
Drug Repurposing, an Attractive Strategy in Pancreatic Cancer Treatment: Preclinical and Clinical Updates

 ,
,  , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
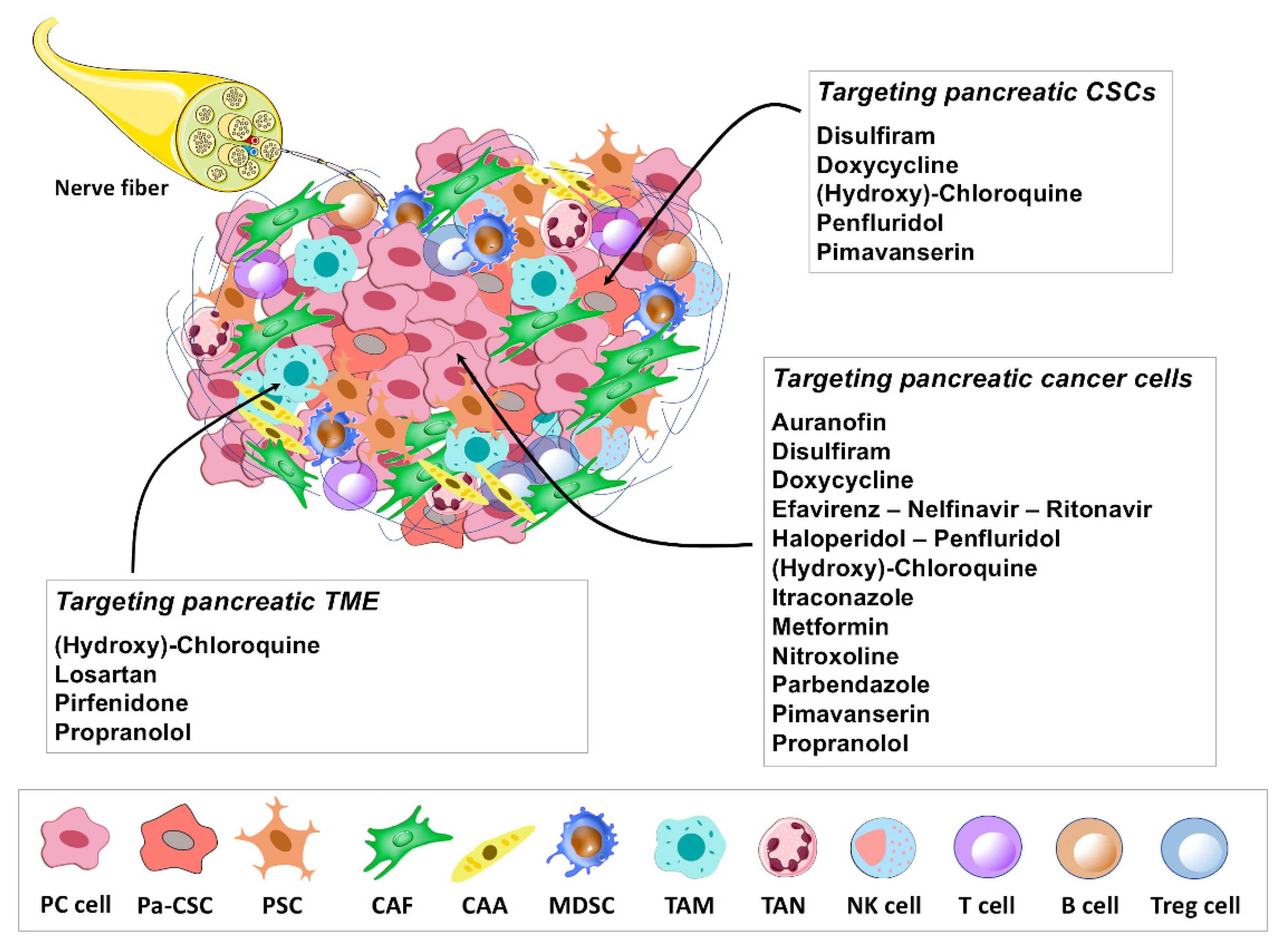
2. Repurposing Drug Candidates in Pancreatic Cancer: Preclinical and Clinical Findings
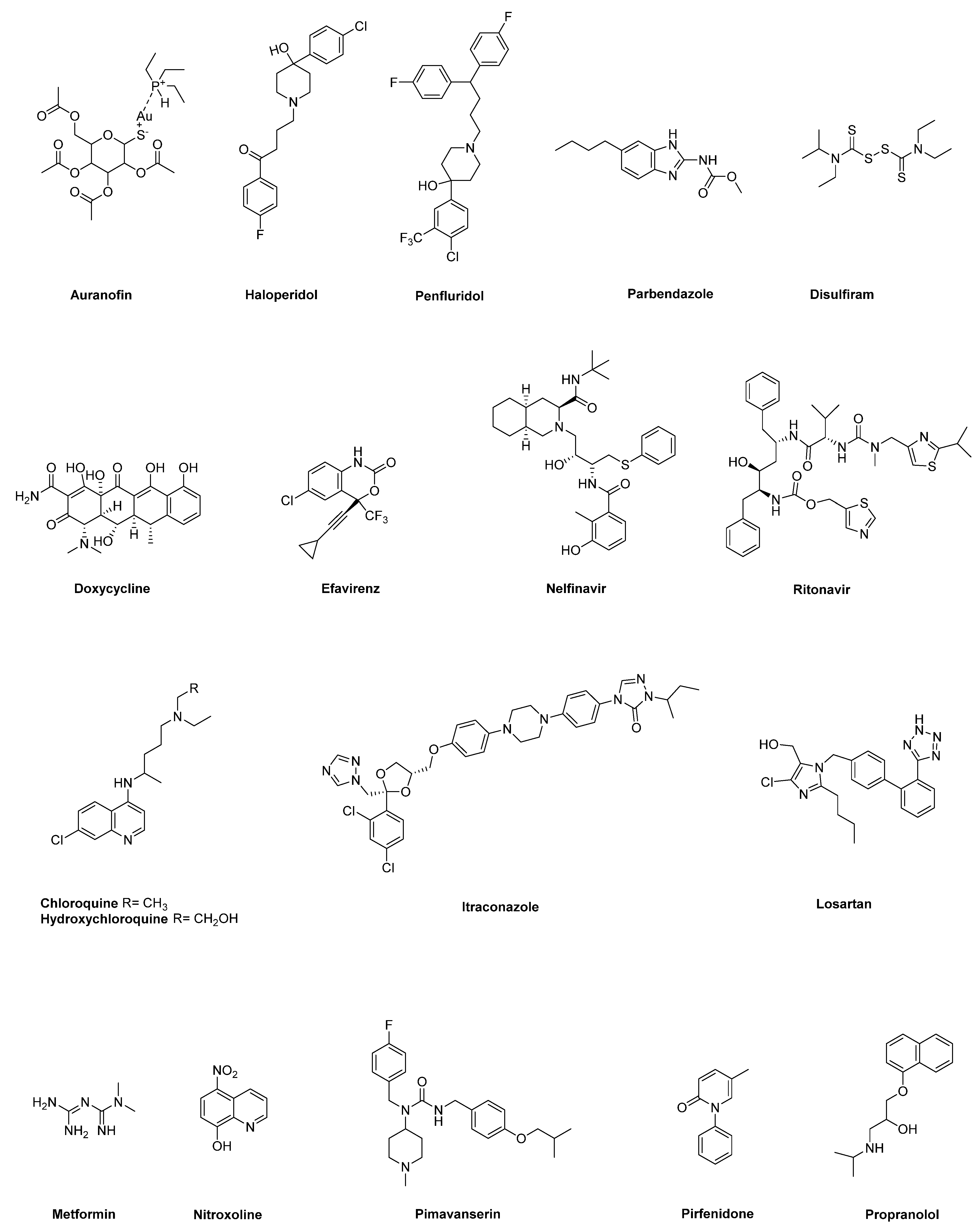
2.1. Auranofin
2.2. Anti-Psychotic Drugs (Haloperidol, Penfluridol)
2.3. Benzimidazole-Based Anthelminthics
2.4. Disulfiram
2.5. Doxycycline
2.6. HIV Inhibitors (Efavirenz, Nelfinavir, Ritonavir)
2.7. Hydroxychloroquine and Chloroquine
2.8. Itraconazole
2.9. Losartan
2.10. Metformin
2.11. Nitroxoline
2.12. Pimavanserin
2.13. Pirfenidone
2.14. Propranolol
3. Molecular Targets for Repurposed Drug Candidates: Hints from an In Silico Analysis
4. Immune-Modulating Potential of Repurposing Drug Candidates
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef] [PubMed]
- Franck, C.; Müller, C.; Rosania, R.; Croner, R.S.; Pech, M.; Venerito, M. Advanced Pancreatic Ductal Adenocarcinoma: Moving Forward. Cancers 2020, 12, 1955. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Gillen, S.; Schuster, T.; Büschenfelde, C.M.Z.; Friess, H.; Kleeff, J. Preoperative/Neoadjuvant Therapy in Pancreatic Cancer: A Systematic Review and Meta-analysis of Response and Resection Percentages. PLoS Med. 2010, 7, e1000267. [Google Scholar] [CrossRef] [Green Version]
- A decade in drug discovery. Nat. Rev. Drug Discov. 2012, 11, 3. [CrossRef] [Green Version]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef] [PubMed]
- Nosengo, N. Can you teach old drugs new tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Liu, J.O. Recent Advances in Drug Repositioning for the Discovery of New Anticancer Drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Bringhen, S.; Caravita, T.; Merla, E.; Capparella, V.; Callea, V.; Cangialosi, C.; Grasso, M.; Rossini, F.; Galli, M.; et al. Oral melphalan and prednisone chemotherapy plus thalidomide compared with melphalan and prednisone alone in elderly patients with multiple myeloma: Randomised controlled trial. Lancet 2006, 367, 825–831. [Google Scholar] [CrossRef]
- Léauté-Labrèze, C.; de la Roque, E.D.; Hubiche, T.; Boralevi, F.; Thambo, J.-B.; Taïeb, A. Propranolol for Severe Hemangiomas of Infancy. N. Engl. J. Med. 2008, 358, 2649–2651. [Google Scholar] [CrossRef]
- Krowchuk, D.P.; Frieden, I.J.; Mancini, A.J.; Darrow, D.H.; Blei, F.; Greene, A.K.; Annam, A.; Baker, C.N.; Frommelt, P.C.; Hodak, A.; et al. Clinical Practice Guideline for the Management of Infantile Hemangiomas. Pediatrics 2019, 143, e20183475. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.; Verma, S.S.; Aggarwal, S.; Gupta, S.C. Drug repurposing for breast cancer therapy: Old weapon for new battle. Semin. Cancer Biol. 2021, 68, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Verbaanderd, C.; Huys, I.; Bouche, G.; Meheus, L. Repurposing drugs in oncology: From candidate selection to clinical adoption. Semin. Cancer Biol. 2021, 68, 186–191. [Google Scholar] [CrossRef]
- Perez, M.V.R.; Roife, D.; Dai, B.; Pratt, M.; Dobrowolski, R.; Kang, Y.; Li, X.; Augustine, J.J.; Zielinski, R.; Priebe, W.; et al. Antineoplastic effects of auranofin in human pancreatic adenocarcinoma preclinical models. Surg. Open Sci. 2019, 1, 56–63. [Google Scholar] [CrossRef]
- Onodera, T.; Momose, I.; Adachi, H.; Yamazaki, Y.; Sawa, R.; Ohba, S.; Kawada, M. Human pancreatic cancer cells under nutrient deprivation are vulnerable to redox system inhibition. J. Biol. Chem. 2020, 295, 16678–16690. [Google Scholar] [CrossRef]
- Kshattry, S.; Saha, A.; Gries, P.; Tiziani, S.; Stone, E.; Georgiou, G.; DiGiovanni, J. Enzyme-mediated depletion of l-cyst(e)ine synergizes with thioredoxin reductase inhibition for suppression of pancreatic tumor growth. NPJ Precis. Oncol. 2019, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Jandaghi, P.; Najafabadi, H.S.; Bauer, A.S.; Papadakis, A.I.; Fassan, M.; Hall, A.; Monast, A.; von Knebel Doeberitz, M.; Neoptolemos, J.P.; Costello, E.; et al. Expression of DRD2 Is Increased in Human Pancreatic Ductal Adenocarcinoma and Inhibitors Slow Tumor Growth in Mice. Gastroenterology 2016, 151, 1218–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Lee, H.Y.; Yi, H.; Ahn, Y.M.; Kim, Y.S. Haloperidol induces demethylation and expression of the dual specificity phosphatase 6 gene in MIA PaCa-2 human pancreatic cancer cells. Life Sci. 2012, 91, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Srivastava, S.K. Penfluridol suppresses pancreatic tumor growth by autophagy-mediated apoptosis. Sci. Rep. 2016, 6, 26165. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, A.; German, N.; Mikelis, C.; Srivenugopal, K.; Srivastava, S.K. Penfluridol induces endoplasmic reticulum stress leading to autophagy in pancreatic cancer. Tumor Biol. 2017, 39, 101042831770551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, W.; Sun, Q.-Y.; Lee, K.L.; Ding, L.-W.; Wuensche, P.; Torres-Fernandez, L.A.; Tan, S.Z.; Tokatly, I.; Zaiden, N.; Poellinger, L.; et al. Activation of protein phosphatase 2A tumor suppressor as potential treatment of pancreatic cancer. Mol. Oncol. 2015, 9, 889–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandawate, P.; Kaushik, G.; Ghosh, C.; Standing, D.; Sayed, A.A.A.; Choudhury, S.; Subramaniam, D.; Manzardo, A.; Banerjee, T.; Santra, S.; et al. Diphenylbutylpiperidine Antipsychotic Drugs Inhibit Prolactin Receptor Signaling to Reduce Growth of Pancreatic Ductal Adenocarcinoma in Mice. Gastroenterology 2020, 158, 1433–1449.e27. [Google Scholar] [CrossRef]
- Florio, R.; Veschi, S.; di Giacomo, V.; Pagotto, S.; Carradori, S.; Verginelli, F.; Cirilli, R.; Casulli, A.; Grassadonia, A.; Tinari, N.; et al. The Benzimidazole-Based Anthelmintic Parbendazole: A Repurposed Drug Candidate That Synergizes with Gemcitabine in Pancreatic Cancer. Cancers 2019, 11, 2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Liu, L.; Yue, X.; Chang, J.; Shi, W.; Hua, Y. A binuclear complex constituted by diethyldithiocarbamate and copper (I) functions as a proteasome activity inhibitor in pancreatic cancer cultures and xenografts. Toxicol. Appl. Pharmacol. 2013, 273, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Cong, J.; Wang, Y.; Zhang, X.; Zhang, N.; Liu, L.; Soukup, K.; Michelakos, T.; Hong, T.; DeLeo, A.; Cai, L.; et al. A novel chemoradiation targeting stem and nonstem pancreatic cancer cells by repurposing disulfiram. Cancer Lett. 2017, 409, 9–19. [Google Scholar] [CrossRef]
- Dinnen, R.D.; Mao, Y.; Qiu, W.; Cassai, N.; Slavkovich, V.N.; Nichols, G.; Su, G.H.; Brandt-Rauf, P.; Fine, R.L. Redirecting Apoptosis to Aponecrosis Induces Selective Cytotoxicity to Pancreatic Cancer Cells through Increased ROS, Decline in ATP Levels, and VDAC. Mol. Cancer Ther. 2013, 12, 2792–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Hu, P.; Ding, S.-Y.; Sun, T.; Liu, L.; Han, S.; DeLeo, A.B.; Sadagopan, A.; Guo, W.; Wang, X. Induction of autophagy-dependent apoptosis in cancer cells through activation of ER stress: An uncovered anti-cancer mechanism by anti-alcoholism drug disulfiram. Am. J. Cancer Res. 2019, 9, 1266–1281. [Google Scholar] [PubMed]
