
Intermittent and Periodic Fasting, Hormones, and Cancer Prevention


1
Department of Oncology and Hemato-Oncology, University of Milan, 20122 Milan, Italy
2
IFOM, FIRC Institute of Molecular Oncology, 20139 Milan, Italy
3
Department of Surgical, Oncological, and Oral Sciences, University of Palermo, 90127 Palermo, Italy
4
Department of Biological Sciences, Longevity Institute, Leonard Davis School of Gerontology, University of Southern California, Los Angeles, CA 90089, USA
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(18), 4587; https://doi.org/10.3390/cancers13184587
Submission received: 31 August 2021
/
Accepted: 9 September 2021
/
Published: 13 September 2021
(This article belongs to the Special Issue Hormone Involvement in Tissue Development, Physiology, and Oncogenesis)
Abstract
:Simple Summary
Hormonal and growth factor alterations, related to an elevated food consumption and excessive adiposity, affect the regulation of genes involved in cellular processes including proliferation, differentiation and DNA repair, allowing cells to survive and proliferate despite the accumulation of mutations which lead to malignant transformation. The growth hormone/insulin growth factor-1 (GH/IGF-1)/ insulin pathway and its downstream effectors, in fact, are known to promote aging and/or age-related diseases, including cancer, in many model organisms. The restriction of nutrients is established to have strong effects on levels of hormones and growth factors, delaying the incidence of age-related diseases and prolonging lifespan. Here, we summarize the effects caused by different nutrition intervention strategies on cellular damage, aging and cancer.
Abstract
The restriction of proteins, amino acids or sugars can have profound effects on the levels of hormones and factors including growth hormone, IGF-1 and insulin. In turn, these can regulate intracellular signaling pathways as well as cellular damage and aging, but also multisystem regeneration. Both intermittent (IF) and periodic fasting (PF) have been shown to have both acute and long-term effects on these hormones. Here, we review the effects of nutrients and fasting on hormones and genes established to affect aging and cancer. We describe the link between dietary interventions and genetic pathways affecting the levels of these hormones and focus on the mechanisms responsible for the cancer preventive effects. We propose that IF and PF can reduce tumor incidence both by delaying aging and preventing DNA damage and immunosenescence and also by killing damaged, pre-cancerous and cancer cells.
1. Introduction
Many dietary patterns, including the Western diet, are associated with reduced lifespan and health span and appear to affect cancer incidence by two major hormonal axes/pathways: (1) the growth hormone-IGF-1; (2) the insulin signaling [1,2,3,4,5,6,7].
Higher protein intake increases the release of growth hormone releasing hormone, and consequently growth hormone release from the pituitary gland and IGF-1 release primarily from the liver [8]. High IGF-1 has been associated with elevated incidence of a number of cancers [8,9,10,11]. On the other hand, excessive carbohydrate and/or fat intake can result in excess adiposity, which is associated with high oxidative stress, inflammation, alterations in hormones and growth factors’ production, acquisition of insulin resistance and consequently hyperinsulinemia [12,13,14]. Overweight women are reported to frequently present insulin resistance and low plasma levels of sex hormone-binding globulin which lead, as a consequence, to an increase in total and free sex hormone levels [15]. Hyperinsulinemia, in fact, blocks the production of sex hormone-binding globulin by the liver and, moreover, is associated with an increased production of androgens which are reported, together with estrogens, to stimulate the development and growth of several cancers [16]. Furthermore, insulin sustains insulin like growth factor 1 (IGF-1) activity, partly through the reduction of IGF binding protein 1 (IGFBP-1) synthesis, and elevated GH-IGF-1 increases insulin levels and resistance [5,6,17]. Not surprisingly, both elevated concentrations of insulin and IGF-1 are associated with multiple cancer types, including breast, endometrium, pancreas and colon [3,4,7,18]. However, it is not clear whether insulin and IGF-1 may promote cancer directly by promoting growth and preventing apoptosis, or by accelerating the aging process, which is a key risk factor for many cancers.
Nutrient sensing pathways regulate metabolism as well as growth and promote aging, aging-related diseases and genomic instability in several organisms, ranging from the simplest eukaryotes to mammals [19]. Studies in these model organisms show that impairment of genes that promote growth can extend lifespan, suggesting that age-related diseases, including cancer, could be postponed or prevented by switching from a pro-growth mode to a maintenance mode [20,21]. The GH/IGF-1 pathway and its downstream effectors including target of rapamycin (TOR), protein kinase A (PKA) and the ribosomal protein S6 kinase (S6K) are reported to promote aging and age-related diseases, including cancer, in several model organisms [22].
Thus, the understanding of nutrient-response pathways can provide insights on the mechanisms linking food intake to age-related diseases.
1.1. Growth Genes, Longevity, and Cancer: From Yeast to Humans
1.1.1. Growth Genes Aging and DNA Damage in Yeast
Yeast has been successfully used as a model system to understand the molecular basis of some of the most basic and important cellular processes. It is therefore not surprising that the aging research field included this simple unicellular organism among the most important organisms for identifying and studying the central pathways regulating longevity. Its short lifespan, low experimental costs and, more importantly, the availability of powerful and rigorous genetic methodologies and high throughput technologies, render it ideal for molecular genetics of aging studies. In addition, even considering the great evolutionary distance between yeast and mammals, comparative genomics highlighted that about 30% of the human genes involved in diseases have a yeast orthologue [23].
In 1959 Mortimer and Johnston demonstrated that a single yeast cell can divide a limited number of times [24]. This discovery was eventually used to screen and identify genomic mutations capable of affecting the replicative potential of a single yeast cell (replicative lifespan, RLS) [25].
More recently, the ability of post-diauxic yeast cells to survive and form new colonies has been successfully used to measure the yeast chronological lifespan (CLS) [19,26]. Thus, RLS is the yeast counterpart of the Hayflick limit observed in mammalian cell cultures, whose measurement reveals the doubling ability of cultured cells, while the CLS is more closely related to the chronological lifespan of higher eukaryotes. It is worthwhile to note that, even though the two methods could identify different processes, some of the pathways identified, including the Tor and serine-threonine Sch9 kinase (Tor-S6K) and the Ras-adenylate cyclase (AC) and PKA pathway (Ras/AC/PKA), have similar effects on the extension of the two lifespan methods [24,25,27,28]. Studies performed in the yeast Saccharomyces cerevisiae showed that deletion of the gene coding the small G protein Ras2 increases resistance to multiple stresses and doubles chronological lifespan [29]. Many data point to the PKA pathway as the Ras-dependent central regulator of longevity in yeast. In fact, mutations impairing adenylate cyclase (AC), a central effector of PKA pathway, also increase yeast longevity. In addition, the key stress resistance transcription factors Msn2/4 are inhibited by PKA activity and Msn2/4 impairment abolishes the pro-longevity effect of Ras2 deletion confirming thus PKA as central regulator of longevity [30].
Genome wide screen of deletion mutants obtained by transposon mutagenesis discovered Sch9-Tor pathway as perhaps the most potent pro-aging pathway in yeast [20]. SCH9, which was originally isolated as a suppressor of impaired Ras pathway [31], codes a serine-threonine protein kinase ortholog of mammalian S6K whose deletion increases resistance to multiple stresses and lifespan. The partial overlap with the PKA pathway is further confirmed by the phenotype reversion observed in sch9 deletion mutant after PKA hyperactivation. However, the overlap between these two pathways is not complete since the contemporary deletions of RAS2 and SCH9 has a greater effect on stress resistance as well as viability with respect to each single deletion. More comprehensive genome wide analysis performed using each of the aforementioned aging paradigms identified other genes whose deletion positively affected longevity. Deletion of genes involved in protein synthesis, such as protein component of ribosomal subunits, or genes involved in nuclear export of tRNA identified the transcription factor Gcn4 as another longevity regulator [32]. It must also be noted that these effects are not additive to Tor-Sch9 suggesting common aging regulatory pathways. Deletion of the tRNA wobbling regulator TRM9 was also identified as a lifespan regulator but its role was argued to be dependent on lower translation efficiency [33].
It is interesting to note that both Ras/AC/PKA and Tor/Sch9/S6k pathways are activated by nutrients and their depletion results in inhibition of these signal transduction pathways [34]. In yeast, the two macronutrients glucose and amino acids activate Ras and Tor, respectively. Reduced glucose supplementation in the growth media is capable of increasing stress resistance, reducing genomic mutation rate and increasing lifespan in a Ras-dependent manner. On the other hand, the restriction of certain amino acids is capable of increasing lifespan by reducing Tor-Sch9/S6k signaling. Interestingly only specific amino acids (methionine, serine, threonine and valine) can activate the Tor-Sch9 pathway and their restriction (each one of them alone or combined) is capable of increasing lifespan as well as stress resistance and decreasing DNA damage [34].
Moreover, the activation of transcription factors Msn2 and Msn4 in S. cerevisiae deficient in the Ras/cAMP/PKA signaling makes cells more resistant to stress, in part by inducing the expression of genes encoding for several heat shock proteins, catalase (Ctt1), and the DNA damage inducible gene DDR2 [20,35]. These results suggest that the effect of mutations in the Tor-Sch9/S6k and Ras/AC/PKA pathways is partly mediated by the regulation of antioxidant defenses and the reduction of oxidants. In fact, yeasts expressing constitutively active RAS2 oncogene present a lower resistance to oxidants and a decreased lifespan [36].
1.1.2. Growth Genes Aging and DNA Damage in Worms and Flies
The role of nutrient-response pathways was also examined in the worm Caenorabditis elegans and fruit fly Drosophila melanogaster. Studies of the nematode Caenorabditis elegans showed that a reduction of the insulin/insulin-like growth factor signaling pathway (IIS) and the consequent activation of the Forkhead FoxO transcription factor daf-16 which, similarly to Msn2/4 in yeast, regulates genes involved in the cellular stress response and detoxification of xenobiotics and free radicals, extends longevity [37,38,39]. The extension of lifespan in worms also requires the heat shock factor hsf-1, which regulates the expression of heat shock proteins [40]. As observed in yeast, inhibition of TOR-S6 kinase signaling can increase lifespan in worms; in particular, TOR pathway inhibition can activate the process of autophagy and alter the activity of other TOR targets, such as the hypoxia-inducible factor 1 (HIF-1) transcription factor, also independently shown to extend lifespan [39,41].
Moreover, in C. elegans mutated in gld-1, a female germline-specific tumor suppressor gene, germ cells can proliferate uncontrolled and form tumors [42]. Mutations in the IGF-1-receptor-like daf-2 gene can protect gld-1 mutants against tumor formations, by reducing germ cell proliferation and inducing cell death through a mechanism which requires daf-16 and cep-1, the orthologue of the mammalian p53 genes [42]. Furthermore, in worms, a mutation in the age-1 PI3K gene, downstream of the daf-2 receptor, was shown to increase mean lifespan by 65% and maximum lifespan at 25 degrees by 110%, suggesting that daf-2 and age-1 act in the same pathway to reduce thermotolerance and antioxidant defenses apparently through a mechanism which involves the stress resistance transcription factor daf-16 [37,43]. Similarly to ras2, cyr1 and sch9 yeast mutants, which reduce Sod2 expression and require it to extend lifespan, C. elegans daf-2 is reported to decrease oxidative stress resistance, in part by down-regulating mitochondrial Mn-SOD gene expression and several heat shock proteins [44].
Many studies suggest that orthologs of the yeast and worm genes regulate fruit flies’ longevity as well. Drosophila melanogaster impairment of the insulin receptor, in fact, controls the germline stem cell division and cysts growth by a cell autonomous mechanism [45]. The fly daf-16 orthologue FOXO, which is involved in lifespan extension, blocks phosphatidylinositol-3 kinase (PI3K) effect on cell number and the down-regulation of IGF-1 or insulin-AKT/PKB axis, which is reported to promote cancer growth and metastasis in fruit flies, can extend lifespan by up to 85% and inhibit uncontrolled cell growth [46,47,48]. As in S. cerevisiae and C. elegans, in D. melanogaster the inhibition of TOR pathway activity, genetically or by rapamycin treatment, increases lifespan partly by activating autophagy, reducing S6K activity and increasing stress resistance [49]. Analogously to yeast and worms, fruit flies with mutations in the insulin/IGF-1 pathway increase SOD expression and increased survival [47,50]. Furthermore, mutation in the G-protein-coupled receptor homolog MTH gene results in a 35% increase in lifespan and resistance to starvation and superoxide toxicity in Drosophila melanogaster [51].
