
Gram-Negative Bacterial Lipopolysaccharide Promotes Tumor Cell Proliferation in Breast Implant-Associated Anaplastic Large-Cell Lymphoma

 ,
,  , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
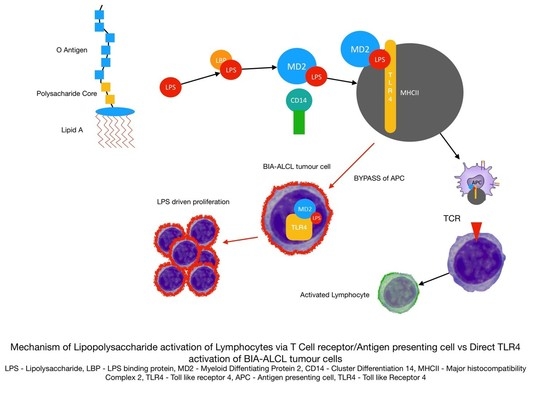
1. Introduction
2. Materials and Methods
2.1. Tumor Cells, Peripheral Blood Mononuclear Cells, Cell Lines
2.1.1. BIA-ALCL Tumor Cells, Cell Lines
2.1.2. Cutaneous ALCL Cell Lines
2.1.3. MT-4 Cell Line
2.1.4. Peripheral Blood Mononuclear Cells (PBMC)
2.1.5. Cell Culture Conditions
2.1.6. Ethics Statement
2.2. Cell Proliferation Assays
2.3. Cell Viability and Apoptosis Assays
2.4. Effect of Implant Surface Texture on BIA-ALCL Cell Proliferation
2.5. TLR4 Detection Assay
2.6. TLR4 Inhibition on Cell Proliferation and TNF-α Production Assays
2.7. Statistical Analysis
3. Results
3.1. Patients’ Clinical Features
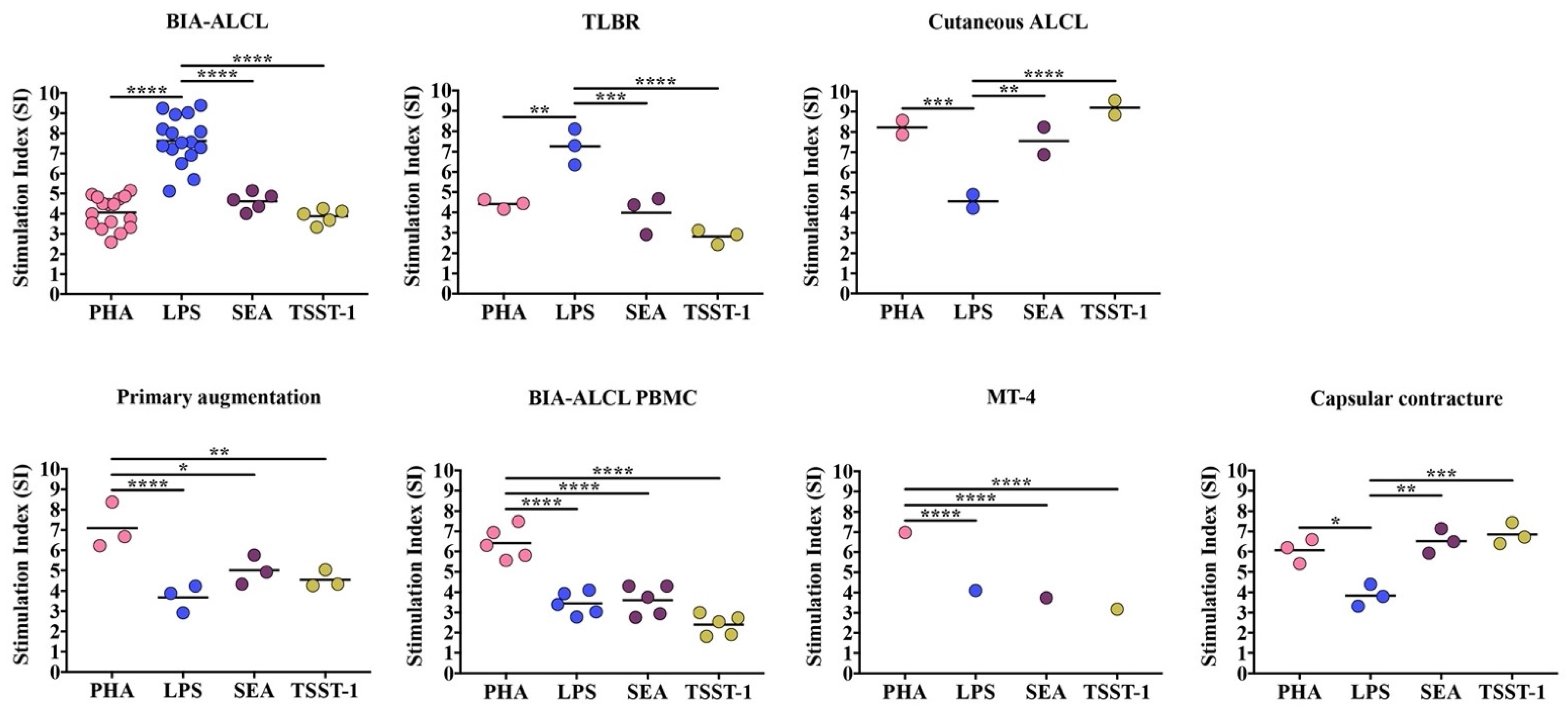
3.2. Cell Proliferation Response to Mitogenic/Antigenic Stimulation
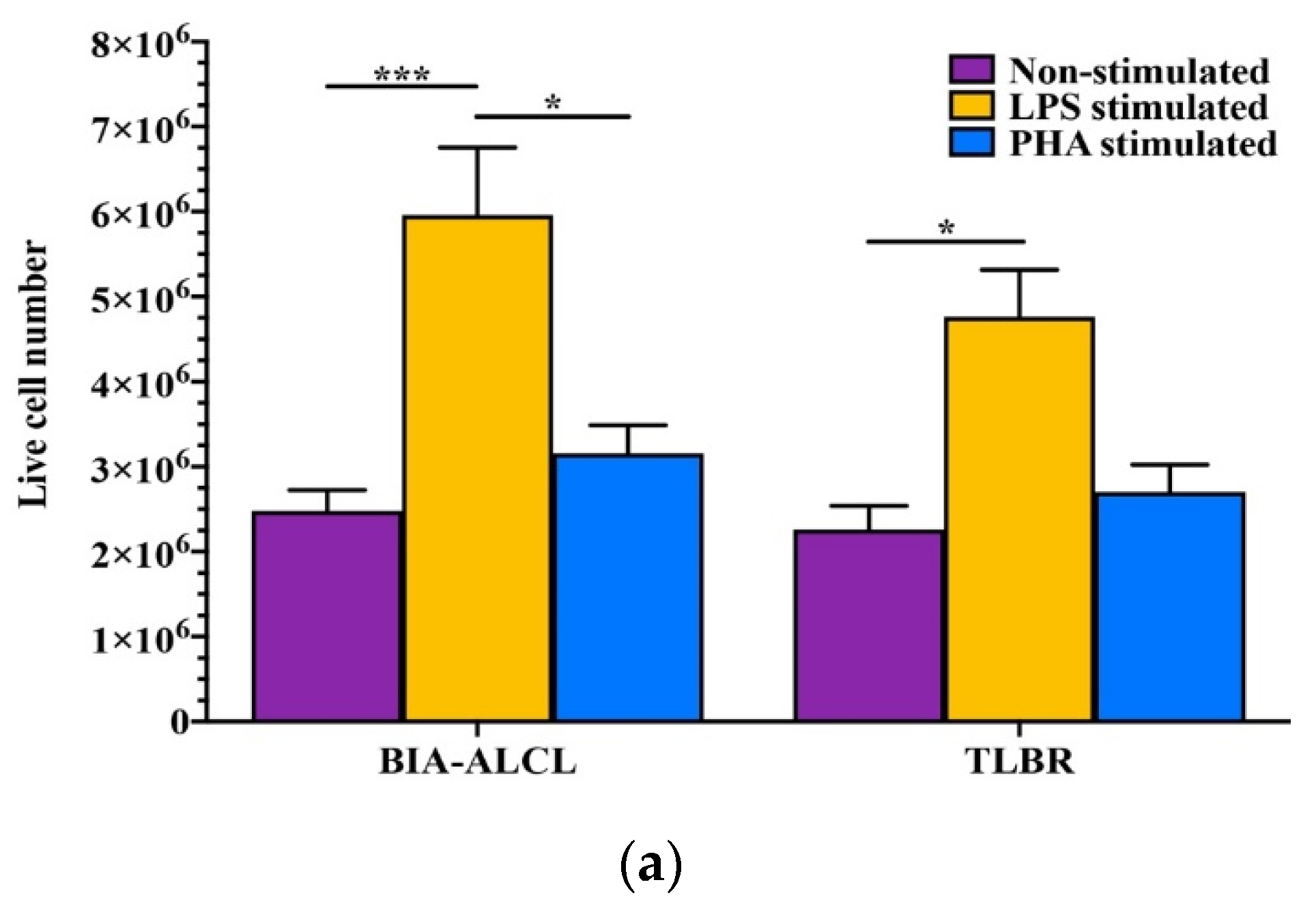
3.3. BIA-ALCL Cell Viability and Apoptosis in Response to Mitogen Stimulation
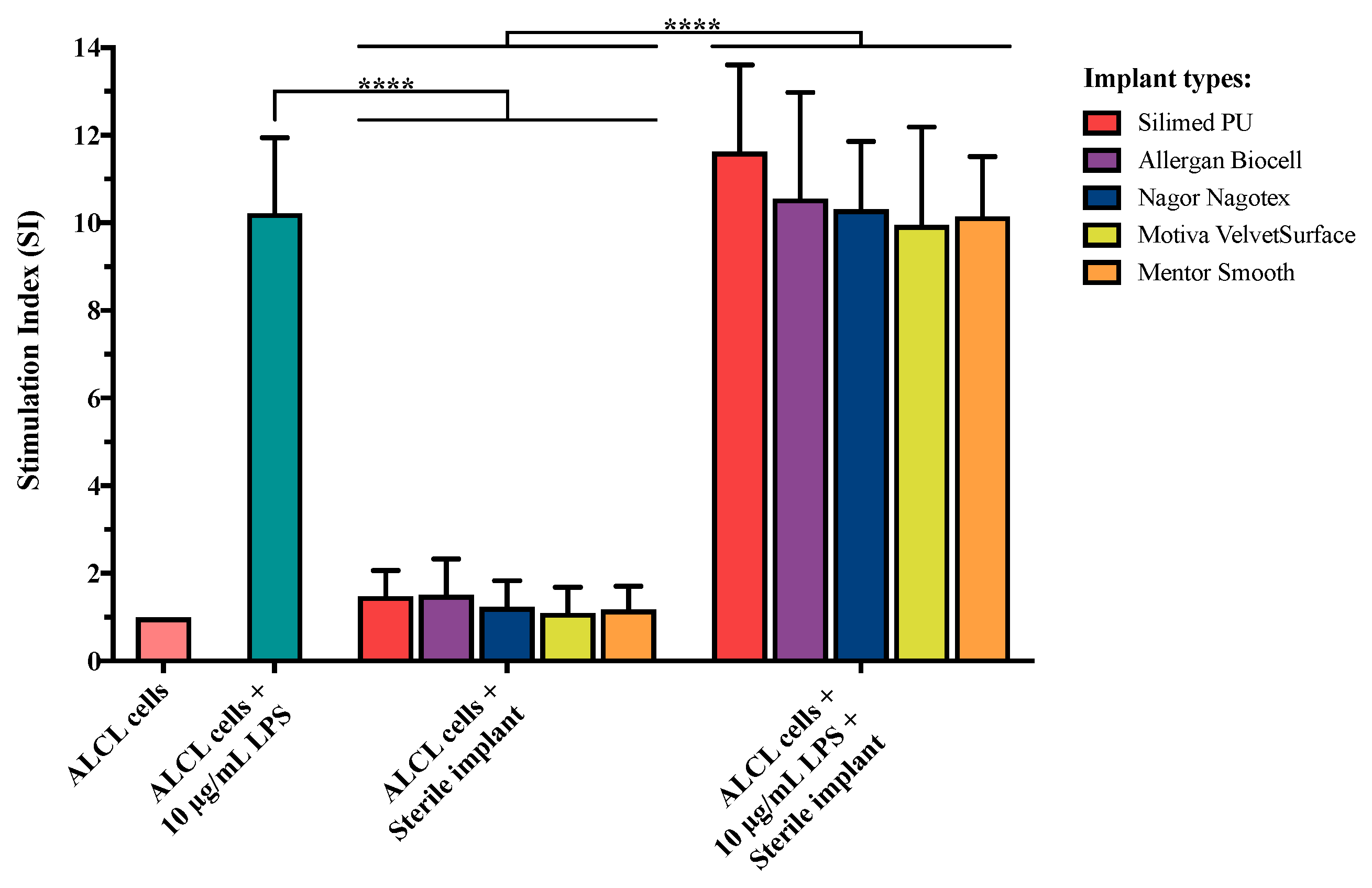
3.4. Effect of Implant Surface Texture on BIA-ALCL Cell Proliferation
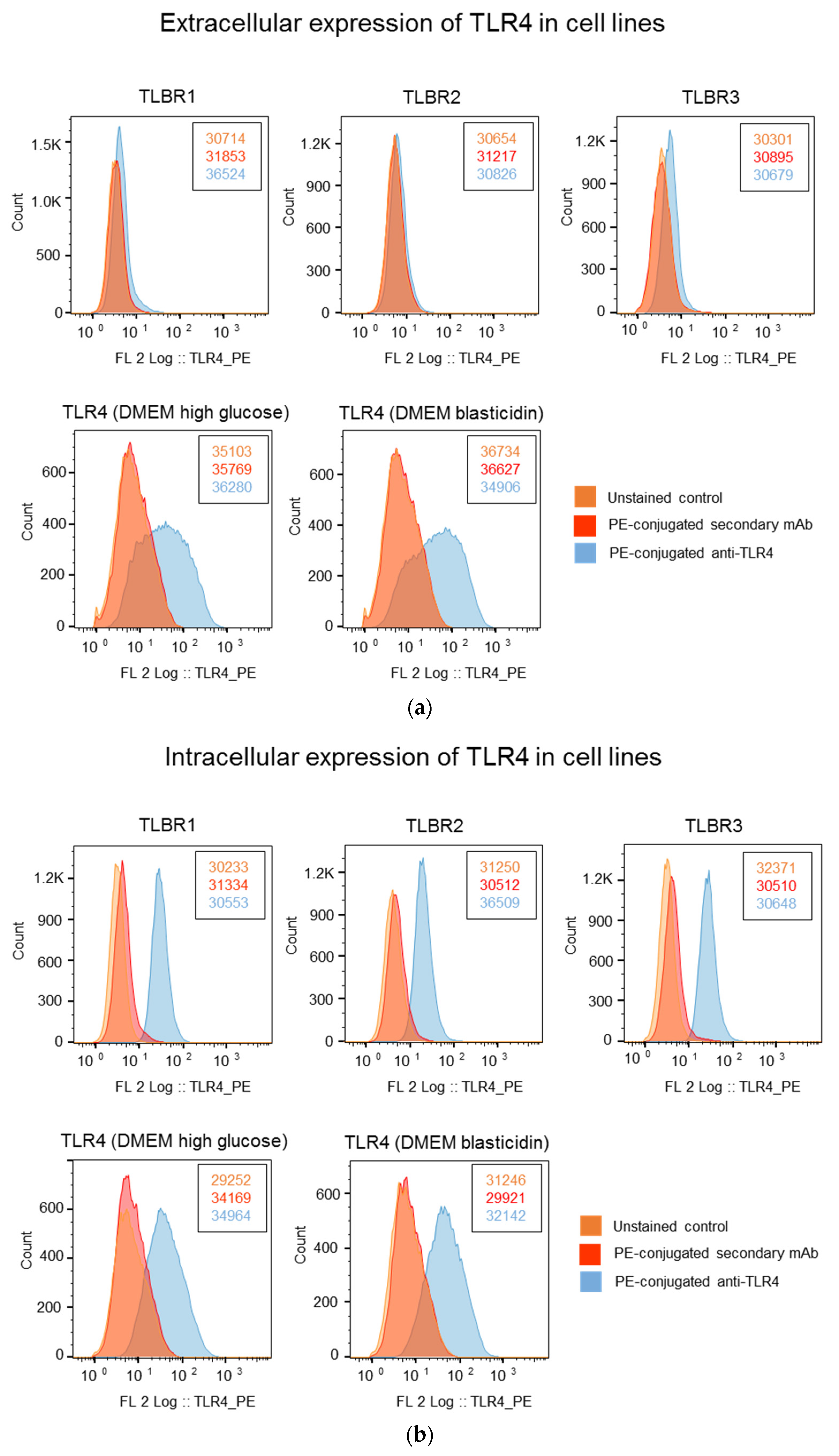
3.5. Higher TLR4 Expression in TLBR Cell Lines
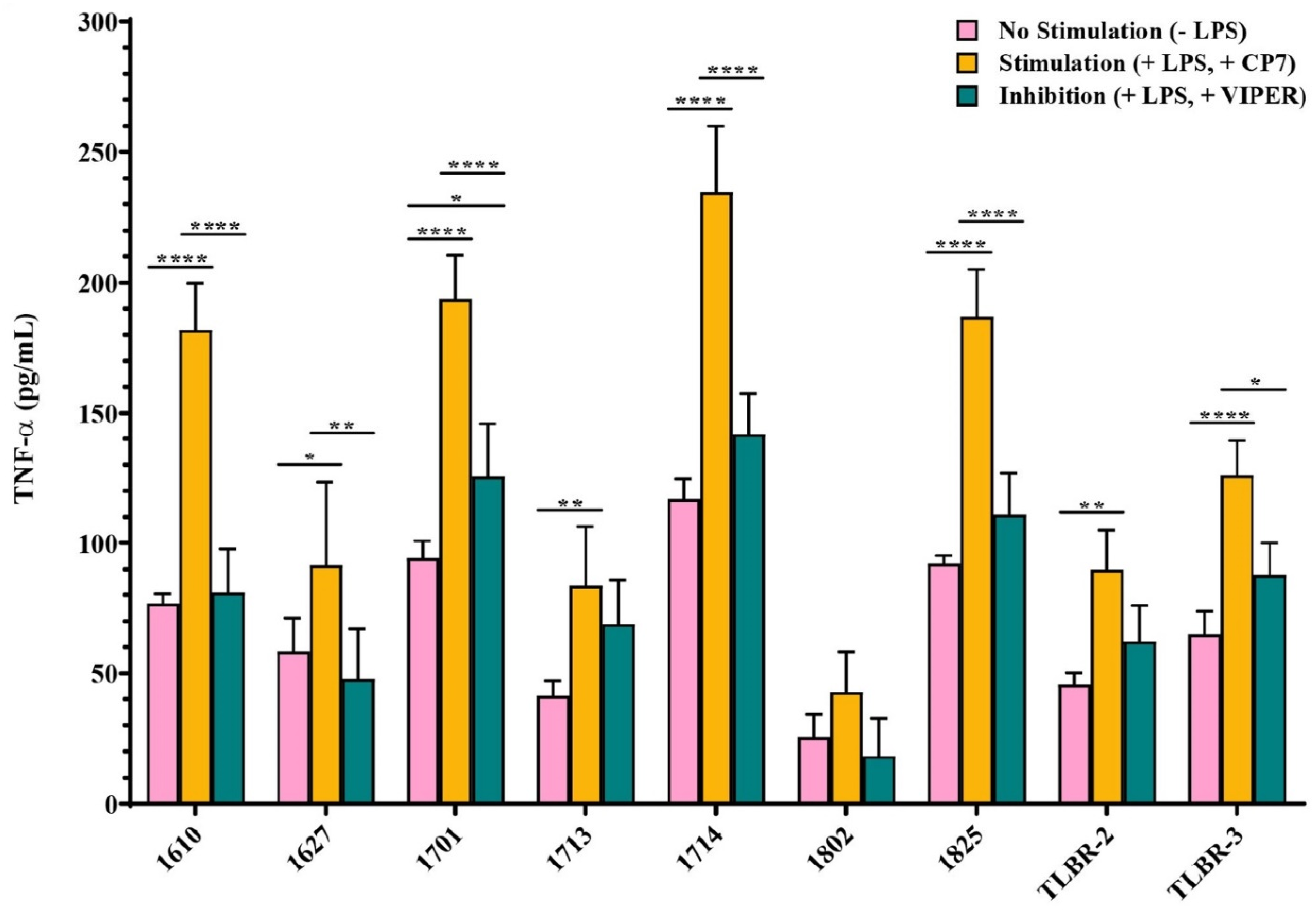
3.6. Effect of TLR4 Inhibition on LPS Stimulation of BIA-ALCL Cells
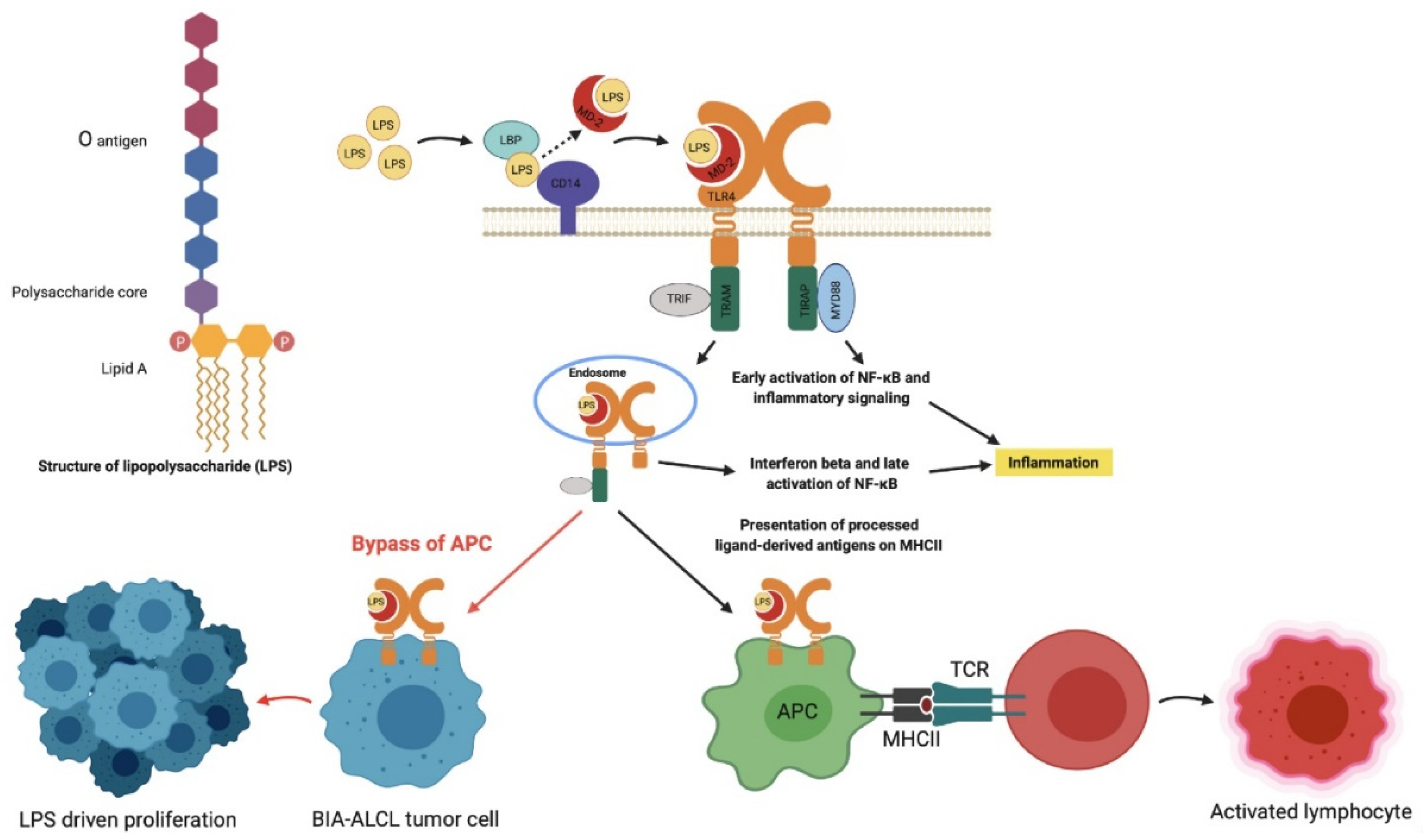
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scheme 1 | Age at Diagnosis (Years) | Duration of Implant (Years) | Implant Type | Clinical Presentation | Stage 1 | Treatment | Samples Analyzed |
|---|---|---|---|---|---|---|---|
| 1610 | 38 | 13 | Silimed PU 2 | Seroma | 1C | Surgery | Tumor cells |
| 1612 | 45 | 5 | Silimed PU | Seroma | 1A | Surgery | Tumor cells |
| 1618 | 51 | 14 | Allergan Biocell | Seroma | 1A | Surgery | Tumor cells |
| 1626 | 45 | 10 1 2 | Allergan Biocell Silimed PU Nagor | Capsular contracture followed by seroma | 1A | Surgery | Tumor cells |
| 1627 | 41 | 4 0.5 | Allergan Biocell Allergan Biocell | Infection followed by revision and incidental mass | 2A | Surgery | Tumor cells |
| 1701 | 33 | 5 | Silimed PU | Seroma | 1A | Surgery | Tumor cells |
| 1713 | 58 | 10 | Allergan Biocell | Seroma | 1A | Surgery | Tumor cells |
| 1714 | 40 | 0.1 4 6 | PIP 3 PIP Silimed PU | Contracture then seroma | 1A | Surgery | Tumor cells |