- Son, K.; Fujioka, S.; Iida, T.; Furukawa, K.; Fujita, T.; Yamada, H.; Chiao, P.J.; Yanaga, K. Doxycycline induces apoptosis in PANC-1 pancreatic cancer cells. Anticancer Res. 2009, 29, 3995–4003. [Google Scholar]
- Mouratidis, P.X.E.; Colston, K.W.; Dalgleish, A.G. Doxycycline induces caspase-dependent apoptosis in human pancreatic cancer cells. Int. J. Cancer 2007, 120, 743–752. [Google Scholar] [CrossRef]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [Green Version]
- Dijk, S.N.; Protasoni, M.; Elpidorou, M.; Kroon, A.M.; Taanman, J.-W. Mitochondria as target to inhibit proliferation and induce apoptosis of cancer cells: The effects of doxycycline and gemcitabine. Sci. Rep. 2020, 10, 4363. [Google Scholar] [CrossRef]
- Liu, H.; Tao, H.; Wang, H.; Yang, Y.; Yang, R.; Dai, X.; Ding, X.; Wu, H.; Chen, S.; Sun, T. Doxycycline Inhibits Cancer Stem Cell-Like Properties via PAR1/FAK/PI3K/AKT Pathway in Pancreatic Cancer. Front. Oncol. 2021, 10, 619317. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Erber, S.; Harrer, T.; Klinker, H.; Roth, T.; Parsch, H.; Fiebig, N.; Fietkau, R.; Distel, L.V. Efavirenz Has the Highest Anti-Proliferative Effect of Non-Nucleoside Reverse Transcriptase Inhibitors against Pancreatic Cancer Cells. PLoS ONE 2015, 10, e0130277. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Harrer, T.; Körber, V.; Sarpong, E.; Moser, F.; Fiebig, N.; Schwegler, M.; Stürzl, M.; Fietkau, R.; Distel, L. Cytotoxic effect of Efavirenz in BxPC-3 pancreatic cancer cells is based on oxidative stress and is synergistic with ionizing radiation. Oncol. Lett. 2017, 15, 1728–1736. [Google Scholar] [CrossRef]
- Kimple, R.J.; Vaseva, A.V.; Cox, A.D.; Baerman, K.M.; Calvo, B.F.; Tepper, J.E.; Shields, J.M.; Sartor, C.I. Radiosensitization of Epidermal Growth Factor Receptor/HER2-Positive Pancreatic Cancer Is Mediated by Inhibition of Akt Independent of Ras Mutational Status. Clin. Cancer Res. 2010, 16, 912–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veschi, S.; De Lellis, L.; Florio, R.; Lanuti, P.; Massucci, A.; Tinari, N.; De Tursi, M.; di Sebastiano, P.; Marchisio, M.; Natoli, C.; et al. Effects of repurposed drug candidates nitroxoline and nelfinavir as single agents or in combination with erlotinib in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 236. [Google Scholar] [CrossRef]
- Batchu, R.; Gruzdyn, O.; Bryant, C.; Qazi, A.; Kumar, S.; Chamala, S.; Kung, S.; Sanka, R.; Puttagunta, U.; Weaver, D.; et al. Ritonavir-Mediated Induction of Apoptosis in Pancreatic Cancer Occurs via the RB/E2F-1 and AKT Pathways. Pharmaceuticals 2014, 7, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated with Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 152, 1492–1506.e24. [Google Scholar] [CrossRef] [Green Version]
- Frieboes, H.B.; Huang, J.S.; Yin, W.C.; McNally, L.R. Chloroquine-mediated cell death in metastatic pancreatic adenocarcinoma through inhibition of autophagy. JOP 2014, 15, 189–197. [Google Scholar] [CrossRef]
- Hashimoto, D.; Bläuer, M.; Hirota, M.; Ikonen, N.H.; Sand, J.; Laukkarinen, J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur. J. Cancer 2014, 50, 1382–1390. [Google Scholar] [CrossRef]
- Fu, Z.; Cheng, X.; Kuang, J.; Feng, H.; Chen, L.; Liang, J.; Shen, X.; Yuen, S.; Peng, C.; Shen, B.; et al. CQ sensitizes human pancreatic cancer cells to gemcitabine through the lysosomal apoptotic pathway via reactive oxygen species. Mol. Oncol. 2018, 12, 529–544. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Balic, A.; Sørensen, M.D.; Trabulo, S.M.; Sainz, B.; Cioffi, M.; Vieira, C.R.; Miranda-Lorenzo, I.; Hidalgo, M.; Kleeff, J.; Erkan, M.; et al. Chloroquine Targets Pancreatic Cancer Stem Cells via Inhibition of CXCR4 and Hedgehog Signaling. Mol. Cancer Ther. 2014, 13, 1758–1771. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Xing, H.; Chen, W.; Du, J.; Ruan, Y.; Lin, A.; Zhou, C. Itraconazole inhibits proliferation of pancreatic cancer cells through activation of Bak-1. J. Cell. Biochem. 2019, 120, 4333–4341. [Google Scholar] [CrossRef]
- Chen, K.; Cheng, L.; Qian, W.; Jiang, Z.; Sun, L.; Zhao, Y.; Zhou, Y.; Zhao, L.; Wang, P.; Duan, W.; et al. Itraconazole inhibits invasion and migration of pancreatic cancer cells by suppressing TGF-β/SMAD2/3 signaling. Oncol. Rep. 2018, 39, 1573–1582. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Toyokawa, H.; Yamao, J.; Satoi, S.; Yanagimoto, H.; Yamamoto, T.; Hirooka, S.; Yamaki, S.; Inoue, K.; Matsui, Y.; et al. Antitumor Effect of Angiotensin II Type 1 Receptor Blocker Losartan for Orthotopic Rat Pancreatic Adenocarcinoma. Pancreas 2014, 43, 886–890. [Google Scholar] [CrossRef]
- Li, Y.; Tang, Y.; Chen, S.; Liu, Y.; Wang, S.; Tian, Y.; Wang, C.; Teng, Z.; Lu, G. Sequential therapy for pancreatic cancer by losartan- and gemcitabine-loaded magnetic mesoporous spheres. RSC Adv. 2019, 9, 19690–19698. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [Green Version]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef] [Green Version]
- Incio, J.; Liu, H.; Suboj, P.; Chin, S.M.; Chen, I.X.; Pinter, M.; Ng, M.R.; Nia, H.T.; Grahovac, J.; Kao, S.; et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016, 6, 852–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, S.A.; Rivera, L.B.; Carbon, J.G.; Toombs, J.E.; Chang, C.-L.; Bradshaw, A.D.; Brekken, R.A. Losartan Slows Pancreatic Tumor Progression and Extends Survival of SPARC-Null Mice by Abrogating Aberrant TGFβ Activation. PLoS ONE 2012, 7, e31384. [Google Scholar] [CrossRef] [Green Version]
- Nair, V.; Sreevalsan, S.; Basha, R.; Abdelrahim, M.; Abudayyeh, A.; Hoffman, A.R.; Safe, S. Mechanism of Metformin-dependent Inhibition of Mammalian Target of Rapamycin (mTOR) and Ras Activity in Pancreatic Cancer. J. Biol. Chem. 2014, 289, 27692–27701. [Google Scholar] [CrossRef] [Green Version]
- Kisfalvi, K.; Eibl, G.; Sinnett-Smith, J.; Rozengurt, E. Metformin Disrupts Crosstalk between G Protein–Coupled Receptor and Insulin Receptor Signaling Systems and Inhibits Pancreatic Cancer Growth. Cancer Res. 2009, 69, 6539–6545. [Google Scholar] [CrossRef] [Green Version]
- Karnevi, E.; Said, K.; Andersson, R.; Rosendahl, A.H. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer 2013, 13, 235. [Google Scholar] [CrossRef] [Green Version]
- Veschi, S.; Ronci, M.; Lanuti, P.; De Lellis, L.; Florio, R.; Bologna, G.; Scotti, L.; Carletti, E.; Brugnoli, F.; Di Bella, M.C.; et al. Integrative proteomic and functional analyses provide novel insights into the action of the repurposed drug candidate nitroxoline in AsPC-1 cells. Sci. Rep. 2020, 10, 2574. [Google Scholar] [CrossRef]