Thus, in yeast, worms and flies, the activation of nutrient signaling pathways is tightly linked to oxidative stress, DNA damage and either increased growth or tumors.
1.1.3. Growth Genes, Aging and Cancer in Mice
Studies in simple model organisms were fundamental to identify conserved nutrient-response pathways that regulate longevity but mouse research on genetics of aging and cancer had also been proceeding in parallel with the studies in simple organisms and reached similar conclusions. Mice studies substantially confirmed the observations made in yeast and other simple aging model systems that certain pro-growth signal transduction pathways activated by nutrients had central roles in aging and cancer. Mutations or deletion in GH or IIS genes, as well as S6K1, can also extend lifespan in mice and reduce age-related diseases including insulin resistance and immune and motor disorders [22,52]. Moreover, a decrease in the PKA signaling pathway in mice extends longevity and reduces the incidence of age-dependent tumors [53]. Mice with homozygous mutations in Prop-1 or Pit1 genes present a deficit in normal pituitary development, which causes a decrease in GH, prolactin and thyroid stimulating hormone (TSH) production; as a consequence, these mice are much smaller than wild-type controls but live ~40% longer [54]. Dwarf mice with a 90% reduction in IGF-1 level or mice carrying heterozygous mutation in IGF-1 receptor (IGF-1R) also present a ~33% increase in life expectancy compared to control mice [55,56]. Several reports provided evidence that a decline in insulin or IGF-1 levels reduces the incidence of spontaneous tumors in mice, thus confirming the central role of IGF-1 as growth promoter and tumorigenesis driver [9]. Transgenic mice expressing IGF-1 are reported to present higher incidences of tumors in mammary glands while transgenic mice overexpressing human GH exhibit hepatic upregulation of GH-signaling mediators which lead to liver neoplasms [57]. Similarly, transgenic mice expressing a constitutively active form of IGF-1R showed aberrant development of the mammary glands and early development of salivary and mammary adenocarcinomas [58]. Conversely, growth hormone receptor binding protein (GHR/BP) knock out mice displayed a lower incidence and a delayed occurrence of neoplastic lesions compared to wild-type littermates, in particular lymphomas and pulmonary carcinomas [59]. Moreover, transgenic animals expressing the GH antagonist G120 GH had lower IGF-1 levels and exhibited decreased tumor incidence in the mammary gland relative to control mice after being treated with the carcinogen 7,12-dimethylbenz(a)anthracene (DMBA) [60]. Accordingly, mice bearing MCF-7 xenografts treated with pegvisomant, which is the clinical version of the G120R GH antagonist able to completely inhibit both GH and IGF-1 signaling in the mammary gland, displayed a 70–80% decreased of circulating IGF-1 and a 30% decrease in tumor size [61]. This effect may be partly due to an increased resistance to oxidative damages and a higher expression of antioxidant enzymes, which are associated with a decrease in IGF-1 levels [19]. Normal levels of GH and IGF-1 could also promote cancer by increasing spontaneous genomic instability through a RAS or AKT hyperactivation dependent mechanism. PTEN inactivation in the mouse prostate causes AKT constitutive activation and is associated with 70% of primary prostate cancers. Moreover, AKT hyperactivation by oncogenic mutations can alter also p53 expression by causing growth-independent hyper-replication and an increase in DNA damage [62].
In summary, in mice there is very strong evidence for the link between high growth hormone and IGF-1 levels, DNA damage and cancer, likely mediated at least in part by the activation of AKT, TOR-S6K and PKA signaling, analogously to what is observed in yeast (Figure 1).
1.1.4. Growth Genes and Cancer in Humans
Alterations in GH-IGF-1 axis have also been studied also in humans. Notably, human cancers are frequently mutated in the IGF-1R (2.48% of all cancers) and in its downstream signaling proteins Ras (19% of all cancers) and Akt (1.8% of all cancers) [64,65,66,67]. In agreement with mouse studies, the modulation of the GH-IGF-1 pathway appears to have a key role in cancer prevention in humans. High levels of IGF-1 are, in fact, associated with an increased incidence of several cancers, including colorectal, prostate and breast cancers, while mutations in the human IGF-1R were found to protect against age-related disorders [9,68]. Recent evidence reports that centenarians most frequently present heterozygous mutations in the IGF-1R gene, associated with low IGF-1 serum levels and a higher IGF-1R activity compared to controls characterized by high IGF-1 serum levels [69]. The role of GH/IGF-1 axis activity on longevity and aging-related diseases in human is also supported by long-term studies of an Ecuadorian cohort affected by Laron syndrome (LS) which is characterized by GHR deficiency and consequently is responsible for a 90% reduction of the IGF-1 levels. Guevara-Aguirre et al., monitoring LS patients for more than 20 years, reported that the relation between pro-growth signaling pathways, oxidative stress, genomic instability and cellular damages shown in several model organisms is also observed in humans and human cells [6]. The results from the Ecuadorian cohort of LS patients showed that these individuals are protected from age-related pathologies, in particular cancer and type 2 diabetes, similarly to the results obtained from GH-deficient mice, which are characterized by a 49% decrease in neoplasms’ incidence and an increase in insulin sensitivity compared to control mice [6,59,70,71,72,73,74]. Analysis performed on human mammary epithelial cells (HMECs) incubated in medium supplemented with 15% of LS patients’ serum showed that these cells are characterized by reduced levels of RAS, PKA and TOR, leading to reduced DNA damage and increase apoptosis upon stimulation with oxidative stress compared to cells incubated with 15% of serum derived from LS patients’ relatives, not affected by GHR deficiency [6]. These data suggest that reduced GH-IGF-1/insulin signaling protects from cancer in part by reducing DNA damage and in part by increasing apoptosis in damaged cells, making the link between decreased activity of growth signaling pathways and DNA protection or repair mechanisms conserved from yeast to humans.
1.2. Calorie Restriction (CR) and Cancer
Calorie restriction (CR), a dietary intervention that reduces calorie intake without inducing malnutrition, is the most studied intervention able to extend lifespan but also well established to postpone or even prevent age-related diseases, including cancer [75] (Table 1). Several studies showed that CR increases lifespan in multiple organisms including yeast, flies, worms, rodents and monkeys, protecting from disorders and decline in functions related to aging [22,30,76,77,78,79,80,81]. Curiously, the first study on calorie restriction was conducted in mammals and not in a simple model system. McCay et al. presented for the first time the effects of CR in retarding aging, by increasing lifespan by ~35%, reducing the incidence of kidney disorders, chronic pneumonia and tumors [82] in mice. Similar results were obtained in several studies performed in different rodent models and the underline mechanisms could be mediated by the decrease in blood glucose, IGF-1 and insulin levels, the increase in glucocorticoids and insulin sensitivity as a result of the homeostatic response to reduced body fat stores and an increase in gluconeogenesis [77,83]. Long-term CR is reported to reduce IGF-1 serum levels in rodents by ~30–40%, protecting them against several types of cancers, while treatments with GH or IGF-1 can reverse this protective effect mediated by CR, confirming the important role of these factors in cancer pathogenesis [84,85,86,87]. The NF-E2-related factor 2 (Nrf2) transcription factor has been shown to mediate, in part, the CR anticancer effects. Nrf2, in fact, when activated, improves the activity of several antioxidant and carcinogen-detoxification enzymes, and the anticancer effects of CR is highly impaired in mice deficient in Nrf2 when exposed to carcinogens [88]. These results further demonstrate the effect of CR in increasing resistance to oxidants and other toxins.
In addition, CR, as well as reduced levels of IGF-1, can decrease genomic instability via Ras- or phosphatidylinositol-3 kinase (PI3K)/Akt/Tor/S6K-dependent mechanisms, which contribute to reducing cancer incidence [89,90] in agreement with what was shown in yeast [20,30].
Studies in different rodent models showed that CR is able to reduce the incidence and delay the onset of spontaneous or chemically induced cancers, while in rhesus monkeys, lifelong CR reduces cancer incidence by 50% [91,92,93]. Many beneficial anticancer effects have been achieved by reducing caloric intake by 10 to 50% in mammals [22]. However, more limited restrictions are also capable of affecting longevity in mice. It was demonstrated that the macronutrients ratio has an important role in longevity and a 30% lifespan increase was observed when the protein to carbohydrate ratio is decreased [94]. In fact, protein intake affects the incidence and growth rate of melanoma and breast tumors, probably by activation of the GHR-IGF-1 signaling described earlier [10].
In addition to continues dietary restrictions, the shortage of food without malnutrition for a period of life could have long-term consequences. At least two events were associated with long-term and severe calorie restrictions in human history: Danes faced a 2-year CR without malnutrition, during the first world war, while Norwegians were forced to CR without malnutrition for 4 years in a row during the Second World War. Even though specific cause mortality is not reported in the first case and only circulatory disease are monitored in the second study, in both cases, CR was associated with a strong reduction of mortality rate, 34% and 30%, respectively, suggesting an effect on cancer incidence as well [95,96]. More recent studies on human spontaneously adhering to CR, report slower metabolism, decreased oxidative damages, enhanced DNA damage repair processes and the reduction of diabetes risk factors, cardiovascular diseases and cancer [97]. Moreover, by reducing body weight, CR improves multiple metabolic and hormonal alterations associated with excessive adiposity, including a decrease in visceral and hepatic fat and a reduction in circulating insulin levels, which, consequently, is associated with an increase in sex hormone binding globulin and free hormones like estrogens and testosterone [13,15,16]. The loss of weight mediated by CR also reduces several markers of oxidative and DNA damages; weight loss in obese man is associated with an increase in telomere length in rectal tissue biopsies, suggesting that CR could contribute to the prevention of telomere shortening [98].
Despite the key role of GH/IGF-1 inhibition in increasing life expectancy and reducing or delaying age-related disorders, mimicking the effect of CR, the two approaches may be acting through mechanisms that are only partially overlapping. CR, in fact, can further extend lifespan in GHR-deficient mice, possibly by reducing side effects such as fat accumulation and disorders related to obesity [19,99].
Notably, even though the lipid profile is improved and oxidative DNA damage is significantly decreased in calorie-restricted patients, the levels of important hormones associated with cancer, such as IGF1, were unchanged or only slightly changed, possibly because many CR subjects consume high levels of proteins, which regulate IGF-1 levels [100]. Notably, CR provides both a wide range of beneficial effects, as well as detrimental effects including low weight and loss of lean body mass as well as immunosuppression and potentially increased susceptibility to certain infections [101,102,103,104]. Not surprisingly, lifelong CR either caused no effect or a small effect on lifespan in monkeys in spite of showing strong effects on age-related disease-dependent mortality, suggesting that the beneficial effects of CR may be counterbalanced by its detrimental ones.
1.3. Fasting and Fasting Mimicking Diets
Because CR can have both very positive and negative effects and is unlikely to be adopted by a significant portion of the populations since it unavoidably causes severe weight loss, intermittent and periodic fasting are emerging as novel interventions which could maintain many of the beneficial effects of CR while reducing the burden and many of the side effects. Fasting, the complete elimination of nutrients from the diet, is the most extreme of the dietary restrictions.
Fasting methods (Table 2). Fasting can be performed for short-term frequent periods, called intermittent fasting (IF), or less frequent but longer periods, known as prolonged and periodic fasting (PF) [63,105]. There are multiple examples of IF diets, including: complete fasting every other day (also called alternate-day fasting ADF); 70% energy restriction every other day; time-restricted feeding (TRF), during which food intake is restricted to 6–12 h per day; and the 5:2 diet, which is achieved by consuming only 500–700 calories for two days a week [106,107,108,109,110]. Thus, IF interventions usually include a phase during which only water is consumed or calorie intake is extremely low, followed by a normal feeding phase which lasts between 12 and 72 h. PF periods, instead, in most cases refer to 2–5 days of water-only fasting or 4–7 days of a fasting mimicking diet (FMD), a diet designed to mimic the metabolic effects induced by fasting [111,112]. Differently from IF, PF does not need to occur at specific intervals and in most cases can be carried out only for few times per year [113]. All these types of fasting cause different degrees of metabolic changes, including the decrease in blood glucose levels, the reduction of glycogen stores, the decrease in circulating leptin levels and the mobilization of fatty acids accompanied by the generation of ketone bodies [63,113]. Moreover, fasting or FMD periods can lead to behavioral changes, including increased awareness, attention, mental acuity, vigilance and feelings of euphoria, thus lowering depressive symptoms [114,115].