| 1715 | 31 | 5 | Silimed PU | Seroma | 1A | Surgery | Tumor cells |
| 1802 | 29 | 9 | Nagor | Seroma | 1A | Surgery | Tumor cells |
| 1803 | 57 | 8 | Silimed PU | Seroma | 1A | Surgery | Tumor cells, PBMC |
| 1808 | 35 | 4 | Nagor | Seroma | 1A | Surgery | Tumor cells, PBMC |
| 1810 | 37 | 3 | Silimed PU | Seroma | 1A | Surgery | Tumor cells, PBMC |
| 1819 | 53 | 14.25 | Mentor Siltex | Seroma | 1A | Surgery | Tumor cells, PBMC |
| 1821 | 52 | 9 | Allergan Biocell | Seroma | 1A | Surgery | Tumor cells, PBMC |
| 1825 | 55 | 20 | McGhan | Seroma | 1A | Surgery | Tumor cells |
| Cell Line | Age at Diagnosis | Implant Type | Clinical Presentation | Treatment |
|---|---|---|---|---|
| TLBR-1 | 42 | Nagor | Seroma | Surgery/Radiation therapy |
| TLBR-2 | 43 | Allergan Biocell | Seroma | Surgery/Chemotherapy and radiation therapy—patient had recurrence and is deceased |
| TLBR-3 | 45 | Allergan Biocell | Seroma | Surgery/Radiation therapy |
| Implant Type | Structure | Process | Surface Area | Roughness | Surface Type |
|---|---|---|---|---|---|
| Silimed Polyurethane (PU) |  | Polyurethane foam | High | High | 4 |
| Allergan Biocell |  | Salt loss | Intermediate | Intermediate | 3 |
| Nagor Nagotex |  | Salt loss | Low | Low | 2 |
| Motiva VelvetSurface |  | Nano | Minimal | Minimal | 1 |
| Mentor Smooth | | Smooth | Minimal | Minimal | 1 |
| Cells | Tukey’s Multiple Comparisons Test | 95% Confidence Interval (CI) of Difference | Adjusted p Value |
|---|---|---|---|
| BIA-ALCL | LPS vs. SEA | 1.490 to 4.093 | <0.0001 |
| LPS vs. TSST-1 | 2.232 to 4.835 | <0.0001 | |
| LPS vs PHA | 2.172 to 4.372 | <0.0001 | |
| PHA vs. SEA | −1.782 to 0.8207 | 0.7654 | |
| PHA vs. TSST-1 | −1.040 to 1.562 | 0.9519 | |
| SEA vs. TSST-1 | −0.7337 to 2.217 | 0.5510 | |
| TLBR | LPS vs. SEA | 1.362 to 5.172 | 0.0001 |
| LPS vs. TSST-1 | 2.527 to 6.337 | <0.001 | |
| LPS vs PHA | 0.9347 to 4.744 | 0.0011 | |
| PHA vs. SEA | −1.477 to 2.332 | 0.9344 | |
| PHA vs. TSST-1 | −0.3125 to 3.497 | 0.1331 | |
| SEA vs. TSST-1 | −0.7400 to 3.070 | 0.3796 | |
| Cutaneous ALCL | PHA vs. LPS | 1.329 to 5.995 | 0.0006 |
| SEA vs. LPS | 0.6654 to 5.331 | 0.0064 | |
| TSST-1 vs. LPS | 2.303 to 6.969 | <0.0001 | |
| PHA vs. SEA | −1.669 to 2.997 | 0.8767 | |
| PHA vs. TSST-1 | −3.307 to 1.359 | 0.6908 | |
| SEA vs. TSST-1 | −3.971 to 0.6950 | 0.2598 | |
| MT-4 | PHA vs. LPS | 1.754 to 3.991 | <0.0001 |