- Ramachandran, S.; Srivastava, S.K. Repurposing Pimavanserin, an Anti-Parkinson Drug for Pancreatic Cancer Therapy. Mol. Ther. Oncolytics 2020, 19, 19–32. [Google Scholar] [CrossRef]
- Usugi, E.; Ishii, K.; Hirokawa, Y.; Kanayama, K.; Matsuda, C.; Uchida, K.; Shiraishi, T.; Watanabe, M. Antifibrotic Agent Pirfenidone Suppresses Proliferation of Human Pancreatic Cancer Cells by Inducing G0/G1 Cell Cycle Arrest. Pharmacology 2019, 103, 250–256. [Google Scholar] [CrossRef]
- Kozono, S.; Ohuchida, K.; Eguchi, D.; Ikenaga, N.; Fujiwara, K.; Cui, L.; Mizumoto, K.; Tanaka, M. Pirfenidone Inhibits Pancreatic Cancer Desmoplasia by Regulating Stellate Cells. Cancer Res. 2013, 73, 2345–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suklabaidya, S.; Das, B.; Ali, S.A.; Jain, S.; Swaminathan, S.; Mohanty, A.K.; Panda, S.K.; Dash, P.; Chakraborty, S.; Batra, S.K.; et al. Characterization and use of HapT1-derived homologous tumors as a preclinical model to evaluate therapeutic efficacy of drugs against pancreatic tumor desmoplasia. Oncotarget 2016, 7, 41825–41842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, C.P.R.; Castro, I.; Caires, H.R.; Ferreira, D.; Cavadas, B.; Pereira, L.; Santos, L.L.; Oliveira, M.J.; Vasconcelos, M.H. Chitinase 3-like-1 and fibronectin in the cargo of extracellular vesicles shed by human macrophages influence pancreatic cancer cellular response to gemcitabine. Cancer Lett. 2021, 501, 210–223. [Google Scholar] [CrossRef]
- Ji, T.; Li, S.; Zhang, Y.; Lang, J.; Ding, Y.; Zhao, X.; Zhao, R.; Li, Y.; Shi, J.; Hao, J.; et al. An MMP-2 Responsive Liposome Integrating Antifibrosis and Chemotherapeutic Drugs for Enhanced Drug Perfusion and Efficacy in Pancreatic Cancer. ACS Appl. Mater. Interfaces 2016, 8, 3438–3445. [Google Scholar] [CrossRef]
- Ji, T.; Lang, J.; Wang, J.; Cai, R.; Zhang, Y.; Qi, F.; Zhang, L.; Zhao, X.; Wu, W.; Hao, J.; et al. Designing Liposomes to Suppress Extracellular Matrix Expression to Enhance Drug Penetration and Pancreatic Tumor Therapy. ACS Nano 2017, 11, 8668–8678. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Wu, J.; Niu, S.; Sun, T.; Li, F.; Bai, Y.; Jin, L.; Lin, L.; Shi, Q.; Zhu, L.-M.; et al. Biodegradable, pH-Sensitive Hollow Mesoporous Organosilica Nanoparticle (HMON) with Controlled Release of Pirfenidone and Ultrasound-Target-Microbubble-Destruction (UTMD) for Pancreatic Cancer Treatment. Theranostics 2019, 9, 6002–6018. [Google Scholar] [CrossRef] [PubMed]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. β2 Adrenergic-Neurotrophin Feedforward Loop Promotes Pancreatic Cancer. Cancer Cell 2018, 33, 75–90.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Ma, Q.; Shen, S.; Hu, H. Inhibition of Pancreatic Cancer Cell Proliferation by Propranolol Occurs Through Apoptosis Induction. Pancreas 2009, 38, 94–100. [Google Scholar] [CrossRef]
- Zhang, D.; Ma, Q.Y.; Hu, H.-T.; Zhang, M. β2-adrenergic antagonists suppress pancreatic cancer cell invasion by inhibiting CREB, NF-κB and AP-1. Cancer Biol. Ther. 2010, 10, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partecke, L.I.; Speerforck, S.; Kading, A.; Seubert, F.; Kühn, S.; Lorenz, E.; Schwandke, S.; Sendler, M.; Keßler, W.; Trung, D.N.; et al. Chronic stress increases experimental pancreatic cancer growth, reduces survival and can be antagonised by beta-adrenergic receptor blockade. Pancreatology 2016, 16, 423–433. [Google Scholar] [CrossRef]
- Li, M.; Xu, H. Fear stress enhanced xenograft pancreatic tumor growth through activating epithelial-mesenchymal transition. Pancreatology 2019, 19, 377–382. [Google Scholar] [CrossRef]
- Al-Wadei, H.A.; Al-Wadei, M.H.; Schuller, H.M. Prevention of pancreatic cancer by the beta-blocker propranolol. Anticancer Drugs 2009, 20, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Samaras, P.; Tusup, M.; Nguyen-Kim, T.D.L.; Seifert, B.; Bachmann, H.; von Moos, R.; Knuth, A.; Pascolo, S. Phase I study of a chloroquine–gemcitabine combination in patients with metastatic or unresectable pancreatic cancer. Cancer Chemother. Pharmacol. 2017, 80, 1005–1012. [Google Scholar] [CrossRef]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.-C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; Liotta, L.A.; et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef]
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef] [Green Version]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel with or Without Hydroxychloroquine on Patients with Advanced Pancreatic Cancer. JAMA Oncol. 2019, 5, 993. [Google Scholar] [CrossRef]
- Brunner, T.B.; Geiger, M.; Grabenbauer, G.G.; Lang-Welzenbach, M.; Mantoni, T.S.; Cavallaro, A.; Sauer, R.; Hohenberger, W.; McKenna, W.G. Phase I Trial of the Human Immunodeficiency Virus Protease Inhibitor Nelfinavir and Chemoradiation for Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2008, 26, 2699–2706. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Verma, V.; Ly, Q.P.; Lazenby, A.; Sasson, A.; Schwarz, J.K.; Meza, J.L.; Are, C.; Li, S.; Wang, S.; et al. Phase I trial of concurrent stereotactic body radiotherapy and nelfinavir for locally advanced borderline or unresectable pancreatic adenocarcinoma. Radiother. Oncol. 2019, 132, 55–62. [Google Scholar] [CrossRef]
- Wilson, J.M.; Fokas, E.; Dutton, S.J.; Patel, N.; Hawkins, M.A.; Eccles, C.; Chu, K.-Y.; Durrant, L.; Abraham, A.G.; Partridge, M.; et al. ARCII: A phase II trial of the HIV protease inhibitor Nelfinavir in combination with chemoradiation for locally advanced inoperable pancreatic cancer. Radiother. Oncol. 2016, 119, 306–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Verma, V.; Lazenby, A.; Ly, Q.P.; Berim, L.D.; Schwarz, J.K.; Madiyalakan, M.; Nicodemus, C.F.; Hollingsworth, M.A.; Meza, J.L.; et al. Phase I/II Trial of Neoadjuvant Oregovomab-based Chemoimmunotherapy Followed by Stereotactic Body Radiotherapy and Nelfinavir for Locally Advanced Pancreatic Adenocarcinoma. Am. J. Clin. Oncol. 2019, 42, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Strauss, V.Y.; Shaw, R.; Virdee, P.S.; Hurt, C.N.; Ward, E.; Tranter, B.; Patel, N.; Bridgewater, J.; Parsons, P.; Radhakrishna, G.; et al. Study protocol: A multi-centre randomised study of induction chemotherapy followed by capecitabine ± nelfinavir with high- or standard-dose radiotherapy for locally advanced pancreatic cancer (SCALOP-2). BMC Cancer 2019, 19, 121. [Google Scholar] [CrossRef]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy with FOLFIRINOX in Combination with Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer. JAMA Oncol. 2019, 5, 1020. [Google Scholar] [CrossRef]
- Braghiroli, M.I.; de Celis Ferrari, A.C.R.; Pfiffer, T.E.; Alex, A.K.; Nebuloni, D.; Carneiro, A.S.; Caparelli, F.; Senna, L.; Lobo, J.; Hoff, P.M.; et al. Phase II trial of metformin and paclitaxel for patients with gemcitabine-refractory advanced adenocarcinoma of the pancreas. Ecancermedicalscience 2015, 9, 563. [Google Scholar] [CrossRef] [Green Version]