Fasting can extend lifespan and protect from age-related disorders, including DNA damage or cancer, in different model organisms [63].
Escherichia coli bacteria cultured in a calorie-free broth instead of a nutrient-rich one present a fourfold increase in lifespan, while yeast Saccharomyces cerevisiae grown in water instead of medium supplemented with glucose present a decrease in Tor/S6K and Ras/AC/PKA nutrient signaling pathways activity and an increased activation of Msn2/4 stress resistance transcription factors [22,30,116]. In the nematode Caenorabditis elegans, food deprivation conditions reached by feeding worms with little or no bacteria increase lifespan through mechanisms which involve AMPK, the stress resistance transcription factor DAF-16 [117,118] and the small GTPase RHEB-1 [119]. In rodents, alterations in metabolic and growth factors turned out to be different in accordance with the different forms of fasting applied and also the age at which cycles of fasting were started [120]. IF cycles applied to middle-aged rats increased lifespan by 30–40% compared to rats subjected to a normal dietary regimen [121]. Moreover, in rodents, IF has been shown to prevent and revert metabolic syndrome aspects, reduce abdominal fat, insulin resistance and protects against renal and liver injuries [52,122,123].
However, there have been reports showing also IF adverse effects in rodent models. A preclinical study showed that rats subjected to IF for one month had improved glucose tolerance, while rats subjected to 8 months of IF achieved impaired glucose tolerance [124]. Moreover, IF has been shown to have adverse effect on glucose metabolism in hypercholesterolemic mice, contradictory to the beneficial effects of IF on lipid and glucose metabolism shown in other rodent and human studies [125,126,127]. Another preclinical study showed that IF, introduced at 1.5 or 10 months of age, was not able to reduce body weight in A/J mice, compared to controls, and did not increase mean and maximum lifespan when started at 1.5 months. In addition, the same study showed that IF, when started at 10 months of age, decreases mean and maximum lifespan compared to control mice [128]. These results suggest that IF effects on body weight and lifespan are variable and depend on the genotype and age on initiation.
On the other hand, PF has been shown to improve glucose tolerance and delay age-associated diseases in mice, in particular by potentiating cellular resistance and affecting the GH/IGF-1 axis [63,113,129]. In mice, 24–72 h of fasting cause a 30% and 40% decrease in circulating insulin and glucose, respectively, lower IGF-1 levels by 70% and cause a major increase in IGFBP-1 [130]. Moreover, PF causes the down-regulation of TOR-SK6 and eAC-PKA nutrient signaling pathways, and decreases the PI3K-AKT pathway activity [129,130]. Additionally, in humans, PF can lead to a major decrease in circulating IGF-1 and a 5- fold increase in IGF binding protein 1(IGFBP1) [131]. These effects are largely mediated by protein restriction and particularly to the restriction of essential amino acids: in fact, in humans, chronic caloric restriction is not associated with reduced IGF-1 levels unless combined with protein restriction [22,97,132].
1.4. Fasting Mimicking Diet, Hormones and Cancer Prevention
Several studies indicate that PF is a much more viable strategy than a continuous CR, for cancer prevention and treatment in humans because: (1) it cause a much more extreme set of metabolic changes than CR, including IGF-1, insulin, leptin and glucose decreases, which can be combined with standard of care drugs to promote strong anticancer effects and cancer-free survival; (2) it stimulates anticancer immune responses; (3) it prevents muscle loss; (4) it is amenable to combination with standard cancer treatments but also cancer preventions since it is only conducted for several days periodically and does not require dietary changes between periodic fasting cycles. Preliminary clinical data report that 48 h of fasting are necessary to obtain relevant clinical effects in oncology, such as preventing DNA damages induced by chemotherapy in healthy tissues and improve quality of life to cancer patients [136,137,138]. However, most patients undergoing water-only fasting during cancer treatment had difficulties with sustaining water fasting and reported side effects such as headache, nausea, light-headedness, anemia and weakness [139]. Thus, water-only fasting and intermittent fasting which are expected to be repeated every other day or twice a week, remain a challenging option for the majority of population, especially frail and older subjects and cancer patients. FMD is a plant-based caloric-restricted alimentary regimen (typically between 300 and 1100 kcal per day) characterized by low proteins and sugars and relatively high unsaturated fats. It was developed to mimic many of the metabolic effects induced by water-only fasting but with reduced nutritional risk and burden [112,138,140,141,142].
1.4.1. Mice Studies
Preclinical studies conducted in rodents show that middle-aged mice subjected bimonthly to 4 days of FMD present a 40% decrease in blood glucose levels and a ~9-fold increase in ketone bodies production, suggesting that this regimen is able to mimic the metabolic switch induced by water fasting [112]. Furthermore, 4 days of FMD lead to a 10-fold decrease in insulin level, reduce IGF-1 level by 45% while increase IGFBP-1 by ~8-fold, similarly to the effects of water fasting. Notably, many of these parameters return to normal levels within 7 days of re-feeding [112] but some, including IGF-1 and leptin, do not, suggesting chronic effects of these periodic interventions [141]. Moreover, bimonthly FMD cycles lasting 4 days, started in middle-aged mice, can extend lifespan and positively affect mice health-span by reducing visceral fat deposits, leading consequently to a reduction in body weight, without affecting lean body mass, and decrease multiple organ weight, possibly promoting their regeneration upon refeeding. Middle-aged mice subjected to FMD cycles twice a month for 4 days display a 45% reduction in neoplasia incidence and a ~50% decrease in inflammation-associated skin lesions occurrence when compared to mice fed ad libitum with standard diet. Notably, the FMD also postponed neoplasm-related death by over 3 months and reduced the number of animals with multiple abnormal lesions, suggesting that the FMD regimen not only prevents neoplasia formation but also delays their onset [112].
Studies carried out on bone-marrow-derived stem and progenitor cells show that FMD can also promote immune system regeneration and rejuvenation thus reducing immune-senescence which could be important for cancer development [112,129].
Fasting/FMD, partly by reducing PKA activity, circulating IGF-1 and glucose levels and by regulating genes involved in DNA repair (REV1) and cell death (p53), enhances chemotherapeutic agents’ efficacy against multiple kind of tumors, while inducing the protection of normal cells from their toxic side effects [111,129,130,143]. Fasting-dependent reduction in IGF-1 levels was found to be fundamental to protect primary glia and neurons, but not glioma and neuroblastoma cells, from chemotherapies and pro-oxidative compounds [130]. In addition, fasting/FMD, by promoting the switch of cancer cell metabolism from aerobic glycolysis to oxidative phosphorylation (OXPHOS), increases ROS production, contributing to making cancer cells more sensitive to chemotherapy, while protecting normal cells [143]. The FMD-dependent reduction in blood insulin, IGF-1 and leptin, which consequently leads to the inhibition of the PI3K/AKT, mTOR pathways, can also enhance the efficacy of estrogen therapies against estrogen receptor positive (ER+) breast cancer [141]. Furthermore, FMD can reduce the expression of Heme-Oxygenase 1 (HO-1), protein which confers protection against oxidative damages and apoptosis, in in vivo xenografts [144]. Through HO-1 downregulation, FMD reverses chemotherapy-induced immunosuppression by increasing CD8+ tumor-infiltrating lymphocyte-dependent cytotoxicity and by reducing tumor-associated Tregs [144].
1.4.2. Human Studies
To investigate the feasibility and impact of FMD in humans, clinical trials were conducted in healthy humans (Figure 2) [112,140]. Participants were subjected to 5 days FMD per month for 3 months in a row and were asked to resume their normal diet after the FMD period. Components and nutrients in the human FMD were selected for their ability to reduce circulating IGF-1 and glucose and to increase IGFBP-1 and the production of ketone bodies, according with the effects of FMD obtained in preclinical studies in rodents. The human FMD diet is composed approximately by at least 11–14% of proteins, 42–43% of carbohydrates and 44–46% of fats and was designed to provide between 34–54% of the normal caloric intake; thus, fat and complex carbohydrates are the higher sources of calories in the FMD regimen [112,140].
Subjects undergoing FMD cycles reported a 11.3% decrease in blood glucose levels and a ~24% reduction in circulating IGF-1, which remained, respectively ~6% and ~15% lower than baseline levels even after the refeeding period. Moreover, participants subjected to FMD showed a 3.7-fold increase in serum ketone bodies and a 1.5-fold increase in IGFBP-1, which returned to baseline levels after resuming the normal diet [112,140]. In addition, FMD decreased systolic blood pressure and reduced bodyweight, waist circumference, total body and trunk fat by 3%, while it increased the relative lean body mass, suggesting that it causes only loss in fat mass [112,140]. Studies conducted in humans report that three FMD cycles reduce the serum level of C-reactive protein (CRP), a marker of inflammation, in subjects classified as at-risk for age-related pathologies than in those subjects who had risk factor values within the normal range, suggesting that FMD also promotes anti-inflammatory effects [112,140]. In general, FMD cycles were well tolerated and no serious adverse events (according to Common Terminology Criteria for Adverse Events) were reported.
Figure 2.
CONSORT diagram. CONSORT diagram of participants selected for the clinical study described by Brandhorst et al., and Wei et al. [112,140]. In the clinical study, 100 participants were enrolled; 48 subjects were randomized to a control group and were asked to continue their normal diet for 3 months and then were subjected to 3 FMD cycles (16 participants withdrew or were excluded throughout the study). Instead, 52 subjects were randomized to the FMD group; FMD was provided for 5 consecutive days per month, for 3 months (13 subjects withdrew or were excluded throughout the study). [112,140].
Figure 2.
CONSORT diagram. CONSORT diagram of participants selected for the clinical study described by Brandhorst et al., and Wei et al. [112,140]. In the clinical study, 100 participants were enrolled; 48 subjects were randomized to a control group and were asked to continue their normal diet for 3 months and then were subjected to 3 FMD cycles (16 participants withdrew or were excluded throughout the study). Instead, 52 subjects were randomized to the FMD group; FMD was provided for 5 consecutive days per month, for 3 months (13 subjects withdrew or were excluded throughout the study). [112,140].

The FMD-dependent reduction in blood glucose and IGF-1 levels is of interest given their key role in age-related diseases, including cancer [6,10,22,68,145]. Collectively, these data indicate that periodic use of cyclic FMD could potentially prevent obesity- or inflammation-related diseases and also reduce cancer risk in humans, as it was shown in rodents [112] (Figure 3).
1.5. Alternative Interventions to Reduce Age-Related Diseases Risk Factors
Aging is frequently associated with impaired glucose tolerance and hyperinsulinemia, due to an increase in insulin secretion as a result of high glucose levels [146]. A decline in glucose tolerance is often associated with an increased risk of developing atherosclerosis or non-insulin dependent diabetes mellitus (NIDDM) [147,148].
Endurance exercise training reduces insulin levels both during fasting and feeding periods. Several studies showed that individuals who practice exercise periodically have improved glucose tolerance and responsiveness to insulin [149,150,151,152,153]. Seals et al., showed that regular exercise prevented the decline in glucose tolerance and hyperinsulinemia development in older people [152]. Moreover, exercise training was reported to normalize glucose tolerance by reducing insulin resistance in patients with mild NIDDM or impaired glucose tolerance (IGT) [154]. Furthermore, consuming a low-calorie and low-protein vegan diet, composed of unprocessed and uncooked plant-derived foods, for at least two years, or performing endurance exercise are associated with a decrease in cardiometabolic risk [155]. In particular, they reduced the plasma concentrations of lipids, lipoproteins, glucose, insulin, C-reactive protein (CRP) and systolic and diastolic blood pressure [155].