| PHA vs. SEA | 2.115 to 4.351 | <0.0001 | |
| PHA vs. TSST-1 | 2.677 to 4.913 | <0.0001 | |
| LPS vs. SEA | −0.7575 to 1.479 | 0.8290 | |
| LPS vs. TSST-1 | −0.1960 to 2.040 | 0.1408 | |
| SEA vs TSST-1 | −0.5567 to 1.680 | 0.5497 | |
| BIA-ALCL PBMC | PHA vs. LPS | 1.498 to 4.449 | <0.0001 |
| PHA vs. SEA | 1.337 to 4.288 | <0.0001 | |
| PHA vs. TSST-1 | 2.549 to 5.500 | <0.0001 | |
| LPS vs. SEA | −1.636 to 1.315 | 0.9917 | |
| LPS vs. TSST-1 | −0.4244 to 2.527 | 0.2479 | |
| SEA vs TSST-1 | −0.2635 to 2.688 | 0.1440 | |
| Capsular contracture PBMC | PHA vs. LPS | 0.3294 to 4.139 | 0.0151 |
| SEA vs. LPS | 0.7831 to 4.593 | 0.0023 | |
| TSST-1 vs. LPS | 1.118 to 4.928 | 0.0005 | |
| PHA vs. SEA | −2.359 to 1.451 | 0.9230 | |
| PHA vs. TSST-1 | −2.694 to 1.116 | 0.6963 | |
| SEA vs. TSST-1 | −2.240 to 1.570 | 0.9667 | |
| Primary augmentation PBMC | PHA vs. LPS | 1.507 to 5.317 | <0.0001 |
| PHA vs. SEA | 0.1812 to 3.991 | 0.0264 | |
| PHA vs. TSST-1 | 0.6430 to 4.453 | 0.0042 | |
| LPS vs. SEA | −3.231 to 0.5790 | 0.2669 | |
| LPS vs. TSST-1 | −2.769 to 1.041 | 0.6323 | |
| SEA vs. TSST-1 | −1.443 to 2.367 | 0.9192 |
| Model | Effect Size (Partial Omega Squared, ω2p) | 95% CI | Test Statistic | p Value |
|---|---|---|---|---|
| Mitogen type | 0.0397 | 0.000 to 0.1323 | F(3, 68) = 6.52 | <0.001 |
| Cell type | 0.2217 | 0.0301 to 0.3558 | F(5, 68) = 19.47 | <0.001 |
| Mitogen type × cell type | 0.5177 | 0.2230 to 0.5810 | F(15, 68) = 15.38 | <0.001 |
References
- Clemens, M.W.; Miranda, R.N. Coming of Age: Breast Implant-Associated Anaplastic Large Cell Lymphoma after 18 Years of Investigation. Clin. Plast. Surg. 2015, 42, 605–613. [Google Scholar] [CrossRef]
- Doren, E.L.; Miranda, R.N.; Selber, J.C.; Garvey, P.B.; Liu, J.; Medeiros, L.J.; Butler, C.E.; Clemens, M.W. United Stated Epidemiology of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Plast. Reconstr. Surg. 2017, 139, 1042–1050. [Google Scholar] [CrossRef]
- Loch-Wilkinson, A.; Beath, K.J.; Knight, R.J.W.; Wessels, W.L.F.; Magnusson, M.; Papadopoulos, T.; Connell, T.; Lofts, J.; Locke, M.; Hopper, I.; et al. Breast Implant-Associated Anaplastic Large Cell Lymphoma in Australia and New Zealand: High-Surface-Area Textured Implants Are Associated with Increased Risk. Plast. Reconstr. Surg. 2017, 140, 645–654. [Google Scholar] [CrossRef]