- Kordes, S.; Pollak, M.N.; Zwinderman, A.H.; Mathôt, R.A.; Weterman, M.J.; Beeker, A.; Punt, C.J.; Richel, D.J.; Wilmink, J.W. Metformin in patients with advanced pancreatic cancer: A double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015, 16, 839–847. [Google Scholar] [CrossRef]
- Reni, M.; Dugnani, E.; Cereda, S.; Belli, C.; Balzano, G.; Nicoletti, R.; Liberati, D.; Pasquale, V.; Scavini, M.; Maggiora, P.; et al. (Ir)relevance of Metformin Treatment in Patients with Metastatic Pancreatic Cancer: An Open-Label, Randomized Phase II Trial. Clin. Cancer Res. 2016, 22, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Hüttner, F.J.; Rooman, I.; Bouche, G.; Knebel, P.; Hüsing, J.; Mihaljevic, A.L.; Hackert, T.; Strobel, O.; Büchler, M.W.; Diener, M.K. Pancreatic resection with perioperative drug repurposing of propranolol and etodolac: Trial protocol of the phase-II randomised placebo controlled PROSPER trial. BMJ Open 2020, 10, e040406. [Google Scholar] [CrossRef] [PubMed]
- Emery, P.; Suarez-Almazor, M. Rheumatoid arthritis. Clin. Evid. 2003, 1349–1371. [Google Scholar]
- Varghese, E.; Büsselberg, D. Auranofin, an Anti-Rheumatic Gold Compound, Modulates Apoptosis by Elevating the Intracellular Calcium Concentration ([Ca2+]i) in MCF-7 Breast Cancer Cells. Cancers 2014, 6, 2243–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an Old Drug for a Golden New Age. Drugs R D 2015, 15, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Hu, J.; Wu, S.; Wang, L.; Cao, X.; Zhang, X.; Dai, B.; Cao, M.; Shao, R.; Zhang, R.; et al. Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells. Oncotarget 2016, 7, 3548–3558. [Google Scholar] [CrossRef] [Green Version]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Jatoi, A.; Breitkopf, C.R.; Foster, N.R.; Block, M.S.; Grudem, M.; Hendrickson, A.W.; Carlson, R.E.; Barrette, B.; Karlin, N.; Fields, A.P. A Mixed-Methods Feasibility Trial of Protein Kinase C Iota Inhibition with Auranofin in Asymptomatic Ovarian Cancer Patients. Oncology 2015, 88, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Snyder, G.L.; Vanover, K.E. Dopamine Targeting Drugs for the Treatment of Schizophrenia: Past, Present and Future. Curr. Top. Med. Chem. 2016, 16, 3385–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendouei, N.; Saghafi, F.; Shadfar, F.; Hosseinimehr, S.J. Molecular mechanisms of anti-psychotic drugs for improvement of cancer treatment. Eur. J. Pharmacol. 2019, 856, 172402. [Google Scholar] [CrossRef]
- Xu, S.; Furukawa, T.; Kanai, N.; Sunamura, M.; Horii, A. Abrogation of DUSP6 by hypermethylation in human pancreatic cancer. J. Hum. Genet. 2005, 50, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef] [Green Version]
- Farrell, A.S.; Allen-Petersen, B.; Daniel, C.J.; Wang, X.; Wang, Z.; Rodriguez, S.; Impey, S.; Oddo, J.; Vitek, M.P.; Lopez, C.; et al. Targeting Inhibitors of the Tumor Suppressor PP2A for the Treatment of Pancreatic Cancer. Mol. Cancer Res. 2014, 12, 924–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.E.; Parker, R.; Tsai, C.M.; Baltz, J.; Miller, M.J.; Shoemaker, R.; Phelps, R.; Bastian, A.; Stocker, J.; Phares, J.; et al. Phase I trial of dihydrolenperone in lung cancer patients: A novel compound within vitro activity against lung cancer. Investig. New Drugs 1993, 11, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Vural, G.; Yardimci, M.; Kocak, M.; Yasar, T.Ö.; Kurt, A.; Harem, I.S.; Carradori, S.; Sciamanna, I.; Siles-Lucas, M.; Fabiani, M.; et al. Efficacy of novel albendazole salt formulations against secondary cystic echinococcosis in experimentally infected mice. Parasitology 2020, 147, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Son, D.-S.; Lee, E.-S.; Adunyah, S.E. The Antitumor Potentials of Benzimidazole Anthelmintics as Repurposing Drugs. Immune Netw. 2020, 20, e29. [Google Scholar] [CrossRef] [PubMed]
- Florio, R.; Carradori, S.; Veschi, S.; Brocco, D.; Di Genni, T.; Cirilli, R.; Casulli, A.; Cama, A.; De Lellis, L. Screening of Benzimidazole-Based Anthelmintics and Their Enantiomers as Repurposed Drug Candidates in Cancer Therapy. Pharmaceuticals 2021, 14, 372. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Weng, Z.; Xu, C. Albendazole suppresses cell proliferation and migration and induces apoptosis in human pancreatic cancer cells. Anticancer Drugs 2020, 31, 431–439. [Google Scholar] [CrossRef]
- Pourgholami, M.H.; Szwajcer, M.; Chin, M.; Liauw, W.; Seef, J.; Galettis, P.; Morris, D.L.; Links, M. Phase I clinical trial to determine maximum tolerated dose of oral albendazole in patients with advanced cancer. Cancer Chemother. Pharmacol. 2010, 65, 597–605. [Google Scholar] [CrossRef]
- Morris, D.L.; Jourdan, J.-L.; Pourgholami, M.H. Pilot Study of Albendazole in Patients with Advanced Malignancy. Oncology 2001, 61, 42–46. [Google Scholar] [CrossRef]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde Dehydrogenase Inhibitors: A Comprehensive Review of the Pharmacology, Mechanism of Action, Substrate Specificity, and Clinical Application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Li, X.; Ren, Y.; Zhang, X. Disulfiram: A novel repurposed drug for cancer therapy. Cancer Chemother. Pharmacol. 2021, 87, 159–172. [Google Scholar] [CrossRef]
- Rinkenbaugh, A.; Baldwin, A. The NF-κB Pathway and Cancer Stem Cells. Cells 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Kelley, K.C.; Grossman, K.F.; Brittain-Blankenship, M.; Thorne, K.M.; Akerley, W.L.; Terrazas, M.C.; Kosak, K.M.; Boucher, K.M.; Buys, S.S.; McGregor, K.A.; et al. A Phase 1 dose-escalation study of disulfiram and copper gluconate in patients with advanced solid tumors involving the liver using S-glutathionylation as a biomarker. BMC Cancer 2021, 21, 510. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, I.; Alfarouk, K.O.; Reshkin, S.J.; Ibrahim, M.E. Doxycycline as Potential Anti-cancer Agent. Anticancer Agents Med. Chem. 2018, 17, 1617–1623. [Google Scholar] [CrossRef]
- Moullan, N.; Mouchiroud, L.; Wang, X.; Ryu, D.; Williams, E.G.; Mottis, A.; Jovaisaite, V.; Frochaux, M.V.; Quiros, P.M.; Deplancke, B.; et al. Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep. 2015, 10, 1681–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, R.; Harrison, H.; Hulit, J.; Smith, D.L.; Lisanti, M.P.; Sotgia, F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget 2014, 5, 11029–11037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huie, M.; Oettel, K.; Van Ummersen, L.; Kim, K.M.; Zhang, Y.; Staab, M.J.; Horvath, D.; Marnocha, R.; Douglas, J.; Drezen, A.; et al. Phase II study of interferon-alpha and doxycycline for advanced renal cell carcinoma. Investig. New Drugs 2006, 24, 255–260. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, W.A.; Jiang, C.; Guan, M. Anti-HIV drugs for cancer therapeutics: Back to the future? Lancet Oncol. 2009, 10, 61–71. [Google Scholar] [CrossRef]