Figure 3.
A periodic diet that mimics fasting promotes longevity and reduces risk factors involved in age-related diseases. The fasting mimicking diet (FMD) is characterized by low proteins and sugars and relatively high unsaturated fats. This dietary regimen is able to mimic the metabolic changes induced by water-only fasting while lowering the burden and risk for side effects [112,140]. Preclinical studies show that periodic cycles of FMD can significantly reduce blood glucose, IGF-1, leptin and insulin levels in mice, while increasing ketone bodies production and IGFBP-1 levels. These metabolic changes promote immune system regeneration and rejuvenation while reducing neoplasia incidence by 45%, inflammation associated skin lesions by 50% and obesity [112]. Analogously to rodents, also in humans, periodic FMD decreases blood glucose, insulin, IGF-1, and leptin levels while increasing ketone bodies and IGFBP-1, metabolic changes partially maintained even after the refeeding period [112,140]. These FMD-dependent effects are of particular interest given their key role in age-related diseases, especially cancer and metabolic disorders. Taken together, these data indicate that periodic cycles of FMD could prevent obesity and reduce cancer risk in humans, as suggested by results obtained in preclinical trials [112].
Figure 3.
A periodic diet that mimics fasting promotes longevity and reduces risk factors involved in age-related diseases. The fasting mimicking diet (FMD) is characterized by low proteins and sugars and relatively high unsaturated fats. This dietary regimen is able to mimic the metabolic changes induced by water-only fasting while lowering the burden and risk for side effects [112,140]. Preclinical studies show that periodic cycles of FMD can significantly reduce blood glucose, IGF-1, leptin and insulin levels in mice, while increasing ketone bodies production and IGFBP-1 levels. These metabolic changes promote immune system regeneration and rejuvenation while reducing neoplasia incidence by 45%, inflammation associated skin lesions by 50% and obesity [112]. Analogously to rodents, also in humans, periodic FMD decreases blood glucose, insulin, IGF-1, and leptin levels while increasing ketone bodies and IGFBP-1, metabolic changes partially maintained even after the refeeding period [112,140]. These FMD-dependent effects are of particular interest given their key role in age-related diseases, especially cancer and metabolic disorders. Taken together, these data indicate that periodic cycles of FMD could prevent obesity and reduce cancer risk in humans, as suggested by results obtained in preclinical trials [112].

In addition, a meta-analysis study showed that physical activity is associated with lower risk of 13 different kinds of cancer, while another study showed that exercise can reduce the risk of cancer recurrence or progression in certain solid tumors [156,157]. However, additional studies are required to deepen the understanding of the association between exercise and cancer prevention.
Metformin is another drug that has been reported to attenuate the progression of aging. Metformin is mostly used for the treatment of type 2 diabetes since it decreases glucose production by the liver and increases insulin sensitivity. Several studies showed that metformin can inhibit several nutrient sensing systems, including the somatotropic axis GH/IGF-1, the mTOR signaling and AMPK [158,159,160,161,162,163,164,165,166]. Moreover, it can also lower oxidative stress via mitochondrial complex 1 inhibition and it is reported to reduce DNA damage by decreasing ROS levels and activating DNA damage response mechanisms [167,168,169]. Thus, metformin is likely to be effective against aging in part by affecting the same genes and pathways that mediate the effects of fasting on longevity and disease.
2. Conclusions
Studies in simple organisms and mice, demonstrate the link between nutrients and particularly protein intake, growth factors, DNA damage and cancer. The effect of growth factors on DNA damage and cancer is mediated, at least in part, by oxidative stress and damage, but in part also by the inhibition of apoptosis. The reduced activity of growth factors and the lowering of oxidation and DNA damage not only decreases cancer but also extends longevity, since aging is the most important factor promoting cancer. Calorie restriction is a powerful anti-aging intervention, but it also forces the organism into an extremely low nourishment state, which may not constitute malnourishment in the short-term but which may do so long-term. Interventions such as IF and PF are emerging as alternatives to CR, with some of them being able to minimize side effects and burden while maximizing efficacy. Studies on PF have also pointed to 2 key processes absent or low in CR and IF: (a) a pronounced breakdown process both at the intracellular (autophagy etc) and cellular (apoptosis) levels requiring 2 or more days and associated with a high ketogenic state, (b) a rebuilding/regeneration process involving stem and progenitor cells in multiple system and associated with the return from PF to normal feeding (re-feeding). The FMD developed and studied by our laboratories is emerging as a viable and effective intervention in the longevity and cancer prevention fields, since it does not require chronic treatment, it does not cause malnourishment or loss of muscle mass and may be effective when performed only a few times a year for 5 days. In the future years it will be important to continue to test different nutritional interventions with the potential to extend the health span and prevent cancer, with a focus on those that are safe and feasible for long-term use in humans.
Author Contributions
G.S., M.G.M. and V.D.L. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by the Associazione Italiana per la Ricerca sul Cancro (AIRC) (IG#21820 to V.D.L.) and the Laurus foundation (to M.G.M).
Conflicts of Interest
V.D.L. has equity interest in L-Nutra, a company that develops medical food.
References
- Sugimura, T. Nutrition and dietary carcinogens. Carcinogenesis 2000, 21, 387–395. [Google Scholar] [CrossRef]
- Kushi, L.H.; Byers, T.; Doyle, C.; Bandera, E.V.; McCullough, M.; McTiernan, A.; Gansler, T.; Andrews, K.S.; Thun, M.J.; American Cancer Society, N.; et al. American Cancer Society Guidelines on Nutrition and Physical Activity for cancer prevention: Reducing the risk of cancer with healthy food choices and physical activity. CA Cancer J. Clin. 2006, 56, 254–281. [Google Scholar] [CrossRef]
- Kaaks, R. Nutrition, hormones, and breast cancer: Is insulin the missing link? Cancer Causes Control 1996, 7, 605–625. [Google Scholar] [CrossRef] [PubMed]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Rohan, T.E.; Manson, J.E.; Li, J.; Ho, G.Y.; Xue, X.; Anderson, G.L.; et al. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 2009, 101, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Guevara-Aguirre, J.; Rosenbloom, A.L.; Balasubramanian, P.; Teran, E.; Guevara-Aguirre, M.; Guevara, C.; Procel, P.; Alfaras, I.; De Cabo, R.; Di Biase, S.; et al. GH Receptor Deficiency in Ecuadorian Adults Is Associated With Obesity and Enhanced Insulin Sensitivity. J. Clin. Endocrinol. Metab. 2015, 100, 2589–2596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef] [Green Version]
- Giovannucci, E. Insulin and colon cancer. Cancer Causes Control 1995, 6, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A.; Sun, L.Y.; Longo, V. Somatotropic signaling: Trade-offs between growth, reproductive development, and longevity. Physiol. Rev. 2013, 93, 571–598. [Google Scholar] [CrossRef]
- Pollak, M.N. Insulin-like growth factors and neoplasia. Novartis Found. Symp. 2004, 262, 84–98. [Google Scholar]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-Aguirre, J.; Wan, J.; et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Anisimov, V.N.; Bartke, A. The key role of growth hormone-insulin-IGF-1 signaling in aging and cancer. Crit. Rev. Oncol. Hematol. 2013, 87, 201–223. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Kaaks, R.; Lukanova, A. Effects of weight control and physical activity in cancer prevention: Role of endogenous hormone metabolism. Ann. N. Y. Acad. Sci. 2002, 963, 268–281. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Berrino, F.; Bellati, C.; Secreto, G.; Camerini, E.; Pala, V.; Panico, S.; Allegro, G.; Kaaks, R. Reducing bioavailable sex hormones through a comprehensive change in diet: The diet and androgens (DIANA) randomized trial. Cancer Epidemiol. Biomark. Prev. 2001, 10, 25–33. [Google Scholar]
- Pugeat, M.; Crave, J.C.; Elmidani, M.; Nicolas, M.H.; Garoscio-Cholet, M.; Lejeune, H.; Dechaud, H.; Tourniaire, J. Pathophysiology of sex hormone binding globulin (SHBG): Relation to insulin. J. Steroid Biochem. Mol. Biol. 1991, 40, 841–849. [Google Scholar] [CrossRef]
- Guevara-Aguirre, J.; Teran, E.; Lescano, D.; Guevara, A.; Guevara, C.; Longo, V.; Gavilanes, A.W.D. Growth hormone receptor deficiency in humans associates to obesity, increased body fat percentage, a healthy brain and a coordinated insulin sensitivity. Growth Horm. IGF Res. 2020, 51, 58–64. [Google Scholar] [CrossRef]
- Weiderpass, E.; Partanen, T.; Kaaks, R.; Vainio, H.; Porta, M.; Kauppinen, T.; Ojajarvi, A.; Boffetta, P.; Malats, N. Occurrence, trends and environment etiology of pancreatic cancer. Scand. J. Work Environ. Health 1998, 24, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Longo, V.D.; Finch, C.E. Evolutionary medicine: From dwarf model systems to healthy centenarians? Science 2003, 299, 1342–1346. [Google Scholar] [CrossRef] [Green Version]
- Fabrizio, P.; Pozza, F.; Pletcher, S.D.; Gendron, C.M.; Longo, V.D. Regulation of longevity and stress resistance by Sch9 in yeast. Science 2001, 292, 288–290. [Google Scholar] [CrossRef] [Green Version]
- Kenyon, C. The plasticity of aging: Insights from long-lived mutants. Cell 2005, 120, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foury, F. Human genetic diseases: A cross-talk between man and yeast. Gene 1997, 195, 1–10. [Google Scholar] [CrossRef]
- Mortimer, R.K.; Johnston, J.R. Life span of individual yeast cells. Nature 1959, 183, 1751–1752. [Google Scholar] [CrossRef] [Green Version]
- Longo, V.D.; Shadel, G.S.; Kaeberlein, M.; Kennedy, B. Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 2012, 16, 18–31. [Google Scholar] [CrossRef] [Green Version]
- Longo, V.D.; Ellerby, L.M.; Bredesen, D.E.; Valentine, J.S.; Gralla, E.B. Human Bcl-2 reverses survival defects in yeast lacking superoxide dismutase and delays death of wild-type yeast. J. Cell Biol. 1997, 137, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Mirisola, M.G.; Longo, V.D. Acetic acid and acidification accelerate chronological and replicative aging in yeast. Cell Cycle 2012, 11, 3532–3533. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wei, M.; Mirisola, M.G.; Longo, V.D. Assessing chronological aging in Saccharomyces cerevisiae. Methods Mol. Biol. 2013, 965, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Longo, V.D. Mutations in signal transduction proteins increase stress resistance and longevity in yeast, nematodes, fruit flies, and mammalian neuronal cells. Neurobiol. Aging 1999, 20, 479–486. [Google Scholar] [CrossRef]