- Srinivasa, D.R.; Miranda, R.N.; Kaura, A.; Francis, A.M.; Campanale, A.; Boldrini, R.; Alexander, J.; Deva, A.K.; Gravina, P.R.; Medeiros, L.J.; et al. Global Adverse Event Reports of Breast Implant-Associated ALCL: An International Review of 40 Government Authority Databases. Plast. Reconstr. Surg. 2017, 139, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Jacombs, A.; Tahir, S.; Hu, H.; Deva, A.K.; Almatroudi, A.; Wessels, W.L.F.; Bradshaw, D.A.; Vickery, K. In vitro and in vivo investigation of the influence of implant surface on the formation of bacterial biofilm in mammary implants. Plast. Reconstr. Surg. 2014, 133, 471e–480e. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Jacombs, A.; Vickery, K.; Merten, S.L.; Pennington, D.G.; Deva, A.K. Chronic biofilm infection in breast implants is associated with an increased T-cell lymphocytic infiltrate: Implications for breast implant-associated lymphoma. Plast. Reconstr. Surg. 2015, 135, 319–329. [Google Scholar] [CrossRef]
- Hu, H.; Johani, K.; Almatroudi, A.; Vickery, K.; van Natta, B.; Kadin, M.E.; Brody, G.; Clemens, M.; Cheah, C.Y.; Lade, S.; et al. Bacterial Biofilm Infection Detected in Breast Implant-Associated Anaplastic Large-Cell Lymphoma. Plast. Reconstr. Surg. 2016, 137, 1659–1669. [Google Scholar] [CrossRef]
- Blombery, P.; Thompson, E.R.; Jones, K.; Arnau, G.M.; Lade, S.; Markham, J.F.; Li, J.; Deva, A.; Johnstone, R.W.; Khot, A.; et al. Whole exome sequencing reveals activating JAK1 and STAT3 mutations in breast-implant associated anaplastic large cell lymphoma. Haematologica 2016, 101, e387–e390. [Google Scholar] [CrossRef] [Green Version]
- Magnusson, M.; Beath, K.; Cooter, R.; Locke, M.; Prince, H.M.; Elder, E.; Deva, A.K. Special update: The epidemiology of breast implant associated anaplastic large cell lymphoma in Australia and New Zealand confirms the highest risk for grade 4 surface breast implants. Plast. Reconstr. Surg. 2019, 143, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Kadin, M.E.; Deva, A.; Xu, H.; Morgan, J.; Khare, P.; MacLeod, R.A.; Van Natta, B.W.; Adams, W.P.; Brody, G.S.; Epstein, A.L. Biomarkers Provide Clues to Early Events in the Pathogenesis of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Aesthetic Surg. J. 2016, 36, 773–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajkos, A.; Deva, A.K.; Vickery, K.; Cope, C.; Chang, L.; Cossart, Y.E. Detection of subclinical infection in significant breast implant capsules. Plast. Reconstr. Surg. 2003, 111, 1605–1611. [Google Scholar] [CrossRef]