- Kaushik, I.; Ramachandran, S.; Prasad, S.; Srivastava, S.K. Drug rechanneling: A novel paradigm for cancer treatment. Semin. Cancer Biol. 2021, 68, 279–290. [Google Scholar] [CrossRef]
- Van den Berg-Wolf, M.; Hullsiek, K.H.; Peng, G.; Kozal, M.J.; Novak, R.M.; Chen, L.; Crane, L.R.; MacArthur, R.D. Virologic, Immunologic, Clinical, Safety, and Resistance Outcomes from a Long-Term Comparison of Efavirenz-Based Versus Nevirapine-Based Antiretroviral Regimens as Initial Therapy in HIV-1—Infected Persons. HIV Clin. Trials 2008, 9, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Landriscina, M.; Fabiano, A.; Altamura, S.; Bagalà, C.; Piscazzi, A.; Cassano, A.; Spadafora, C.; Giorgino, F.; Barone, C.; Cignarelli, M. Reverse Transcriptase Inhibitors Down-Regulate Cell Proliferation in Vitro and in Vivo and Restore Thyrotropin Signaling and Iodine Uptake in Human Thyroid Anaplastic Carcinoma. J. Clin. Endocrinol. Metab. 2005, 90, 5663–5671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Zhang, F.; Sun, Z.; Liu, P.; Zhang, X.; Ye, Y.; Cai, B.; Walsh, M.J.; Ren, X.; Hao, X.; et al. LINE-1 Retrotransposition Promotes the Development and Progression of Lung Squamous Cell Carcinoma by Disrupting the Tumor-Suppressor Gene FGGY. Cancer Res. 2019, 79, 4453–4465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.-A.; Bae, S.-S.; Fernandes, A.; Wu, J.; Muschel, R.J.; McKenna, W.G.; Birnbaum, M.J.; Bernhard, E.J. Selective Inhibition of Ras, Phosphoinositide 3 Kinase, and Akt Isoforms Increases the Radiosensitivity of Human Carcinoma Cell Lines. Cancer Res. 2005, 65, 7902–7910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, T.B.; Cengel, K.A.; Hahn, S.M.; Wu, J.; Fraker, D.L.; McKenna, W.G.; Bernhard, E.J. Pancreatic Cancer Cell Radiation Survival and Prenyltransferase Inhibition: The Role of K-Ras. Cancer Res. 2005, 65, 8433–8441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.K.; Cerniglia, G.J.; Mick, R.; McKenna, W.G.; Muschel, R.J. HIV Protease Inhibitors Block Akt Signaling and Radiosensitize Tumor Cells Both In vitro and In vivo. Cancer Res. 2005, 65, 8256–8265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauschenbach, L.; Wieland, A.; Reinartz, R.; Kebir, S.; Till, A.; Oppong, M.D.; Dobersalske, C.; Ullrich, V.; Ahmad, A.; Jabbarli, R.; et al. Drug repositioning of antiretroviral ritonavir for combinatorial therapy in glioblastoma. Eur. J. Cancer 2020, 140, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Hisatake, Y.; Takeuchi, T.; Ohtsuki, Y.; Yang, Y.; Said, J.W.; Taguchi, H.; Koeffler, H.P. HIV-1 Protease Inhibitor, Ritonavir. Cancer Res. 2004, 64, 7426–7431. [Google Scholar] [CrossRef] [Green Version]
- Isono, M.; Sato, A.; Asano, T.; Okubo, K.; Asano, T. Delanzomib Interacts with Ritonavir Synergistically to Cause Endoplasmic Reticulum Stress in Renal Cancer Cells. Anticancer Res. 2018, 38, 3493–3500. [Google Scholar] [CrossRef]
- Blumenthal, G.M.; Gills, J.J.; Ballas, M.S.; Bernstein, W.B.; Komiya, T.; Dechowdhury, R.; Morrow, B.; Root, H.; Chun, G.; Helsabeck, C.; et al. A phase I trial of the HIV protease inhibitor nelfinavir in adults with solid tumors. Oncotarget 2014, 5, 8161–8172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Virdee, P.; Shaw, R.; Bridgewater, J.; Radhakrishna, G.; Falk, S.; Scott-Brown, M.; Strauss, V.Y.; Brooks, C.; Gillmore, R.; et al. SCALOP-2: A multi-centre randomised trial of induction chemotherapy followed by capecitabine +/-nelfinavir with high or standard dose radiotherapy for locally advanced pancreatic cancer (LAPC): Results of stage 1 the non-randomised dose-finding component. Ann. Oncol. 2018, 29, viii243. [Google Scholar] [CrossRef]
- Nirk, E.L.; Reggiori, F.; Mauthe, M. Hydroxychloroquine in rheumatic autoimmune disorders and beyond. EMBO Mol. Med. 2020, 12, e12476. [Google Scholar] [CrossRef] [PubMed]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)—chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renz, B.; D’Haese, J.; Werner, J.; Westphalen, C.; Ilmer, M. Repurposing Established Compounds to Target Pancreatic Cancer Stem Cells (CSCs). Med. Sci. 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.; Lee, S. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and Pharmacodynamic Study of Autophagy Inhibition Using Hydroxychloroquine in Patients With Metastatic Pancreatic Adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef] [Green Version]
- Bao, P.; Potter, D.; Eisenberg, D.P.; Lenzner, D.; Zeh, H.J.; Lee, K.K.W.; Hughes, S.J.; Sanders, M.K.; Young, J.L.; Moser, A.J. Validation of a prediction rule to maximize curative (R0) resection of early-stage pancreatic adenocarcinoma. HPB 2009, 11, 606–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- De Beule, K.; Van Gestel, J. Pharmacology of Itraconazole. Drugs 2001, 61, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Pounds, R.; Leonard, S.; Dawson, C.; Kehoe, S. Repurposing itraconazole for the treatment of cancer. Oncol. Lett. 2017, 14, 2587–2597. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a Commonly Used Antifungal that Inhibits Hedgehog Pathway Activity and Cancer Growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef] [Green Version]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Castillo, C.F.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.-C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsubamoto, H.; Ueda, T.; Inoue, K.; Sakata, K.; Shibahara, H.; Sonoda, T. Repurposing itraconazole as an anticancer agent. Oncol. Lett. 2017, 14, 1240–1246. [Google Scholar] [CrossRef] [Green Version]
- Tsubamoto, H.; Sonoda, T.; Ikuta, S.; Tani, S.; Inoue, K.; Yamanaka, N. Combination Chemotherapy with Itraconazole for Treating Metastatic Pancreatic Cancer in the Second-line or Additional Setting. Anticancer Res. 2015, 35, 4191–4196. [Google Scholar] [PubMed]
- Rahma, O.E.; Duffy, A.; Liewehr, D.J.; Steinberg, S.M.; Greten, T.F. Second-line treatment in advanced pancreatic cancer: A comprehensive analysis of published clinical trials. Ann. Oncol. 2013, 24, 1972–1979. [Google Scholar] [CrossRef] [PubMed]
- Simpson, K.L.; McClellan, K.J. Losartan. Drugs Aging 2000, 16, 227–250. [Google Scholar] [CrossRef]
- Leung, P.S.; Chappell, M.C. A local pancreatic renin-angiotensin system: Endocrine and exocrine roles. Int. J. Biochem. Cell Biol. 2003, 35, 838–846. [Google Scholar] [CrossRef]
- George, A.J.; Thomas, W.G.; Hannan, R.D. The renin–angiotensin system and cancer: Old dog, new tricks. Nat. Rev. Cancer 2010, 10, 745–759. [Google Scholar] [CrossRef]