- Wei, M.; Fabrizio, P.; Hu, J.; Ge, H.; Cheng, C.; Li, L.; Longo, V.D. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 2008, 4, e13. [Google Scholar] [CrossRef] [Green Version]
- Toda, T.; Cameron, S.; Sass, P.; Wigler, M. SCH9, a gene of Saccharomyces cerevisiae that encodes a protein distinct from, but functionally and structurally related to, cAMP-dependent protein kinase catalytic subunits. Genes Dev. 1988, 2, 517–527. [Google Scholar] [CrossRef] [Green Version]
- McCormick, M.A.; Delaney, J.R.; Tsuchiya, M.; Tsuchiyama, S.; Shemorry, A.; Sim, S.; Chou, A.C.; Ahmed, U.; Carr, D.; Murakami, C.J.; et al. A Comprehensive Analysis of Replicative Lifespan in 4698 Single-Gene Deletion Strains Uncovers Conserved Mechanisms of Aging. Cell Metab. 2015, 22, 895–906. [Google Scholar] [CrossRef] [Green Version]
- Fabrizio, P.; Hoon, S.; Shamalnasab, M.; Galbani, A.; Wei, M.; Giaever, G.; Nislow, C.; Longo, V.D. Genome-wide screen in Saccharomyces cerevisiae identifies vacuolar protein sorting, autophagy, biosynthetic, and tRNA methylation genes involved in life span regulation. PLoS Genet. 2010, 6, e1001024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirisola, M.G.; Taormina, G.; Fabrizio, P.; Wei, M.; Hu, J.; Longo, V.D. Serine- and threonine/valine-dependent activation of PDK and Tor orthologs converge on Sch9 to promote aging. PLoS Genet. 2014, 10, e1004113. [Google Scholar] [CrossRef] [Green Version]
- Thevelein, J.M.; de Winde, J.H. Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 1999, 33, 904–918. [Google Scholar] [CrossRef]
- Hlavata, L.; Aguilaniu, H.; Pichova, A.; Nystrom, T. The oncogenic RAS2(val19) mutation locks respiration, independently of PKA, in a mode prone to generate ROS. EMBO J. 2003, 22, 3337–3345. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.E. Increased life-span of age-1 mutants in Caenorhabditis elegans and lower Gompertz rate of aging. Science 1990, 249, 908–912. [Google Scholar] [CrossRef]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef]
- Hansen, M.; Chandra, A.; Mitic, L.L.; Onken, B.; Driscoll, M.; Kenyon, C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.E. Caenorhabditis elegans 2007: The premier model for the study of aging. Exp. Gerontol. 2008, 43, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, R.; Steinkraus, K.A.; Sutphin, G.L.; Ramos, F.J.; Shamieh, L.S.; Huh, A.; Davis, C.; Chandler-Brown, D.; Kaeberlein, M. Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science 2009, 324, 1196–1198. [Google Scholar] [CrossRef] [Green Version]
- Pinkston, J.M.; Garigan, D.; Hansen, M.; Kenyon, C. Mutations that increase the life span of C. elegans inhibit tumor growth. Science 2006, 313, 971–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, V.D.; Fabrizio, P. Regulation of longevity and stress resistance: A molecular strategy conserved from yeast to humans? Cell Mol. Life Sci. 2002, 59, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Honda, S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999, 13, 1385–1393. [Google Scholar] [CrossRef] [Green Version]
- LaFever, L.; Drummond-Barbosa, D. Direct control of germline stem cell division and cyst growth by neural insulin in Drosophila. Science 2005, 309, 1071–1073. [Google Scholar] [CrossRef]
- Clancy, D.J.; Gems, D.; Harshman, L.G.; Oldham, S.; Stocker, H.; Hafen, E.; Leevers, S.J.; Partridge, L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 2001, 292, 104–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatar, M.; Kopelman, A.; Epstein, D.; Tu, M.P.; Yin, C.M.; Garofalo, R.S. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 2001, 292, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Igaki, T.; Pagliarini, R.A.; Xu, T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 2006, 16, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010, 11, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Orr, W.C.; Sohal, R.S. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 1994, 263, 1128–1130. [Google Scholar] [CrossRef]
- Lin, Y.J.; Seroude, L.; Benzer, S. Extended life-span and stress resistance in the Drosophila mutant methuselah. Science 1998, 282, 943–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Enns, L.C.; Morton, J.F.; Treuting, P.R.; Emond, M.J.; Wolf, N.S.; Dai, D.F.; McKnight, G.S.; Rabinovitch, P.S.; Ladiges, W.C. Disruption of protein kinase A in mice enhances healthy aging. PLoS ONE 2009, 4, e5963. [Google Scholar] [CrossRef] [Green Version]
- Brown-Borg, H.M.; Borg, K.E.; Meliska, C.J.; Bartke, A. Dwarf mice and the ageing process. Nature 1996, 384, 33. [Google Scholar] [CrossRef]
- Coschigano, K.T.; Clemmons, D.; Bellush, L.L.; Kopchick, J.J. Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology 2000, 141, 2608–2613. [Google Scholar] [CrossRef] [PubMed]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Miquet, J.G.; Gonzalez, L.; Matos, M.N.; Hansen, C.E.; Louis, A.; Bartke, A.; Turyn, D.; Sotelo, A.I. Transgenic mice overexpressing GH exhibit hepatic upregulation of GH-signaling mediators involved in cell proliferation. J. Endocrinol. 2008, 198, 317–330. [Google Scholar] [CrossRef]
- Carboni, J.M.; Lee, A.V.; Hadsell, D.L.; Rowley, B.R.; Lee, F.Y.; Bol, D.K.; Camuso, A.E.; Gottardis, M.; Greer, A.F.; Ho, C.P.; et al. Tumor development by transgenic expression of a constitutively active insulin-like growth factor I receptor. Cancer Res. 2005, 65, 3781–3787. [Google Scholar] [CrossRef] [Green Version]
- Ikeno, Y.; Hubbard, G.B.; Lee, S.; Cortez, L.A.; Lew, C.M.; Webb, C.R.; Berryman, D.E.; List, E.O.; Kopchick, J.J.; Bartke, A. Reduced incidence and delayed occurrence of fatal neoplastic diseases in growth hormone receptor/binding protein knockout mice. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 522–529. [Google Scholar] [CrossRef]
- Pollak, M.; Blouin, M.J.; Zhang, J.C.; Kopchick, J.J. Reduced mammary gland carcinogenesis in transgenic mice expressing a growth hormone antagonist. Br. J. Cancer 2001, 85, 428–430. [Google Scholar] [CrossRef]
- Divisova, J.; Kuiatse, I.; Lazard, Z.; Weiss, H.; Vreeland, F.; Hadsell, D.L.; Schiff, R.; Osborne, C.K.; Lee, A.V. The growth hormone receptor antagonist pegvisomant blocks both mammary gland development and MCF-7 breast cancer xenograft growth. Breast Cancer Res. Treat. 2006, 98, 315–327. [Google Scholar] [CrossRef]
- Longo, V.D.; Lieber, M.R.; Vijg, J. Turning anti-ageing genes against cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms and clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Viciana, P.; Tetsu, O.; Oda, K.; Okada, J.; Rauen, K.; McCormick, F. Cancer targets in the Ras pathway. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Yoeli-Lerner, M.; Toker, A. Akt/PKB signaling in cancer: A function in cell motility and invasion. Cell Cycle 2006, 5, 603–605. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Consortium, A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Renehan, A.G.; Zwahlen, M.; Minder, C.; O’Dwyer, S.T.; Shalet, S.M.; Egger, M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 2004, 363, 1346–1353. [Google Scholar] [CrossRef]
- Suh, Y.; Atzmon, G.; Cho, M.O.; Hwang, D.; Liu, B.; Leahy, D.J.; Barzilai, N.; Cohen, P. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc. Natl. Acad. Sci. USA 2008, 105, 3438–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominici, F.P.; Arostegui Diaz, G.; Bartke, A.; Kopchick, J.J.; Turyn, D. Compensatory alterations of insulin signal transduction in liver of growth hormone receptor knockout mice. J. Endocrinol. 2000, 166, 579–590. [Google Scholar] [CrossRef]
- Dominici, F.P.; Hauck, S.; Argentino, D.P.; Bartke, A.; Turyn, D. Increased insulin sensitivity and upregulation of insulin receptor, insulin receptor substrate (IRS)-1 and IRS-2 in liver of Ames dwarf mice. J. Endocrinol. 2002, 173, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A. Insulin resistance and cognitive aging in long-lived and short-lived mice. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 133–134. [Google Scholar] [CrossRef]
- Masternak, M.M.; Panici, J.A.; Bonkowski, M.S.; Hughes, L.F.; Bartke, A. Insulin sensitivity as a key mediator of growth hormone actions on longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 516–521. [Google Scholar] [CrossRef] [Green Version]
- Steuerman, R.; Shevah, O.; Laron, Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur. J. Endocrinol. 2011, 164, 485–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hursting, S.D.; Lavigne, J.A.; Berrigan, D.; Perkins, S.N.; Barrett, J.C. Calorie restriction, aging, and cancer prevention: Mechanisms of action and applicability to humans. Annu. Rev. Med. 2003, 54, 131–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrows, C.H., Jr.; Kokkonen, G. The effect of various dietary restricted regimes on biochemical variables in the mouse. Growth 1978, 42, 71–85. [Google Scholar]
- Weindruch, R.; Walford, R.L.; Fligiel, S.; Guthrie, D. The retardation of aging in mice by dietary restriction: Longevity, cancer, immunity and lifetime energy intake. J. Nutr. 1986, 116, 641–654. [Google Scholar] [CrossRef] [Green Version]
- Lane, M.A.; Mattison, J.; Ingram, D.K.; Roth, G.S. Caloric restriction and aging in primates: Relevance to humans and possible CR mimetics. Microsc. Res. Tech. 2002, 59, 335–338. [Google Scholar] [CrossRef]
- Anderson, R.M.; Shanmuganayagam, D.; Weindruch, R. Caloric restriction and aging: Studies in mice and monkeys. Toxicol. Pathol. 2009, 37, 47–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandison, R.C.; Piper, M.D.; Partridge, L. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 2009, 462, 1061–1064. [Google Scholar] [CrossRef] [Green Version]
- Colman, R.J.; Beasley, T.M.; Kemnitz, J.W.; Johnson, S.C.; Weindruch, R.; Anderson, R.M. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 2014, 5, 3557. [Google Scholar] [CrossRef]
- McCay, C.M.; Crowell, M.F.; Maynard, L.A. The effect of retarded growth upon the length of life span and upon the ultimate body size. 1935. Nutrition 1989, 5, 155–171. [Google Scholar]
- Patel, N.V.; Finch, C.E. The glucocorticoid paradox of caloric restriction in slowing brain aging. Neurobiol. Aging 2002, 23, 707–717. [Google Scholar] [CrossRef]
- Hursting, S.D.; Switzer, B.R.; French, J.E.; Kari, F.W. The growth hormone: Insulin-like growth factor 1 axis is a mediator of diet restriction-induced inhibition of mononuclear cell leukemia in Fischer rats. Cancer Res. 1993, 53, 2750–2757. [Google Scholar] [PubMed]
- Tomas, F.M.; Chandler, C.S.; Coyle, P.; Bourgeois, C.S.; Burgoyne, J.L.; Rofe, A.M. Effects of insulin and insulin-like growth factors on protein and energy metabolism in tumour-bearing rats. Biochem. J. 1994, 301 Pt 3, 769–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, S.E.; Kari, F.W.; French, J.; Leininger, J.R.; Travlos, G.; Wilson, R.; Barrett, J.C. Dietary restriction reduces insulin-like growth factor I levels, which modulates apoptosis, cell proliferation, and tumor progression in p53-deficient mice. Cancer Res. 1997, 57, 4667–4672. [Google Scholar]