- Trickett, A.; Kwan, Y.L. T cell stimulation and expansion using anti-CD3/CD28 beads. J. Immunol. Methods 2003, 275, 251–255. [Google Scholar] [CrossRef]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [Green Version]
- Bryant, C.E.; Spring, D.R.; Gangloff, M.; Gay, N.J. The molecular basis of the host response to lipopolysaccharide. Nat. Rev. Microbiol. 2010, 8, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Mcaleer, J.P.; Vella, A.T. Understanding how lipopolysaccharide impacts CD4 T-cell immunity. Crit. Rev. Immunol. 2008, 28, 281–299. [Google Scholar] [CrossRef] [Green Version]
- Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Staphylococcal enterotoxins. Toxins 2010, 2, 2177–2197. [Google Scholar] [CrossRef] [Green Version]
- Buonpane, R.A.; Moza, B.; Sundberg, E.J.; Kranz, D.M. Characterization of T cell receptors engineered for high affinity against toxic shock syndrome toxin-1. J. Mol. Biol. 2005, 353, 308–321. [Google Scholar] [CrossRef]
- Kelly, M.G.; Alvero, A.B.; Chen, R.; Silasi, D.A.; Abrahams, V.M.; Chan, S.; Visintin, I.; Rutherford, T.; Mor, G. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006, 66, 3859–3868. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Stamatos, N.M.; Dragan, A.I.; Medvedev, A.; Whitford, M.; Zhang, L.; Song, C.; Rallabhandi, P.; Cole, L.; Nhu, Q.M.; et al. Sialyl residues modulate LPS-mediated signaling through the Toll-like receptor 4 complex. PLoS ONE 2012, 7, e32359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, M.G.; Megiel, C.; Church, C.H.; Angell, T.E.; Russell, S.M.; Sevell, R.B.; Jang, J.K.; Brody, G.S.; Epstein, A.L. Survival signals and targets for therapy in breast implant-associated ALK—Anaplastic large cell lymphoma. Clin. Cancer Res. 2012, 18, 4549–4559. [Google Scholar] [CrossRef] [Green Version]
- Kadin, M.E.; Cavaille-Coll, M.W.; Gertz, R.; Massague, J.; Cheifetz, S.; George, D. Loss of receptors for transforming growth factor beta in human T-cell malignancies. Proc. Natl. Acad. Sci. USA 1994, 91, 6002–6006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marti, R.M.; Wasik, M.A.; Kadin, M.E. Constitutive secrection of GM-CSF by three different cell lines derived from a single patient with a progressive cutaneous lymphoproliferative disorder. Cytokine 1996, 8, 323–329. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Miyoshi, I.; Hinuma, Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc. Natl. Acad. Sci. USA 1982, 79, 2031–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vickery, K.; Pajkos, A.; Cossart, Y. In vitro response to mitogens by duck splenic mononuclear cells. Res. Vet. Sci. 1995, 59, 242–246. [Google Scholar] [CrossRef]
