. Dermatol. 2015, 135, 2040–2048. [Google Scholar] [CrossRef] [Green Version]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Lin, J.Y.; Alloo, A.; Wilson, B.J.; Schatton, T.; Zhan, Q.; Murphy, G.F.; Waaga-Gasser, A.M.; Gasser, M.; Stephen Hodi, F.; et al. Isolation of tumorigenic circulating melanoma cells. Biochem. Biophys. Res. Commun. 2010, 402, 711–717. [Google Scholar] [CrossRef] [Green Version]
- Aalto, Y.; Eriksson, L.; Seregard, S.; Larsson, O.; Knuutila, S. Concomitant loss of chromosome 3 and whole arm losses and gains of chromosome 1, 6, or 8 in metastasizing primary uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2001, 42, 313–317. [Google Scholar]
- Damato, B.; Dopierala, J.A.; Coupland, S.E. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin. Cancer Res. 2010, 16, 6083–6092. [Google Scholar] [CrossRef] [Green Version]
- Kamalidehghan, B.; Houshmand, M.; Kamalidehghan, F.; Jafarzadeh, N.; Azari, S.; Akmal, S.N.; Rosli, R. Establishment and characterization of two human breast carcinoma cell lines by spontaneous immortalization: Discordance between Estrogen, Progesterone and HER2/neu receptors of breast carcinoma tissues with derived cell lines. Cancer Cell Int. 2012, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Warkiani, M.E.; Khoo, B.L.; Wu, L.; Tay, A.K.; Bhagat, A.A.; Han, J.; Lim, C.T. Ultra-fast, label-free isolation of circulating tumor cells from blood using spiral microfluidics. Nat. Protoc. 2016, 11, 134–148. [Google Scholar] [CrossRef]
- Sarioglu, A.F.; Aceto, N.; Kojic, N.; Donaldson, M.C.; Zeinali, M.; Hamza, B.; Engstrom, A.; Zhu, H.; Sundaresan, T.K.; Miyamoto, D.T.; et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat. Methods 2015, 12, 685–691. [Google Scholar] [CrossRef]
- Yeo, T.; Tan, S.J.; Lim, C.L.; Lau, D.P.; Chua, Y.W.; Krisna, S.S.; Iyer, G.; Tan, G.S.; Lim, T.K.; Tan, D.S.; et al. Microfluidic enrichment for the single cell analysis of circulating tumor cells. Sci. Rep. 2016, 6, 22076. [Google Scholar] [CrossRef] [Green Version]
- Khoja, L.; Shenjere, P.; Hodgson, C.; Hodgetts, J.; Clack, G.; Hughes, A.; Lorigan, P.; Dive, C. Prevalence and heterogeneity of circulating tumour cells in metastatic cutaneous melanoma. Melanoma Res. 2014, 24, 40–46. [Google Scholar] [CrossRef]
- De Giorgi, V.; Pinzani, P.; Salvianti, F.; Panelos, J.; Paglierani, M.; Janowska, A.; Grazzini, M.; Wechsler, J.; Orlando, C.; Santucci, M.; et al. Application of a filtration- and isolation-by-size technique for the detection of circulating tumor cells in cutaneous melanoma. J. Investig. Dermatol. 2010, 130, 2440–2447. [Google Scholar] [CrossRef]
- Aya-Bonilla, C.; Marsavela, G.; Freeman, J.B.; Lomma, C.; Frank, M.H.; Khattak, M.A.; Meniawy, T.M.; Millward, M.; Warkiani, M.E.; Gray, E.S.; et al. Isolation and detection of circulating tumour cells from metastatic melanoma patients using a slanted spiral microfluidic device. Oncotarget 2017, 8, 67355–67368. [Google Scholar] [CrossRef] [Green Version]