- Hursting, S.D.; Slaga, T.J.; Fischer, S.M.; DiGiovanni, J.; Phang, J.M. Mechanism-based cancer prevention approaches: Targets, examples, and the use of transgenic mice. J. Natl. Cancer Inst. 1999, 91, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Pearson, K.J.; Lewis, K.N.; Price, N.L.; Chang, J.W.; Perez, E.; Cascajo, M.V.; Tamashiro, K.L.; Poosala, S.; Csiszar, A.; Ungvari, Z.; et al. Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc. Natl. Acad. Sci. USA 2008, 105, 2325–2330. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Zhu, Z.; Thompson, H.J. Dietary energy restriction modulates the activity of AMP-activated protein kinase, Akt, and mammalian target of rapamycin in mammary carcinomas, mammary gland, and liver. Cancer Res. 2008, 68, 5492–5499. [Google Scholar] [CrossRef] [Green Version]
- Moore, T.; Beltran, L.; Carbajal, S.; Strom, S.; Traag, J.; Hursting, S.D.; DiGiovanni, J. Dietary energy balance modulates signaling through the Akt/mammalian target of rapamycin pathways in multiple epithelial tissues. Cancer Prev. Res. 2008, 1, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Albanes, D. Total calories, body weight, and tumor incidence in mice. Cancer Res. 1987, 47, 1987–1992. [Google Scholar]
- Klurfeld, D.M.; Welch, C.B.; Lloyd, L.M.; Kritchevsky, D. Inhibition of DMBA-induced mammary tumorigenesis by caloric restriction in rats fed high-fat diets. Int. J. Cancer 1989, 43, 922–925. [Google Scholar] [CrossRef]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Solon-Biet, S.M.; McMahon, A.C.; Ballard, J.W.; Ruohonen, K.; Wu, L.E.; Cogger, V.C.; Warren, A.; Huang, X.; Pichaud, N.; Melvin, R.G.; et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014, 19, 418–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strom, A.; Jensen, R.A. Mortality from circulatory diseases in Norway 1940–1945. Lancet 1951, 1, 126–129. [Google Scholar] [CrossRef]
- Hindhede, M. The Effect of Food Restriction During War on Mortality in Copenhagen. JAMA 1920, 74, 381–382. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Klein, S. Aging, adiposity, and calorie restriction. JAMA 2007, 297, 986–994. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, N.J.; Clifton, P.M.; Noakes, M.; Fenech, M. Weight loss in obese men is associated with increased telomere length and decreased abasic sites in rectal mucosa. Rejuvenation Res. 2009, 12, 169–176. [Google Scholar] [CrossRef]
- Bartke, A. Mutations prolong life in flies; implications for aging in mammals. Trends Endocrinol. Metab. 2001, 12, 233–234. [Google Scholar] [CrossRef]
- Heilbronn, L.K.; de Jonge, L.; Frisard, M.I.; DeLany, J.P.; Larson-Meyer, D.E.; Rood, J.; Nguyen, T.; Martin, C.K.; Volaufova, J.; Most, M.M.; et al. Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: A randomized controlled trial. JAMA 2006, 295, 1539–1548. [Google Scholar] [CrossRef]
- Dirks, A.J.; Leeuwenburgh, C. Caloric restriction in humans: Potential pitfalls and health concerns. Mech. Ageing Dev. 2006, 127, 1–7. [Google Scholar] [CrossRef]
- Holloszy, J.O.; Fontana, L. Caloric restriction in humans. Exp. Gerontol. 2007, 42, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L. Promoting health and longevity through diet: From model organisms to humans. Cell 2015, 161, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.K.; Roberts, S.B.; Bhapkar, M.V.; Villareal, D.T.; Fontana, L.; Martin, C.K.; Racette, S.B.; Fuss, P.J.; Kraus, W.E.; Wong, W.W.; et al. Body-composition changes in the Comprehensive Assessment of Long-term Effects of Reducing Intake of Energy (CALERIE)-2 study: A 2-y randomized controlled trial of calorie restriction in nonobese humans. Am. J. Clin. Nutr. 2017, 105, 913–927. [Google Scholar] [CrossRef]
- Trepanowski, J.F.; Canale, R.E.; Marshall, K.E.; Kabir, M.M.; Bloomer, R.J. Impact of caloric and dietary restriction regimens on markers of health and longevity in humans and animals: A summary of available findings. Nutr. J. 2011, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Bruce-Keller, A.J.; Umberger, G.; McFall, R.; Mattson, M.P. Food restriction reduces brain damage and improves behavioral outcome following excitotoxic and metabolic insults. Ann. Neurol. 1999, 45, 8–15. [Google Scholar] [CrossRef]
- Johnson, J.B.; Summer, W.; Cutler, R.G.; Martin, B.; Hyun, D.H.; Dixit, V.D.; Pearson, M.; Nassar, M.; Telljohann, R.; Maudsley, S.; et al. Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic. Biol. Med. 2007, 42, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Harvie, M.N.; Pegington, M.; Mattson, M.P.; Frystyk, J.; Dillon, B.; Evans, G.; Cuzick, J.; Jebb, S.A.; Martin, B.; Cutler, R.G.; et al. The effects of intermittent or continuous energy restriction on weight loss and metabolic disease risk markers: A randomized trial in young overweight women. Int. J. Obes. 2011, 35, 714–727. [Google Scholar] [CrossRef] [Green Version]
- Regmi, P.; Chaudhary, R.; Page, A.J.; Hutchison, A.T.; Vincent, A.D.; Liu, B.; Heilbronn, L. Early or delayed time-restricted feeding prevents metabolic impact of obesity in mice. J. Endocrinol. 2021, 248, 75–86. [Google Scholar] [CrossRef]
- Chaix, A.; Lin, T.; Le, H.D.; Chang, M.W.; Panda, S. Time-Restricted Feeding Prevents Obesity and Metabolic Syndrome in Mice Lacking a Circadian Clock. Cell Metab. 2019, 29, 303–319.e304. [Google Scholar] [CrossRef]
- Raffaghello, L.; Lee, C.; Safdie, F.M.; Wei, M.; Madia, F.; Bianchi, G.; Longo, V.D. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc. Natl. Acad. Sci. USA 2008, 105, 8215–8220. [Google Scholar] [CrossRef] [Green Version]
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015, 22, 86–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, V.D.; Panda, S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016, 23, 1048–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fond, G.; Macgregor, A.; Leboyer, M.; Michalsen, A. Fasting in mood disorders: Neurobiology and effectiveness. A review of the literature. Psychiatry Res. 2013, 209, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Maniaci, G.; La Cascia, C.; Giammanco, A.; Ferraro, L.; Chianetta, R.; Di Peri, R.; Sardella, Z.; Citarrella, R.; Mannella, Y.; Larcan, S.; et al. Efficacy of a fasting-mimicking diet in functional therapy for depression: A randomised controlled pilot trial. J. Clin. Psychol. 2020, 76, 1807–1817. [Google Scholar] [CrossRef]
- Gonidakis, S.; Finkel, S.E.; Longo, V.D. Genome-wide screen identifies Escherichia coli TCA-cycle-related mutants with extended chronological lifespan dependent on acetate metabolism and the hypoxia-inducible transcription factor ArcA. Aging Cell 2010, 9, 868–881. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.D.; Wilson, M.A.; Zhu, M.; Wolkow, C.A.; de Cabo, R.; Ingram, D.K.; Zou, S. Dietary deprivation extends lifespan in Caenorhabditis elegans. Aging Cell 2006, 5, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Dowlatshahi, D.; Banko, M.R.; Villen, J.; Hoang, K.; Blanchard, D.; Gygi, S.P.; Brunet, A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 2007, 17, 1646–1656. [Google Scholar] [CrossRef] [Green Version]
- Honjoh, S.; Yamamoto, T.; Uno, M.; Nishida, E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature 2009, 457, 726–730. [Google Scholar] [CrossRef]
- Mattson, M.P.; Longo, V.D.; Harvie, M. Impact of intermittent fasting on health and disease processes. Ageing Res. Rev. 2017, 39, 46–58. [Google Scholar] [CrossRef]
- Singh, R.; Manchanda, S.; Kaur, T.; Kumar, S.; Lakhanpal, D.; Lakhman, S.S.; Kaur, G. Middle age onset short-term intermittent fasting dietary restriction prevents brain function impairments in male Wistar rats. Biogerontology 2015, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Wan, R.; Camandola, S.; Mattson, M.P. Intermittent fasting and dietary supplementation with 2-deoxy-D-glucose improve functional and metabolic cardiovascular risk factors in rats. FASEB J. 2003, 17, 1133–1134. [Google Scholar] [CrossRef] [PubMed]
- Castello, L.; Froio, T.; Maina, M.; Cavallini, G.; Biasi, F.; Leonarduzzi, G.; Donati, A.; Bergamini, E.; Poli, G.; Chiarpotto, E. Alternate-day fasting protects the rat heart against age-induced inflammation and fibrosis by inhibiting oxidative damage and NF-kB activation. Free Radic. Biol. Med. 2010, 48, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, F.M.; da Cunha, F.M.; Caldeira da Silva, C.C.; Chausse, B.; Romano, R.L.; Garcia, C.C.; Colepicolo, P.; Medeiros, M.H.; Kowaltowski, A.J. Long-term intermittent feeding, but not caloric restriction, leads to redox imbalance, insulin receptor nitration, and glucose intolerance. Free Radic. Biol. Med. 2011, 51, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Varady, K.A.; Bhutani, S.; Church, E.C.; Klempel, M.C. Short-term modified alternate-day fasting: A novel dietary strategy for weight loss and cardioprotection in obese adults. Am. J. Clin. Nutr. 2009, 90, 1138–1143. [Google Scholar] [CrossRef] [Green Version]
- Varady, K.A.; Hudak, C.S.; Hellerstein, M.K. Modified alternate-day fasting and cardioprotection: Relation to adipose tissue dynamics and dietary fat intake. Metabolism 2009, 58, 803–811. [Google Scholar] [CrossRef]
- Dorighello, G.G.; Rovani, J.C.; Luhman, C.J.; Paim, B.A.; Raposo, H.F.; Vercesi, A.E.; Oliveira, H.C. Food restriction by intermittent fasting induces diabetes and obesity and aggravates spontaneous atherosclerosis development in hypercholesterolaemic mice. Br. J. Nutr. 2014, 111, 979–986. [Google Scholar] [CrossRef] [Green Version]
- Goodrick, C.L.; Ingram, D.K.; Reynolds, M.A.; Freeman, J.R.; Cider, N. Effects of intermittent feeding upon body weight and lifespan in inbred mice: Interaction of genotype and age. Mech. Ageing Dev. 1990, 55, 69–87. [Google Scholar] [CrossRef]
- Cheng, C.W.; Adams, G.B.; Perin, L.; Wei, M.; Zhou, X.; Lam, B.S.; Da Sacco, S.; Mirisola, M.; Quinn, D.I.; Dorff, T.B.; et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell 2014, 14, 810–823. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Safdie, F.M.; Raffaghello, L.; Wei, M.; Madia, F.; Parrella, E.; Hwang, D.; Cohen, P.; Bianchi, G.; Longo, V.D. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res. 2010, 70, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- Cotterill, A.M.; Holly, J.M.; Wass, J.A. The regulation of insulin-like growth factor binding protein (IGFBP)-1 during prolonged fasting. Clin. Endocrinol. 1993, 39, 357–362. [Google Scholar] [CrossRef]
- Thissen, J.P.; Ketelslegers, J.M.; Underwood, L.E. Nutritional regulation of the insulin-like growth factors. Endocr. Rev. 1994, 15, 80–101. [Google Scholar] [CrossRef]
- Giovannucci, E.; Pollak, M.; Platz, E.A.; Willett, W.C.; Stampfer, M.J.; Majeed, N.; Colditz, G.A.; Speizer, F.E.; Hankinson, S.E. Insulin-like growth factor I (IGF-I), IGF-binding protein-3 and the risk of colorectal adenoma and cancer in the Nurses’ Health Study. Growth Horm. IGF Res. 2000, 10 (Suppl. A), S30–S31. [Google Scholar] [CrossRef]
- Lee, C.; Longo, V.D. Fasting vs. dietary restriction in cellular protection and cancer treatment: From model organisms to patients. Oncogene 2011, 30, 3305–3316. [Google Scholar] [CrossRef] [Green Version]