- Adams, W.P., Jr. Discussion: The Epidemiology of Breast Implant–Associated Anaplastic Large Cell Lymphoma in Australia and New Zealand Confirms the Highest Risk for Grade 4 Surface Breast Implants. Plast. Reconstr. Surg. 2019, 143, 1293–1294. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Redbino, M.R.; Bultman, S.J. The role of the microbiome in Cancer Development and Therapy. CA Cancer J. Clin. 2017, 67, 326–344. [Google Scholar] [CrossRef] [Green Version]
- Genth-Zotz, S.; von Haehling, S.; Bolger, A.P.; Kalra, P.R.; Wensel, R.; Coats, A.J.; Volk, H.D.; Anker, S.D. The anti-CD14 antibody IC14 suppresses ex vivo endotoxin stimulated tumor necrosis factor-alpha in patients with chronic heart failure. Eur. J. Heart Fail. 2006, 8, 366–372. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Kagan, J.C. Microbe-inducible trafficking pathways that control Toll-like receptor signaling. Traffic 2017, 18, 6–17. [Google Scholar] [CrossRef]
- Murase, M.; Kawasaki, T.; Hakozaki, R.; Sueyoshi, T.; Putri, D.D.P.; Kitai, Y.; Sato, S.; Ikawa, M.; Kawai, T. Intravesicular Acidification Regulates Lipopolysaccharide Inflammation and Tolerance through TLR4 Trafficking. J. Immunol. 2018, 200, 2798–2808. [Google Scholar] [CrossRef]
- Lucas, K.; Maes, M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: Possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 2013, 48, 190–204. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.M. Dynamic lipopolysaccharide transfer cascade to TLR4/MD2 complex via LBP and CD14. BMB Rep. 2017, 50, 55–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blombery, P.; Thompson, E.; Ryland, G.L.; Joyce, R.; Byrne, D.J.; Khoo, C.; Lade, S.; Hertzberg, M.; Hapgood, G.; Marlton, P.; et al. Frequent activating STAT3 mutations and novel recurrent genomic abnormalities detected in breast implant-associated anaplastic large cell lymphoma. Oncotarget 2018, 9, 36126–36136. [Google Scholar] [CrossRef] [Green Version]