- Tamarat, R.; Silvestre, J.-S.; Durie, M.; Levy, B.I. Angiotensin II angiogenic effect in vivo involves vascular endothelial growth factor- and inflammation-related pathways. Lab. Investig. 2002, 82, 747–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arafat, H.A.; Gong, Q.; Chipitsyna, G.; Rizvi, A.; Saa, C.T.; Yeo, C.J. Antihypertensives as Novel Antineoplastics: Angiotensin-I-Converting Enzyme Inhibitors and Angiotensin II Type 1 Receptor Blockers in Pancreatic Ductal Adenocarcinoma. J. Am. Coll. Surg. 2007, 204, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Hauge, A.; Rofstad, E.K. Antifibrotic therapy to normalize the tumor microenvironment. J. Transl. Med. 2020, 18, 207. [Google Scholar] [CrossRef] [PubMed]
- Khoshghamat, N.; Jafari, N.; Toloue-pouya, V.; Azami, S.; Mirnourbakhsh, S.H.; Khazaei, M.; Ferns, G.A.; Rajabian, M.; Avan, A. The therapeutic potential of renin-angiotensin system inhibitors in the treatment of pancreatic cancer. Life Sci. 2021, 270, 119118. [Google Scholar] [CrossRef]
- Yang, J.; Yang, X.; Gao, L.; Zhang, J.; Yi, C.; Huang, Y. The role of the renin-angiotensin system inhibitors in malignancy: A review. Am. J. Cancer Res. 2021, 11, 884–897. [Google Scholar]
- Liu, W.-B. Effects of angiotensin II receptor antagonist, Losartan on the apoptosis, proliferation and migration of the human pancreatic stellate cells. World J. Gastroenterol. 2005, 11, 6489. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koay, E.J.; Truty, M.J.; Cristini, V.; Thomas, R.M.; Chen, R.; Chatterjee, D.; Kang, Y.; Bhosale, P.R.; Tamm, E.P.; Crane, C.H.; et al. Transport properties of pancreatic cancer describe gemcitabine delivery and response. J. Clin. Investig. 2014, 124, 1525–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Naxerova, K.; Pinter, M.; Incio, J.; Lee, H.; Shigeta, K.; Ho, W.W.; Crain, J.A.; Jacobson, A.; Michelakos, T.; et al. Use of Angiotensin System Inhibitors Is Associated with Immune Activation and Longer Survival in Nonmetastatic Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 5959–5969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Mir, M.-Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide Inhibits Cell Respiration via an Indirect Effect Targeted on the Respiratory Chain Complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.-H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Ray, L.B.; Sturgill, T.W. Insulin-stimulated microtubule associated protein kinase is detectable by analytical gel chromatography as a 35-kDa protein in myocytes, adipocytes, and hepatocytes. Arch. Biochem. Biophys. 1988, 262, 307–313. [Google Scholar] [CrossRef]
- Lipner, M.B.; Marayati, R.; Deng, Y.; Wang, X.; Raftery, L.; O’Neil, B.H.; Yeh, J.J. Metformin Treatment Does Not Inhibit Growth of Pancreatic Cancer Patient-Derived Xenografts. PLoS ONE 2016, 11, e0147113. [Google Scholar] [CrossRef]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, R.; Tomosugi, M.; Horinaka, M.; Sowa, Y.; Sakai, T. Metformin Causes G1-Phase Arrest via Down-Regulation of MiR-221 and Enhances TRAIL Sensitivity through DR5 Up-Regulation in Pancreatic Cancer Cells. PLoS ONE 2015, 10, e0125779. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Permert, J.; Adrian, T.E.; Jacobsson, P.; Jorfelt, L.; Fruin, A.B.; Larsson, J. Is profound peripheral insulin resistance in patients with pancreatic cancer caused by a tumor-associated factor? Am. J. Surg. 1993, 165, 61–67. [Google Scholar] [CrossRef]
- Currie, C.J.; Poole, C.D.; Jenkins-Jones, S.; Gale, E.A.M.; Johnson, J.A.; Morgan, C.L. Mortality after Incident Cancer in People with and without Type 2 Diabetes: Impact of metformin on survival. Diabetes Care 2012, 35, 299–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadeghi, N.; Abbruzzese, J.L.; Yeung, S.-C.J.; Hassan, M.; Li, D. Metformin Use Is Associated with Better Survival of Diabetic Patients with Pancreatic Cancer. Clin. Cancer Res. 2012, 18, 2905–2912. [Google Scholar] [CrossRef] [Green Version]
- Hwang, A.L.; Haynes, K.; Hwang, W.-T.; Yang, Y.-X. Metformin and Survival in Pancreatic Cancer. Pancreas 2013, 42, 1054–1059. [Google Scholar] [CrossRef] [Green Version]
- Cerullo, M.; Gani, F.; Chen, S.Y.; Canner, J.; Pawlik, T.M. Metformin Use Is Associated with Improved Survival in Patients Undergoing Resection for Pancreatic Cancer. J. Gastrointest. Surg. 2016, 20, 1572–1580. [Google Scholar] [CrossRef]
- Chaiteerakij, R.; Petersen, G.M.; Bamlet, W.R.; Chaffee, K.G.; Zhen, D.B.; Burch, P.A.; Leof, E.R.; Roberts, L.R.; Oberg, A.L. Metformin Use and Survival of Patients With Pancreatic Cancer: A Cautionary Lesson. J. Clin. Oncol. 2016, 34, 1898–1904. [Google Scholar] [CrossRef]
- Beg, M.S.; Gupta, A.; Sher, D.; Ali, S.; Khan, S.; Gao, A.; Stewart, T.; Ahn, C.; Berry, J.; Mortensen, E.M. Impact of Concurrent Medication Use on Pancreatic Cancer Survival—SEER-Medicare Analysis. Am. J. Clin. Oncol. 2018, 41, 766–771. [Google Scholar] [CrossRef]
- Frouws, M.A.; Mulder, B.G.S.; Bastiaannet, E.; Zanders, M.M.J.; van Herk-Sukel, M.P.P.; de Leede, E.M.; Bonsing, B.A.; Mieog, J.S.D.; Van de Velde, C.J.H.; Liefers, G.-J. No association between metformin use and survival in patients with pancreatic cancer. Medicine 2017, 96, e6229. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Scragg, R.; Pandol, S.J.; Goodarzi, M.O.; Petrov, M.S. Antidiabetic Medications and Mortality Risk in Individuals With Pancreatic Cancer–Related Diabetes and Postpancreatitis Diabetes: A Nationwide Cohort Study. Diabetes Care 2019, 42, 1675–1683. [Google Scholar] [CrossRef] [PubMed]
- Toriola, A.T.; Luo, S.; Thomas, T.S.; Drake, B.F.; Chang, S.-H.; Sanfilippo, K.M.; Carson, K.R. Metformin Use and Pancreatic Cancer Survival among Non-Hispanic White and African American U.S. Veterans with Diabetes Mellitus. Cancer Epidemiol. Biomark. Prev. 2020, 29, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, T.; Liu, Z.; Gou, S.; Wang, C. The effect of metformin on survival of patients with pancreatic cancer: A meta-analysis. Sci. Rep. 2017, 7, 5825. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.-T.; Li, B.; Liu, F.-R.; Zhang, M.-C.; Wang, Q.; Li, Y.-Y.; Xu, C.; Liu, Y.-H.; Yao, Y.; Li, D. Metformin is associated with survival benefit in pancreatic cancer patients with diabetes: A systematic review and meta-analysis. Oncotarget 2017, 8, 25242–25250. [Google Scholar] [CrossRef] [PubMed]
- Jian-Yu, E.; Graber, J.M.; Lu, S.-E.; Lin, Y.; Lu-Yao, G.; Tan, X.-L. Effect of Metformin and Statin Use on Survival in Pancreatic Cancer Patients: A Systematic Literature Review and Meta-analysis. Curr. Med. Chem. 2018, 25, 2595–2607. [Google Scholar] [CrossRef]
- Morgillo, F.; Sasso, F.C.; Della Corte, C.M.; Vitagliano, D.; D’Aiuto, E.; Troiani, T.; Martinelli, E.; De Vita, F.; Orditura, M.; De Palma, R.; et al. Synergistic Effects of Metformin Treatment in Combination with Gefitinib, a Selective EGFR Tyrosine Kinase Inhibitor, in LKB1 Wild-type NSCLC Cell Lines. Clin. Cancer Res. 2013, 19, 3508–3519. [Google Scholar] [CrossRef] [Green Version]