- Murphy, N.; Carreras-Torres, R.; Song, M.; Chan, A.T.; Martin, R.M.; Papadimitriou, N.; Dimou, N.; Tsilidis, K.K.; Banbury, B.; Bradbury, K.E.; et al. Circulating Levels of Insulin-like Growth Factor 1 and Insulin-like Growth Factor Binding Protein 3 Associate With Risk of Colorectal Cancer Based on Serologic and Mendelian Randomization Analyses. Gastroenterology 2020, 158, 1300–1312.e20. [Google Scholar] [CrossRef] [Green Version]
- Safdie, F.M.; Dorff, T.; Quinn, D.; Fontana, L.; Wei, M.; Lee, C.; Cohen, P.; Longo, V.D. Fasting and cancer treatment in humans: A case series report. Aging 2009, 1, 988–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Groot, S.; Vreeswijk, M.P.; Welters, M.J.; Gravesteijn, G.; Boei, J.J.; Jochems, A.; Houtsma, D.; Putter, H.; van der Hoeven, J.J.; Nortier, J.W.; et al. The effects of short-term fasting on tolerance to (neo) adjuvant chemotherapy in HER2-negative breast cancer patients: A randomized pilot study. BMC Cancer 2015, 15, 652. [Google Scholar] [CrossRef] [Green Version]
- Bauersfeld, S.P.; Kessler, C.S.; Wischnewsky, M.; Jaensch, A.; Steckhan, N.; Stange, R.; Kunz, B.; Bruckner, B.; Sehouli, J.; Michalsen, A. The effects of short-term fasting on quality of life and tolerance to chemotherapy in patients with breast and ovarian cancer: A randomized cross-over pilot study. BMC Cancer 2018, 18, 476. [Google Scholar] [CrossRef]
- Dorff, T.B.; Groshen, S.; Garcia, A.; Shah, M.; Tsao-Wei, D.; Pham, H.; Cheng, C.W.; Brandhorst, S.; Cohen, P.; Wei, M.; et al. Safety and feasibility of fasting in combination with platinum-based chemotherapy. BMC Cancer 2016, 16, 360. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Brandhorst, S.; Shelehchi, M.; Mirzaei, H.; Cheng, C.W.; Budniak, J.; Groshen, S.; Mack, W.J.; Guen, E.; Di Biase, S.; et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Caffa, I.; Spagnolo, V.; Vernieri, C.; Valdemarin, F.; Becherini, P.; Wei, M.; Brandhorst, S.; Zucal, C.; Driehuis, E.; Ferrando, L.; et al. Author Correction: Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 2020, 588, E33. [Google Scholar] [CrossRef]
- de Groot, S.; Lugtenberg, R.T.; Cohen, D.; Welters, M.J.P.; Ehsan, I.; Vreeswijk, M.P.G.; Smit, V.; de Graaf, H.; Heijns, J.B.; Portielje, J.E.A.; et al. Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nat. Commun. 2020, 11, 3083. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Raffaghello, L.; Brandhorst, S.; Safdie, F.M.; Bianchi, G.; Martin-Montalvo, A.; Pistoia, V.; Wei, M.; Hwang, S.; Merlino, A.; et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci. Transl. Med. 2012, 4, 124ra27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.W.; Cacciottolo, M.; Martin-Montalvo, A.; de Cabo, R.; Wei, M.; et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocks, T.; Rapp, K.; Bjorge, T.; Manjer, J.; Ulmer, H.; Selmer, R.; Lukanova, A.; Johansen, D.; Concin, H.; Tretli, S.; et al. Blood glucose and risk of incident and fatal cancer in the metabolic syndrome and cancer project (me-can): Analysis of six prospective cohorts. PLoS Med. 2009, 6, e1000201. [Google Scholar] [CrossRef]
- Davidson, M.B. The effect of aging on carbohydrate metabolism: A review of the English literature and a practical approach to the diagnosis of diabetes mellitus in the elderly. Metabolism 1979, 28, 688–705. [Google Scholar] [CrossRef]
- Pyorala, K. Relationship of glucose tolerance and plasma insulin to the incidence of coronary heart disease: Results from two population studies in Finland. Diabetes Care 1979, 2, 131–141. [Google Scholar] [CrossRef]
- Stout, R.W. The relationship of abnormal circulating insulin levels to atherosclerosis. Atherosclerosis 1977, 27, 1–13. [Google Scholar] [CrossRef]
- Bjorntorp, P.; De Jounge, K.; Sjostrom, L.; Sullivan, L. The effect of physical training on insulin production in obesity. Metabolism 1970, 19, 631–638. [Google Scholar] [CrossRef]
- LeBlanc, J.; Nadeau, A.; Richard, D.; Tremblay, A. Studies on the sparing effect of exercise on insulin requirements in human subjects. Metabolism 1981, 30, 1119–1124. [Google Scholar] [CrossRef]
- Heath, G.W.; Gavin, J.R., 3rd; Hinderliter, J.M.; Hagberg, J.M.; Bloomfield, S.A.; Holloszy, J.O. Effects of exercise and lack of exercise on glucose tolerance and insulin sensitivity. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1983, 55, 512–517. [Google Scholar] [CrossRef]
- Seals, D.R.; Hagberg, J.M.; Allen, W.K.; Hurley, B.F.; Dalsky, G.P.; Ehsani, A.A.; Holloszy, J.O. Glucose tolerance in young and older athletes and sedentary men. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 56, 1521–1525. [Google Scholar] [CrossRef]
- De Groote, E.; Britto, F.A.; Balan, E.; Warnier, G.; Thissen, J.P.; Nielens, H.; Sylow, L.; Deldicque, L. Effect of hypoxic exercise on glucose tolerance in healthy and prediabetic adults. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E43–E54. [Google Scholar] [CrossRef]
- Holloszy, J.O.; Schultz, J.; Kusnierkiewicz, J.; Hagberg, J.M.; Ehsani, A.A. Effects of exercise on glucose tolerance and insulin resistance. Brief review and some preliminary results. Acta Med. Scand. Suppl. 1986, 711, 55–65. [Google Scholar] [CrossRef]
- Fontana, L.; Meyer, T.E.; Klein, S.; Holloszy, J.O. Long-term low-calorie low-protein vegan diet and endurance exercise are associated with low cardiometabolic risk. Rejuvenation Res. 2007, 10, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.C.; Lee, I.M.; Weiderpass, E.; Campbell, P.T.; Sampson, J.N.; Kitahara, C.M.; Keadle, S.K.; Arem, H.; Berrington de Gonzalez, A.; Hartge, P.; et al. Association of Leisure-Time Physical Activity With Risk of 26 Types of Cancer in 1.44 Million Adults. JAMA Intern. Med. 2016, 176, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Friedenreich, C.M.; Neilson, H.K.; Farris, M.S.; Courneya, K.S. Physical Activity and Cancer Outcomes: A Precision Medicine Approach. Clin. Cancer Res. 2016, 22, 4766–4775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarfstein, R.; Friedman, Y.; Attias-Geva, Z.; Fishman, A.; Bruchim, I.; Werner, H. Metformin downregulates the insulin/IGF-I signaling pathway and inhibits different uterine serous carcinoma (USC) cells proliferation and migration in p53-dependent or -independent manners. PLoS ONE 2013, 8, e61537. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wang, J.L.; Ji, M.; Yuan, Z.F.; Peng, Z.; Zhang, Y.; Wen, J.G.; Shi, H.R. Regulation of insulin-like growth factor signaling by metformin in endometrial cancer cells. Oncol. Lett. 2014, 8, 1993–1999. [Google Scholar] [CrossRef] [Green Version]
- Klement, R.J.; Fink, M.K. Dietary and pharmacological modification of the insulin/IGF-1 system: Exploiting the full repertoire against cancer. Oncogenesis 2016, 5, e193. [Google Scholar] [CrossRef] [Green Version]
- Onken, B.; Driscoll, M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 2010, 5, e8758. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef] [Green Version]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, S.; Lux, A.; O’Callaghan, F. The journey of metformin from glycaemic control to mTOR inhibition and the suppression of tumour growth. Br. J. Clin. Pharmacol. 2019, 85, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Kane, D.A.; Anderson, E.J.; Price, J.W., 3rd; Woodlief, T.L.; Lin, C.T.; Bikman, B.T.; Cortright, R.N.; Neufer, P.D. Metformin selectively attenuates mitochondrial H2O2 emission without affecting respiratory capacity in skeletal muscle of obese rats. Free Radic. Biol. Med. 2010, 49, 1082–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, M.; Cheki, M.; Rezapoor, S.; Geraily, G.; Motevaseli, E.; Carnovale, C.; Clementi, E.; Shirazi, A. Metformin: Prevention of genomic instability and cancer: A review. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2018, 827, 1–8. [Google Scholar] [CrossRef]
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O’Neill, L.A. Metformin Inhibits the Production of Reactive Oxygen Species from NADH:Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1beta (IL-1beta) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-activated Macrophages. J. Biol. Chem. 2015, 290, 20348–20359. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Conserved nutrient signaling pathways in yeast, worms, flies and mammals. A schematic model of the conserved nutrient-signaling pathways that regulate stress response mechanisms, DNA damage, longevity and cancer in different model organisms [22]. In Saccharomyces cerevisiae, glucose and amino acids activate Ras/AC/PKA and TOR/Sch9 pathways, respectively. Their activation leads to serine-threonine kinase Rim15 inhibition and consequently to a lowering in Msn2/Msn4 stress resistance transcription factors. These mechanisms promote aging in part by decreasing cell stress response and repair thus increasing DNA damage [20]. In Caenorhabditis elegans, insulin/IGF-1 receptor like (daf-2) signaling, through the activation of AKT/PKB pathway, inactivates the Forkhead FoxO transcription factor daf-16, which is involved in the regulation of genes implicated in the cellular stress response and protection against free radicals. As in yeast, also in worms, amino acids can activate the TOR/S6K pathway, accelerating the aging process [44]. Analogously to worms, in Drosophila melanogaster growth factors and amino acids activate AKT/PKB and TOR/S6K pathways, respectively [47]. The activation of TOR/S6K, PI3K/AKT and Ras/AC/PKA pathways, mediated by glucose, amino acids and IGF-1 like signaling, is also maintained in rodents and other mammals, suggesting that these nutrient-sensing pathways, involved in longevity and stress-response mechanisms, are conserved, in part, from the simplest model organism to humans [22,63].
Figure 1.
Conserved nutrient signaling pathways in yeast, worms, flies and mammals. A schematic model of the conserved nutrient-signaling pathways that regulate stress response mechanisms, DNA damage, longevity and cancer in different model organisms [22]. In Saccharomyces cerevisiae, glucose and amino acids activate Ras/AC/PKA and TOR/Sch9 pathways, respectively. Their activation leads to serine-threonine kinase Rim15 inhibition and consequently to a lowering in Msn2/Msn4 stress resistance transcription factors. These mechanisms promote aging in part by decreasing cell stress response and repair thus increasing DNA damage [20]. In Caenorhabditis elegans, insulin/IGF-1 receptor like (daf-2) signaling, through the activation of AKT/PKB pathway, inactivates the Forkhead FoxO transcription factor daf-16, which is involved in the regulation of genes implicated in the cellular stress response and protection against free radicals. As in yeast, also in worms, amino acids can activate the TOR/S6K pathway, accelerating the aging process [44]. Analogously to worms, in Drosophila melanogaster growth factors and amino acids activate AKT/PKB and TOR/S6K pathways, respectively [47]. The activation of TOR/S6K, PI3K/AKT and Ras/AC/PKA pathways, mediated by glucose, amino acids and IGF-1 like signaling, is also maintained in rodents and other mammals, suggesting that these nutrient-sensing pathways, involved in longevity and stress-response mechanisms, are conserved, in part, from the simplest model organism to humans [22,63].