- Malcolm, T.I.; Villarese, P.; Fairbairn, C.J.; Lamant, L.; Trinquand, A.; Hook, C.E.; Burke, G.A.; Brugières, L.; Hughes, K.; Payet, D.; et al. Anaplastic large cell lymphoma arises in thymocytes and requires transient TCR expression for thymic egress. Nat. Commun. 2016, 7, 10087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadin, M.E.; Adams, W.P., Jr.; Inghirami, G.; Di Napoli, A. Does Breast Implant–Associated ALCL Begin as a Lymphoproliferative Disorder? Plast. Reconstr. Surg. 2020, 145, 30e–38e. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G. Gram-positive and gram-negative bacterial toxins in sepsis—A brief review. Virulence 2014, 5, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tevis, S.E.; Hunt, K.K.; Miranda, R.N.; Lange, C.; Butler, C.E.; Clemens, M.W. Differences in Human Leukocyte Antigen Expression between Breast Implant-Associated Anaplastic Large Cell Lymphoma Patients and the General Population. Aesthetic Surg. J. 2019, 39, 1065–1070. [Google Scholar] [CrossRef]
- Kooy-Winkelaar, Y.M.; Bouwer, D.; Janssen, G.M.; Thompson, A.; Brugman, M.H.; Schmitz, F.; de Ru, A.H.; van Gils, T.; Bouma, G.; van Rood, J.J.; et al. CD4 T-cell cytokines synergize to induce proliferation of malignant and nonmalignant innate intraepithelial lymphocytes. Proc. Natl. Acad. Sci. USA 2017, 114, E980–E989. [Google Scholar] [CrossRef] [Green Version]
- Willerslev-Olsen, A.; Krejsgaard, T.; Lindahl, L.M.; Litvinov, I.V.; Fredholm, S.; Petersen, D.L.; Nastasi, C.; Gniadecki, R.; Mongan, N.P.; Sasseville, D.; et al. Staphylococcal enterotoxin A (SEA) stimulates STAT3 activation and IL-17 expression in cutaneous T-cell lymphoma. Blood 2016, 127, 1287–1296. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [Green Version]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE 2013, 8, e56964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, C.L.; Garrett, W.S. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014, 15, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Noto, J.M.; Peek, R.M., Jr. The gastric microbiome, its interaction with Helicobacter pylori, and its potential role in the progression to stomach cancer. PLoS Pathog. 2017, 13, e1006573. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Tan, Q.; Fu, Q.; Zhou, Y.; Hu, Y.; Tang, S.; Zhou, Y.; Zhang, J.; Qiu, J.; Lv, Q. Gastrointestinal microbiome and breast cancer: Correlations, mechanisms and potential clinical implications. Breast Cancer 2017, 24, 220–228. [Google Scholar] [CrossRef]