- Aya-Bonilla, C.A.; Morici, M.; Hong, X.; McEvoy, A.C.; Sullivan, R.J.; Freeman, J.; Calapre, L.; Khattak, M.A.; Meniawy, T.; Millward, M.; et al. Detection and prognostic role of heterogeneous populations of melanoma circulating tumour cells. Br. J. Cancer 2020, 122, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, C.; Pinzani, P.; Salvianti, F.; Scatena, C.; Paglierani, M.; Ucci, F.; Pazzagli, M.; Massi, D. Circulating tumor cells detection and counting in uveal melanomas by a filtration-based method. Cancers 2014, 6, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Roszik, J.; Gombos, D.; Upshaw, J.; Sarli, V.; Meas, S.; Lucci, A.; Hall, C.; Patel, S. Pilot Study of Circulating Tumor Cells in Early-Stage and Metastatic Uveal Melanoma. Cancers 2019, 11, 856. [Google Scholar] [CrossRef] [Green Version]
- Chew, A.L.; Spilsbury, K.; Isaacs, T.W. Survival from uveal melanoma in Western Australia 1981–2005. Clin. Exp. Ophthalmol. 2015, 43, 422–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diener-West, M.; Reynolds, S.M.; Agugliaro, D.J.; Caldwell, R.; Cumming, K.; Earle, J.D.; Green, D.L.; Hawkins, B.S.; Hayman, J.; Jaiyesimi, I.; et al. Screening for Metastasis From Choroidal Melanoma: The Collaborative Ocular Melanoma Study Group Report 23. J. Clin. Oncol. 2004, 22, 2438–2444. [Google Scholar] [CrossRef]
- Gragoudas, E.S.; Egan, K.M.; Seddon, J.M.; Glynn, R.J.; Walsh, S.M.; Finn, S.M.; Munzenrider, J.E.; Spar, M.D. Survival of patients with metastases from uveal melanoma. Ophthalmology 1991, 98, 383–389. [Google Scholar] [CrossRef]
- Rietschel, P.; Panageas, K.S.; Hanlon, C.; Patel, A.; Abramson, D.H.; Chapman, P.B. Variates of Survival in Metastatic Uveal Melanoma. J. Clin. Oncol. 2005, 23, 8076–8080. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Auer, M.; Gasch, C.; Pichler, M.; Ulz, P.; Hoffmann, E.M.; Lax, S.; Waldispuehl-Geigl, J.; Mauermann, O.; Lackner, C.; et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013, 73, 2965–2975. [Google Scholar] [CrossRef] [Green Version]
- Tura, A.; Merz, H.; Reinsberg, M.; Luke, M.; Jager, M.J.; Grisanti, S.; Luke, J. Analysis of monosomy-3 in immunomagnetically isolated circulating melanoma cells in uveal melanoma patients. Pigment Cell Melanoma Res. 2016, 29, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Filshie, R.J.; Zannettino, A.C.; Makrynikola, V.; Gronthos, S.; Henniker, A.J.; Bendall, L.J.; Gottlieb, D.J.; Simmons, P.J.; Bradstock, K.F. MUC18, a member of the immunoglobulin superfamily, is expressed on bone marrow fibroblasts and a subset of hematological malignancies. Leukemia 1998, 12, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Dye, D.E.; Karlen, S.; Rohrbach, B.; Staub, O.; Braathen, L.R.; Eidne, K.A.; Coombe, D.R. hShroom1 links a membrane bound protein to the actin cytoskeleton. Cell. Mol. Life Sci. 2009, 66, 681–696. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Age * | Cell Morphology (Callender) | TMA Cell Morphology (Callender) | Tumour Size Basal × Height (mm) | Location | Metastatic Disease: Interval Time (Months) | MLPA |
|---|---|---|---|---|---|---|---|
| PUM1 | 38 | M (Predominantly S) | M | 12 × 9 | Left choroidal | No: 51 | NC ** |
| PUM2 | 51 | M | S/M | 12 × 8 | Right choroidal | Yes: 18 | L3p, L3q, G8q |
| PUM3 | 65 | M | E/M | 10 × 6 | Left choroidal | Yes: 33 | L3p, G8q |