- Lau, Y.-K.I.; Du, X.; Rayannavar, V.; Hopkins, B.; Shaw, J.; Bessler, E.; Thomas, T.; Pires, M.M.; Keniry, M.; Parsons, R.E.; et al. Metformin and erlotinib synergize to inhibit basal breast cancer. Oncotarget 2014, 5, 10503–10517. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Lanza-Jacoby, S. Metformin decreases growth of pancreatic cancer cells by decreasing reactive oxygen species: Role of NOX4. Biochem. Biophys. Res. Commun. 2015, 465, 41–46. [Google Scholar] [CrossRef]
- Broadhurst, P.J.; Hart, A.R. Metformin as an Adjunctive Therapy for Pancreatic Cancer: A Review of the Literature on Its Potential Therapeutic Use. Dig. Dis. Sci. 2018, 63, 2840–2852. [Google Scholar] [CrossRef] [Green Version]
- Suissa, S.; Azoulay, L. Metformin and the Risk of Cancer: Time-related biases in observational studies. Diabetes Care 2012, 35, 2665–2673. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, O.M.; Groenwold, R.H.H. When observational studies can give wrong answers: The potential of immortal time bias. Eur. J. Endocrinol. 2021, 184, E1–E4. [Google Scholar] [CrossRef]
- Dong, Y.-W.; Shi, Y.-Q.; He, L.-W.; Cui, X.-Y.; Su, P.-Z. Effects of metformin on survival outcomes of pancreatic cancer: A meta-analysis. Oncotarget 2017, 8, 55478–55488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.-Q.; Zhou, X.-C.; Du, P.; Yin, M.-Y.; Xu, L.; Chen, W.-J.; Xu, C.-F. Relationships are between metformin use and survival in pancreatic cancer patients concurrent with diabetes. Medicine 2020, 99, e21687. [Google Scholar] [CrossRef]
- Armando, R.; Gomez, D.M.; Gomez, D. New drugs are not enough-drug repositioning in oncology: An update. Int. J. Oncol. 2020, 56, 651–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kos, J.; Mitrović, A. Nitroxoline: Repurposing its antimicrobial to antitumor application. Acta Biochim. Pol. 2019, 66, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, S.; Yang, D.; Pan, K.; Li, L.; Yuan, S. Preclinical pharmacodynamic evaluation of antibiotic nitroxoline for anticancer drug repurposing. Oncol. Lett. 2016, 11, 3265–3272. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Matsui, Y.; Bhat, S.; Nacev, B.A.; Xu, J.; Bhang, H.C.; Dhara, S.; Han, K.C.; Chong, C.R.; Pomper, M.G.; et al. Effect of Nitroxoline on Angiogenesis and Growth of Human Bladder Cancer. J. Natl. Cancer Inst. 2010, 102, 1855–1873. [Google Scholar] [CrossRef] [Green Version]
- Veschi, S.; Carradori, S.; De Lellis, L.; Florio, R.; Brocco, D.; Secci, D.; Guglielmi, P.; Spano, M.; Sobolev, A.P.; Cama, A. Synthesis and evaluation of a large library of nitroxoline derivatives as pancreatic cancer antiproliferative agents. J. Enzym. Inhib. Med. Chem. 2020, 35, 1331–1344. [Google Scholar] [CrossRef]
- Dashtipour, K.; Gupta, F.; Hauser, R.A.; Karunapuzha, C.A.; Morgan, J.C. Pimavanserin Treatment for Parkinson’s Disease Psychosis in Clinical Practice. Parkinsons. Dis. 2021, 2021, 2603641. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, R.; Bachet, J.-B.; Calomme, A.; Demetter, P.; Delpero, J.R.; Svrcek, M.; Cros, J.; Bardier-Dupas, A.; Puleo, F.; Monges, G.; et al. Sonic Hedgehog and Gli1 Expression Predict Outcome in Resected Pancreatic Adenocarcinoma. Clin. Cancer Res. 2015, 21, 1215–1224. [Google Scholar] [CrossRef] [Green Version]
- Azuma, A. Pirfenidone treatment of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2012, 6, 107–114. [Google Scholar] [CrossRef]
- Iyer, S.N.; Gurujeyalakshmi, G.; Giri, S.N. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1999, 291, 367–373. [Google Scholar]
- Conte, E.; Gili, E.; Fagone, E.; Fruciano, M.; Iemmolo, M.; Vancheri, C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur. J. Pharm. Sci. 2014, 58, 13–19. [Google Scholar] [CrossRef] [PubMed]
- García, L.; Hernández, I.; Sandoval, A.; Salazar, A.; Garcia, J.; Vera, J.; Grijalva, G.; Muriel, P.; Margolin, S.; Armendariz-Borunda, J. Pirfenidone effectively reverses experimental liver fibrosis. J. Hepatol. 2002, 37, 797–805. [Google Scholar] [CrossRef]
- Marwitz, S.; Turkowski, K.; Nitschkowski, D.; Weigert, A.; Brandenburg, J.; Reiling, N.; Thomas, M.; Reck, M.; Drömann, D.; Seeger, W.; et al. The Multi-Modal Effect of the Anti-fibrotic Drug Pirfenidone on NSCLC. Front. Oncol. 2020, 9, 1550. [Google Scholar] [CrossRef] [PubMed]
- Merika, E.E.; Syrigos, K.N.; Saif, M.W. Desmoplasia in Pancreatic Cancer. Can We Fight It? Gastroenterol. Res. Pract. 2012, 2012, 781765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.-J. The Role of the Extracellular Matrix in Cancer Stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef]
- De Lellis, L.; Florio, R.; Di Bella, M.C.; Brocco, D.; Guidotti, F.; Tinari, N.; Grassadonia, A.; Lattanzio, R.; Cama, A.; Veschi, S. Exosomes as Pleiotropic Players in Pancreatic Cancer. Biomedicines 2021, 9, 275. [Google Scholar] [CrossRef]
- Babovic-Vuksanovic, D.; Ballman, K.; Michels, V.; McGrann, P.; Lindor, N.; King, B.; Camp, J.; Micic, V.; Babovic, N.; Carrero, X.; et al. Phase II trial of pirfenidone in adults with neurofibromatosis type 1. Neurology 2006, 67, 1860–1862. [Google Scholar] [CrossRef]
- Widemann, B.C.; Babovic-Vuksanovic, D.; Dombi, E.; Wolters, P.L.; Goldman, S.; Martin, S.; Goodwin, A.; Goodspeed, W.; Kieran, M.W.; Cohen, B.; et al. Phase II trial of pirfenidone in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Pediatr. Blood Cancer 2014, 61, 1598–1602. [Google Scholar] [CrossRef]
- Borden, P.; Houtz, J.; Leach, S.D.; Kuruvilla, R. Sympathetic Innervation during Development Is Necessary for Pancreatic Islet Architecture and Functional Maturation. Cell Rep. 2013, 4, 287–301. [Google Scholar] [CrossRef]
- Qiao, G.; Chen, M.; Bucsek, M.J.; Repasky, E.A.; Hylander, B.L. Adrenergic Signaling: A Targetable Checkpoint Limiting Development of the Antitumor Immune Response. Front. Immunol. 2018, 9, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forget, P.; Aguirre, J.A.; Bencic, I.; Borgeat, A.; Cama, A.; Condron, C.; Eintrei, C.; Eroles, P.; Gupta, A.; Hales, T.G.; et al. How Anesthetic, Analgesic and Other Non-Surgical Techniques During Cancer Surgery Might Affect Postoperative Oncologic Outcomes: A Summary of Current State of Evidence. Cancers 2019, 11, 592. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.; Shi, S.; Xu, J.; Zhang, B.; Qin, Y.; Ji, S.; Xu, W.; Liu, J.; Liu, L.; Liu, C.; et al. New insights into perineural invasion of pancreatic cancer: More than pain. Biochim. Biophys. Acta Rev. Cancer 2016, 1865, 111–122. [Google Scholar] [CrossRef]
- Ceyhan, G.O.; Bergmann, F.; Kadihasanoglu, M.; Altintas, B.; Demir, I.E.; Hinz, U.; Müller, M.W.; Giese, T.; Büchler, M.W.; Giese, N.A.; et al. Pancreatic Neuropathy and Neuropathic Pain—A Comprehensive Pathomorphological Study of 546 Cases. Gastroenterology 2009, 136, 177–186.e1. [Google Scholar] [CrossRef] [PubMed]