- Chan, A.A.; Bashir, M.; Rivas, M.N.; Duvall, K.; Sieling, P.A.; Pieber, T.R.; Vaishampayan, P.A.; Love, S.M.; Lee, D.J. Characterization of the microbiome of nipple aspirate fluid of breast cancer survivors. Sci. Rep. 2016, 6, 28061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw029. [Google Scholar] [CrossRef]
- Kakegawa, T.; Bae, Y.; Ito, T.; Uchida, K.; Sekine, M.; Nakajima, Y.; Furukawa, A.; Suzuki, Y.; Kumagai, J.; Akashi, T.; et al. Frequency of Propionibacterium acnes Infection in Prostate Glands with Negative Biopsy Results Is an Independent Risk Factor for Prostate Cancer in Patients with Increased Serum PSA Titers. PLoS ONE 2017, 12, e0169984. [Google Scholar] [CrossRef]
- Wang, L.; Ganly, I. The oral microbiome and oral cancer. Clin. Lab. Med. 2014, 34, 711–719. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Holley, D.; Collins, L.B.; Montgomery, S.A.; Whitmore, A.C.; Hillhouse, A.; Curry, K.P.; Renner, S.W.; Greenwalt, A.; Ryan, E.P.; et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014, 4, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Mempin, M.; Hu, H.; Chowdhury, D.; Foley, M.; Cooter, R.; Adams, W.P., Jr.; Vickery, K.; Deva, A.K. The functional influence of breast implant outer shell morphology on bacterial attachment and growth. Plast. Reconstr. Surg. 2018, 142, 837–849. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mempin, M.; Hu, H.; Vickery, K.; Kadin, M.E.; Prince, H.M.; Kouttab, N.; Morgan, J.W.; Adams, W.P., Jr.; Deva, A.K. Gram-Negative Bacterial Lipopolysaccharide Promotes Tumor Cell Proliferation in Breast Implant-Associated Anaplastic Large-Cell Lymphoma. Cancers 2021, 13, 5298. https://doi.org/10.3390/cancers13215298
Mempin M, Hu H, Vickery K, Kadin ME, Prince HM, Kouttab N, Morgan JW, Adams WP Jr., Deva AK. Gram-Negative Bacterial Lipopolysaccharide Promotes Tumor Cell Proliferation in Breast Implant-Associated Anaplastic Large-Cell Lymphoma. Cancers. 2021; 13(21):5298. https://doi.org/10.3390/cancers13215298
Chicago/Turabian StyleMempin, Maria, Honghua Hu, Karen Vickery, Marshall E. Kadin, H. Miles Prince, Nicola Kouttab, John W. Morgan, William P. Adams, Jr., and Anand K. Deva. 2021. "Gram-Negative Bacterial Lipopolysaccharide Promotes Tumor Cell Proliferation in Breast Implant-Associated Anaplastic Large-Cell Lymphoma" Cancers 13, no. 21: 5298. https://doi.org/10.3390/cancers13215298