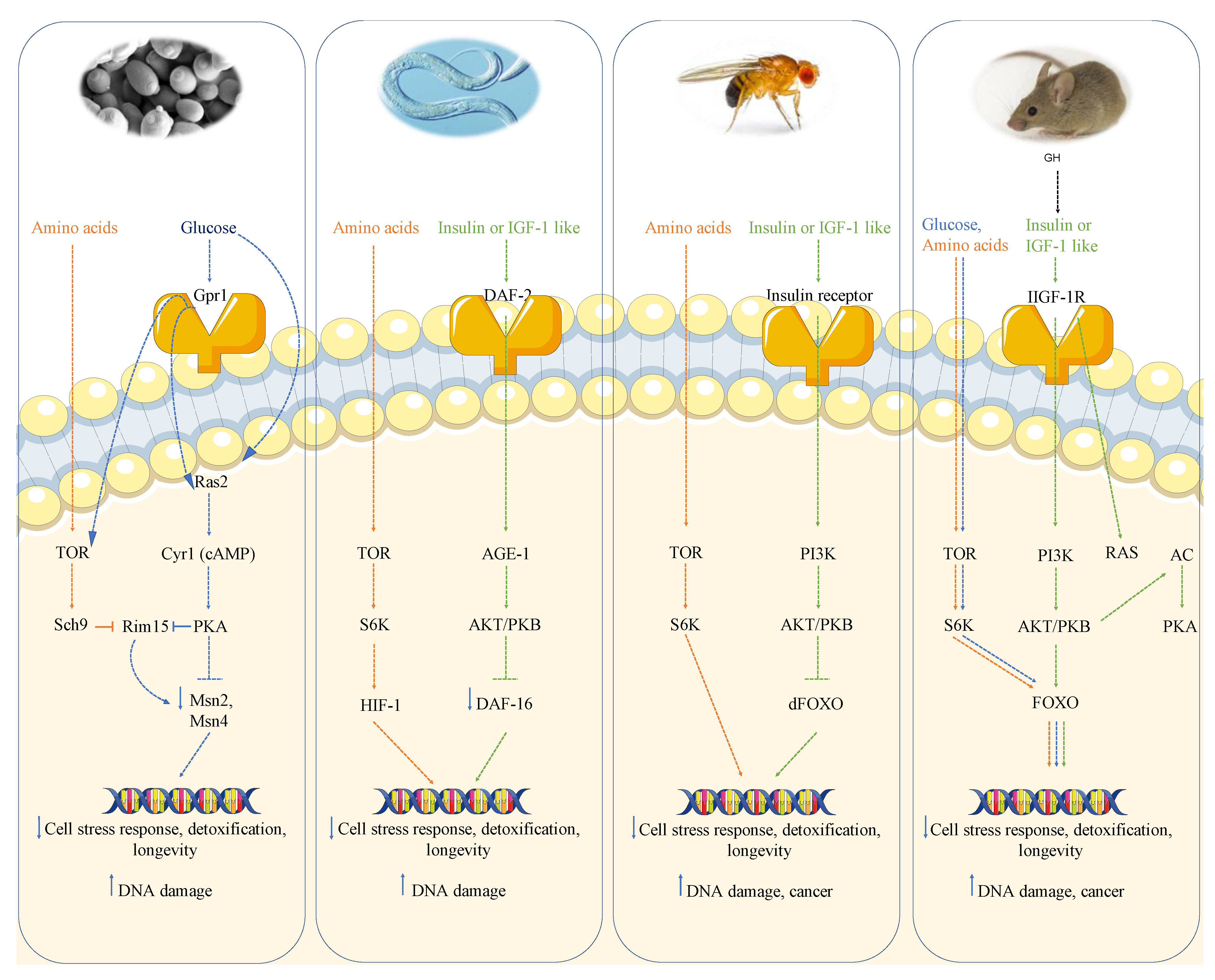{kind=link}
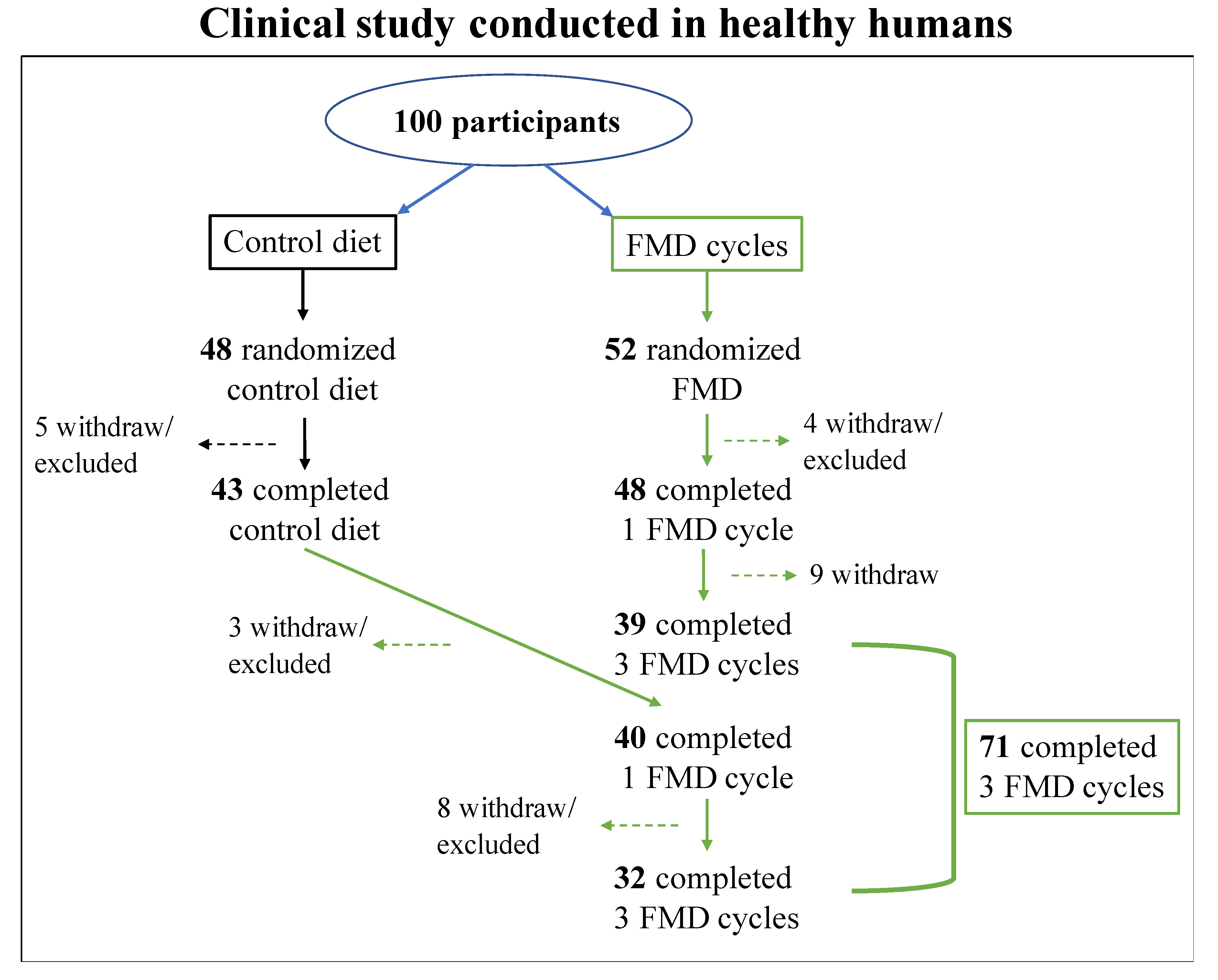{kind=link}
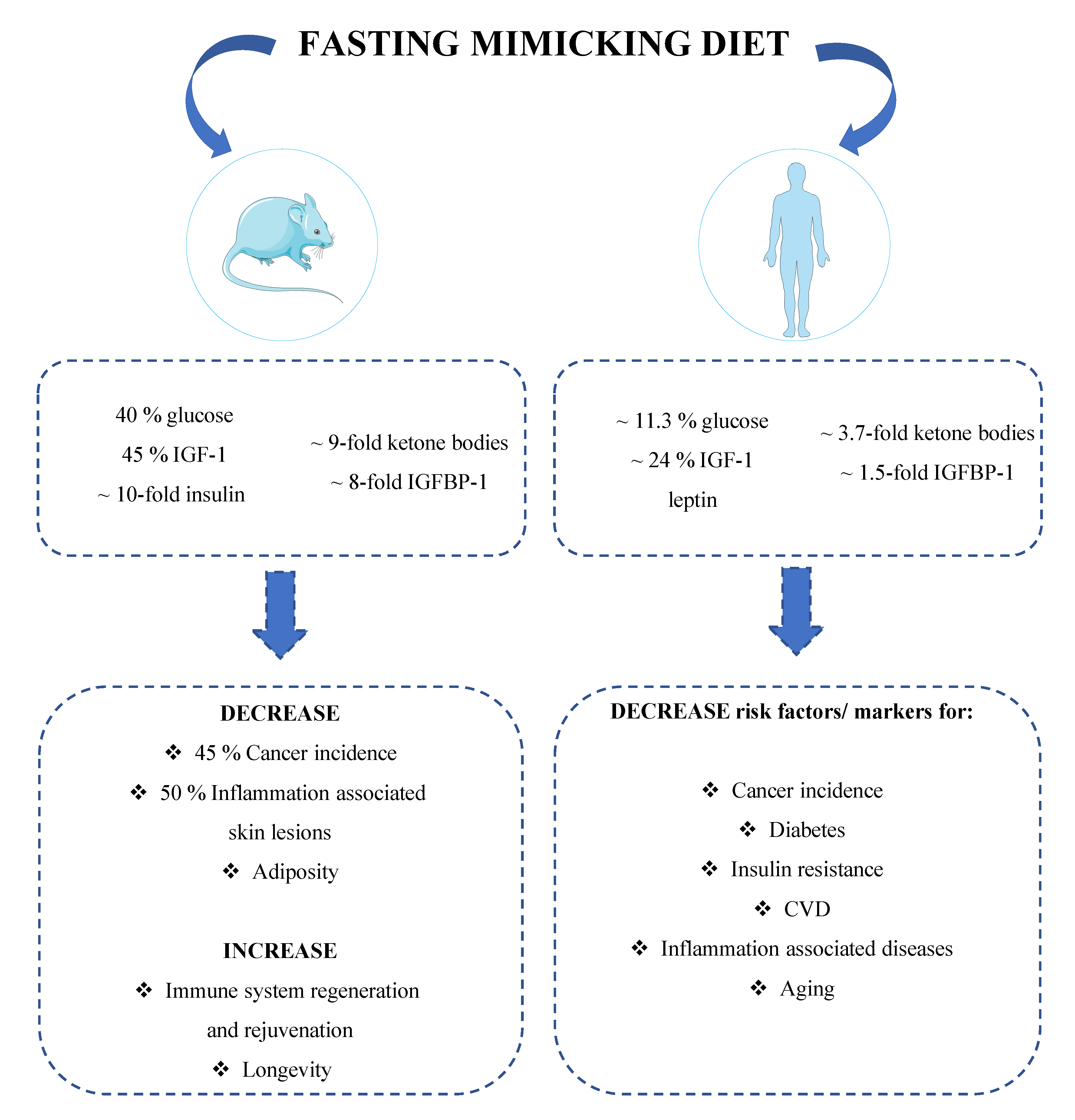{kind=link}
Table 1.
Metabolic, molecular and cellular mechanisms induced by CR to prevent cancer.
| Calorie Restriction (CR) | Metabolic Adaptations | Molecular Adaptations | Cellular Adaptations |
| ↓ IGF-1 ↓ Insulin ↓ Oxidative stress ↓ Inflammation ↑ Cortisol | ↓ PI3K/Akt/S6K ↓ mTOR ↓ Ras/MAPK ↑ Nrf2 ↑ FOXO ↑ PTEN | ↓ Cell proliferation ↓ Oxidative damage ↑ DNA repair ↑ Genome instability |
Table 2.
Dietary approaches to promote health span.
| Type of Fasting | Schedule | Description | |
|---|---|---|---|
| Intermittent Fasting (IF) | ADF | 24 h fast/ 24 h eating period | Water only fasting every other day |
| 5:2 | 2 days fast or very low calorie consumption (500–700 kcal)/ 5 days eating period | Alternation of 2 days of very low-calorie consumption with a 5 days ad libitum re-feeding period | |
| TRF | 12- to 18 h fast/ 6- to 12 h eating period | Food intake resctricted to 6–12 h per day | |
| Periodic Fasting (PF) | Prolonged fasting | 2–5 days of water fast/ 7 days eating period (or longer) | Water only fasting period followed by an ad libitum re-feeding period |
| Prolonged FMD | 4- to 7 days FMD/ 10- to 25 days eating period | 30–50% of the normal caloric intake using a fasting mimicking diet for 4–7 days followed by an ad libitum re-feeding period |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Salvadori, G.; Mirisola, M.G.; Longo, V.D. Intermittent and Periodic Fasting, Hormones, and Cancer Prevention. Cancers 2021, 13, 4587. https://doi.org/10.3390/cancers13184587
AMA Style
Salvadori G, Mirisola MG, Longo VD. Intermittent and Periodic Fasting, Hormones, and Cancer Prevention. Cancers. 2021; 13(18):4587. https://doi.org/10.3390/cancers13184587
Chicago/Turabian StyleSalvadori, Giulia, Mario Giuseppe Mirisola, and Valter D. Longo. 2021. "Intermittent and Periodic Fasting, Hormones, and Cancer Prevention" Cancers 13, no. 18: 4587. https://doi.org/10.3390/cancers13184587
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.