| PUM4 | 47 | M | E/M | 11 × 12 | Right choroidal | Yes: −3 | L3p, L3q, G8q |
| PUM5 | 59 | M | S/M | 9 × 4 | Right choroidal | No: 42 | G8q |
| PUM6 | 31 | M (E < 10%) | E/M | 15 × 12 | Left choroidal | Yes: 12 | G8q, some evidence of 3p loss, but not strong |
| PUM7 | 42 | S | S | 16 × 9 | Left choroidal | Unknown | NC |
| PUM8 | 80 | M | S | 11 × 12 | Left choroidal | Unknown | loss 1p, 3p, 3q, gain 8q |
| PUM9 | 59 | M | E/M | 14 × 8 | Right choroidal | Yes: 3 | loss 3p, 3q, gain 8q |
| PUM10 | 79 | M | E | 20 × 15 | Right choroidal | No: 19 | NC ** |
| Average Marker Intensity Score | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | ABCB5 | MART1 | gp100 | S100β | Nestin | 5HT2B | MCAM | RANK | MCSP |
| PUM5 | 3 | 3 | 1 | 2 | 2 | 1 | 1.5 | 0 | 0 |
| PUM6 | 3 | 1 | 2 | 1.5 | 0 | 1 | 1 | 0 | 0 |
| PUM10 | 2 | 3 | 1 | 2 | 2 | 1 | 2 | 2 | 0 |
| PUM9 | 2 | 2 | 1 | 0 | 2 | 1.5 | 2 | 1 | 0 |
| PUM2 | 3 | 2.8 | 1 | 2 | 1.5 | 1 | 0 | 0 | 0 |
| PUM3 | 3 | 2.5 | 2 | 1 | 2 | 0 | 0 | 1 | 0 |
| PUM4 | 2 | 2.6 | 1.3 | 0 | 0 | 1 | 0 | 1.3 | 0 |
| PUM8 | 3 | 3 | 1.5 | 0 | 0 | 0 | 0 | 1 | 0 |
| PUM7 | 3 | 3 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| PUM1 | 0 | 2 | 2 | 1 | 0 | 0 | 0 | 0 | 0 |
| PID | Age | Sex | Eye | Anatomical Location | Apical Height (mm) | Largest Basal Diameter (mm) | Volume (mm3) | Callendar Classification | BAP1 Status | Tissue Mutation | Genetic Features | CTCs | FU (Weeks) | PFS (Weeks) | COD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 556 | 59 | F | Left | Choroid | 4.6 | 9.8 | 462 | Mixed | - | - | - | 4 | 72 | ||
| 565 | 72 | M | Left | Choroid | 4 | 8.3 | 288 | - | - | - | NC | 2 | 221 | ||
| 580 | 80 | F | Left | Choroid | 4.6 | 10 | 481 | 2 | 225 | ||||||
| 646 | 33 | F | Right | Choroid | 2 | 5 | 52 | 0 | 210 | ||||||
| 694 | 68 | M | Left | Choroid | 4.3 | 10 | 450 | 1 | 141 | ||||||
| 695 | 80 | F | Left | Choroid | 7.9 | 17.4 | 2503 | 4 | 37 | 32 | UM | ||||
| 703 | 96 | F | Left | Choroid | 10 | 15 | 2355 | Mixed | L | GNAQ Q209P | L1p, L3, L6q, G8 | 7 | 66 | ||
| 712 | 57 | F | Right | Choroid | 1.7 | 10 | 178 | 24 | 13 | ||||||
| 716 | 72 | F | Left | Iris | - | - | - | Spindle | - | GNAQ Q209L | NC | 1 | 166 | ||
| 721 | 49 | F | Left | Choroid | 2 | 15 | 471 | Mixed | P | GNA11 Q209L | L1p, L3, L8p, G8q | 3 | 127 | 70 | UM |
| 763 | 63 | M | Right | Ciliary | 2.8 | 6 | 106 | 2 | 105 | ||||||
| 805 | 82 | M | Left | Choroid | 2 | 9 | 170 | Mixed | 2 | 181 | |||||
| 840 | 81 | M | Right | Choroid | 11.3 | 16 | 3028 | Mixed | L | - | L3, G8q | 25 | 104 | 67 | UM |
| 872 | 74 | M | Right | Choroid | 7.6 | 17.7 | 2492 | Mixed | - | GNA11 Q209L | - | 1 | 125 | ||
| 879 | 35 | F | Right | Ciliary | 12.6 | 13.5 | 2404 | Epithelioid | - | GNAQ Q209P | pL3, G8q | 10 | 145 | ||
| 995 | 76 | M | Left | Choroid | 9 | 10 | 942 | - | GNAQ Q209L | - | 2 | 0 | |||
| 1022 | 71 | F | Right | Iris | - | - | - | 5 | 83 | ||||||
| 1041 | 26 | M | Right | Choroid | 4.8 | 12 | 723 | Mixed | L | GNAQ R183Q | L3, L6q, L8p, G8q | 31 | 115 | ||
| 1059 | 69 | M | Right | Choroid | 9 | 20 | 3768 | Mixed | - | GNAQ Q209P | - | 3 | 112 | ||
| 1160 | 62 | F | Left | Iris | 1.6 | 37 | 2293 | Mixed | P | pG6p | 2 | 58 | |||
| 1163 | 48 | M | Left | Choroid | 2.2 | 10 | 230 | - | - | - | NC | 2 | 102 | ||
| 1180 | 53 | M | Left | Choroid | 5 | 17 | 1512 | Mixed | P | GNAQ Q209P | NC | 6 | 83 | ||
| 1217 | 70 | F | Right | Choroid | 4.6 | 15.7 | 1187 | 2 | 51 | ||||||
| 1218 | 63 | F | Left | Choroid | 7.4 | 12.7 | 1249 | Spindle | P | - | - | 4 | 51 | ||
| 1227 | 40 | F | Left | Ciliary | 7.5 | 10 | 785 | 4 | 88 | ||||||
| 1231 | 31 | F | Right | Choroid | 2.7 | 9 | 229 | 2 | 95 | ||||||
| 1237 | 62 | F | Right | Choroid | 3.3 | 15 | 777 | - | - | GNAQ Q209P | L1p, pLBAP1, pL6p | 5 | 80 | ||
| 1238 | 80 | M | Right | Choroid | 2.2 | 12 | 332 | 6 | 21 | ||||||
| 1258 | 54 | M | Right | Choroid | 10 | 18 | 3391 | Mixed | P | GNAQ Q209L | G6p, G8q | 5 | 81 | ||
| 1284 | 54 | F | Right | Choroid | 2 | 10 | 209 | Mixed | - | - | L1p, G6p | 2 | 31 | ||
| 1285 | 70 | F | Left | Choroid | 9 | 14 | 1846 | Mixed | L | GNAQ R183Q | L1p, L3, G8q | 0 | 58 | ||
| 1286 | 52 | M | Right | Choroid | 3.4 | 12.5 | 556 | Epithelioid | L | - | - | 0 | 37 | ||
| 1287 | 30 | M | Right | Choroid | 4.3 | 10.9 | 535 | Epithelioid | P | - | - | 89 | 37 | ||
| 1297 | 69 | F | Left | Choroid | 6.5 | 14.1 | 1353 | 9 | 8 | ||||||
| 1298 | 75 | M | Left | Choroid | 6 | 12 | 904 | - | - | GNA11 Q209L | G6p | 0 | 80 | ||
| 1307 | 56 | M | Left | Iris | - | - | - | 16 | 94 | 48 | |||||
| 1308 | 73 | M | Right | Ciliary | 14 | 8 | 938 | - | - | GNAQ Q209L | L1p, L3, G8 | 1 | 50 | ||
| 1338 | 72 | M | Right | Choroid | 6.5 | 13.5 | 1240 | - | - | - | L1p, L3, G8 | 0 | 5 | ||
| 1370 | 60 | M | Left | Choroid | 2.7 | 10 | 283 | Spindle | P | GNAQ Q209P | G6p | 2 | 0 | ||
| 1387 | 55 | M | Right | Choroid | 11.4 | 20 | 4773 | Mixed | L | - | L3, G6p, L6q, G8q | 13 | 5 | ||
| 1401 | 53 | F | Right | Choroid | 4.8 | 12 | 723 | 21 | 0 | ||||||
| 1405 | 60 | F | Right | Choroid | 1.2 | 7.6 | 73 | 3 | 0 | ||||||
| 1408 | 86 | M | Left | Ciliary | 15 | 20 | 6280 | Mixed | - | GNAQ Q209L | L1p, L3, pL6p, G8q | 0 | 37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beasley, A.B.; Isaacs, T.W.; Vermeulen, T.; Freeman, J.; DeSousa, J.-L.; Bhikoo, R.; Hennessy, D.; Reid, A.; Chen, F.K.; Bentel, J.; et al. Analysis of Circulating Tumour Cells in Early-Stage Uveal Melanoma: Evaluation of Tumour Marker Expression to Increase Capture. Cancers 2021, 13, 5990. https://doi.org/10.3390/cancers13235990
Beasley AB, Isaacs TW, Vermeulen T, Freeman J, DeSousa J-L, Bhikoo R, Hennessy D, Reid A, Chen FK, Bentel J, et al. Analysis of Circulating Tumour Cells in Early-Stage Uveal Melanoma: Evaluation of Tumour Marker Expression to Increase Capture. Cancers. 2021; 13(23):5990. https://doi.org/10.3390/cancers13235990
Chicago/Turabian StyleBeasley, Aaron B., Timothy W. Isaacs, Tersia Vermeulen, James Freeman, Jean-Louis DeSousa, Riyaz Bhikoo, Doireann Hennessy, Anna Reid, Fred K. Chen, Jacqueline Bentel, and et al. 2021. "Analysis of Circulating Tumour Cells in Early-Stage Uveal Melanoma: Evaluation of Tumour Marker Expression to Increase Capture" Cancers 13, no. 23: 5990. https://doi.org/10.3390/cancers13235990