- Liebl, F.; Demir, I.E.; Mayer, K.; Schuster, T.; D’Haese, J.G.; Becker, K.; Langer, R.; Bergmann, F.; Wang, K.; Rosenberg, R.; et al. The Impact of Neural Invasion Severity in Gastrointestinal Malignancies. Ann. Surg. 2014, 260, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Bapat, A.A.; Munoz, R.M.; Von Hoff, D.D.; Han, H. Blocking Nerve Growth Factor Signaling Reduces the Neural Invasion Potential of Pancreatic Cancer Cells. PLoS ONE 2016, 11, e0165586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoni, M.H.; Lutgendorf, S.K.; Cole, S.W.; Dhabhar, F.S.; Sephton, S.E.; McDonald, P.G.; Stefanek, M.; Sood, A.K. The influence of bio-behavioural factors on tumour biology: Pathways and mechanisms. Nat. Rev. Cancer 2006, 6, 240–248. [Google Scholar] [CrossRef]
- Clark, K.L.; Loscalzo, M.; Trask, P.C.; Zabora, J.; Philip, E.J. Psychological distress in patients with pancreatic cancer-an understudied group. Psycho-Oncology 2010, 19, 1313–1320. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [Green Version]
- Wrobel, L.J.; Bod, L.; Lengagne, R.; Kato, M.; Prévost-Blondel, A.; Le Gal, F.-A. Propranolol induces a favourable shift of anti-tumor immunity in a murine spontaneous model of melanoma. Oncotarget 2016, 7, 77825–77837. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Bao, X.; Hu, M.; Chang, H.; Jiao, M.; Cheng, J.; Xie, L.; Huang, Q.; Li, F.; Li, C.-Y. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 2020, 588, 693–698. [Google Scholar] [CrossRef]
- Sun, W.; Sanderson, P.E.; Zheng, W. Drug combination therapy increases successful drug repositioning. Drug Discov. Today 2016, 21, 1189–1195. [Google Scholar] [CrossRef] [Green Version]
- Breckenridge, A.; Jacob, R. Overcoming the legal and regulatory barriers to drug repurposing. Nat. Rev. Drug Discov. 2019, 18, 1–2. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug Name | Original Indications | Type of Study | Main Results of the Study (Proposed Molecular Targets/Mechanisms of Action in PC) | Refs. |
|---|---|---|---|---|
| Auranofin | Rheumatoid arthritis | In vitro/ animal models | Inhibition of TrxR1 and HIF1α leading to decreased antioxidant activity within PC cells, followed by apoptosis; antitumor effect at the primary site and suppression of distant organ metastasis in PC orthotopic mouse models | [20] |
| In vitro/ animal models | Preferential cytotoxicity towards PC cells under nutrient-deprived conditions through ROS accumulation and subsequent induction of apoptosis by caspase 3/7 activation and proteolytic PARP cleavage; suppression of tumor growth in a PC xenograft model | [21] | ||
| In vitro/ animal models | Increase in mitochondrial ROS and apoptosis, together with inhibition of autophagic flux when used in combination with an engineered human cyst(e)inase; suppression of tumor growth in PC xenografts by combined treatment | [22] | ||
| Haloperidol | Psychosis | In vitro/ animal models | Blockade of DRD2 leading to inhibition of proliferation by promoting ER stress, impairment of migration and cell cycle progression, induction of apoptosis in PC cells; reduction in tumor size and metastatic dissemination in mice with orthotopic xenograft PC tumors | [23] |
| In vitro | Inhibition of PC cell viability by restoring the expression of DUSP6 gene through epigenetic modification of its transcriptional regulation | [24] | ||
| Penfluridol | Psychosis | In vitro/ animal models | Inhibition of PC cell proliferation by inducing ER stress, leading to autophagy and apoptosis; reduction in tumor growth in xenograft and orthotopic PC models | [25,26] |
| In vitro | Inhibition of proliferation, promotion of cell cycle arrest and apoptosis in PC cells through the activation of PP2A; synergistic effects on the viability of gemcitabine-resistant and gemcitabine-sensitive PC cells when used in combination with gemcitabine | [27] | ||
| In vitro/ animal models | Inhibition of JAK2–STAT3 and ERK/AKT signaling, through binding with the JAK2-binding site in PRLR, leading to suppression of colony and spheroid formation, together with induction of autophagy; slowdown of tumor growth in different xenograft mouse models of PC | [28] | ||
| Parbendazole | Intestinal parasitic infections | In vitro | Inhibition of proliferation, clonogenicity and migration of PC cells, together with promotion of apoptosis, cell cycle perturbation and DNA damage response through the alteration of microtubule organization, formation of irregular mitotic spindles and appearance of polyploid cells; synergistic effects on PC cell viability when used in combination with gemcitabine | [29] |
| Disulfiram | Chronic alcoholism | In vitro/ animal models | A complex formed by a disulfiram metabolite bound to copper (DDTC-Cu) induces inhibition of PC cell proliferation through the impairment of proteasome activity; reduction in tumor growth in xenograft mouse models of PC, with accumulation of ubiquitinated proteins, up-regulation of p27 and down-regulation of NF-kB expression in tumor tissues | [30] |
| In vitro/ animal models | A complex formed by a disulfiram metabolite bound to copper (DDTC-Cu) induces depletion of pre-existing CSCs and radiation-induced CSCs in PC cells through NF-kB-stemness gene pathway downregulation; antitumor effects in syngeneic mouse PC models, with reduced sphere formation, when used in combination with 5-FU and radiotherapy | [31] | ||
| In vitro/ animal models | Promotion of aponecrosis death pathways in K-Ras mutant PC cell lines, through intracellular ATP depletion and ROS release, when used in combination with arsenic trioxide and ascorbic acid; reduction in tumor growth in mice with PANC-1 xenografts by using the three-drug combination | [32] | ||
| In vitro | Induction of autophagy-dependent apoptosis in PC cells through the activation of the ER stress/IRE1α-XBP1 pathway, by a direct interaction with IRE1α, or by an indirect mechanism involving the inhibition of the NPL4 cofactor of the p97/VCP segregase and of proteasome, along with the production of ROS | [33] | ||
| Doxycycline | Bacterial infections | In vitro/ animal models | Inhibition of PC cell proliferation by activating proapoptotic genes and by suppressing antiapoptotic genes, perturbating cell cycle and inhibiting the expression of the proangiogenic IL-8; reduction in tumor growth by 80% in a xenograft mouse model of PC | [34,35] |
| In vitro | Inhibition of tumorsphere formation in PC cells, without affecting the viability of both bulk cancer cells and normal fibroblasts | [36] | ||
| In vitro | Inhibition of mitochondrial protein synthesis leading to decreased PC cell proliferation, through ATP depletion, when used in combination with gemcitabine; enhancement of gemcitabine-induced apoptosis by decreasing mitochondrial membrane potential and fostering ROS production | [37] | ||
| In vitro/ animal models | Inhibition of PC cell growth, migration, invasion and tumorsphere formation through the down-regulation of PAR1/FAK/PI3K/AKT signaling pathway; synergistic effects on PC cell viability and reduction in tumor growth by 80.5% in subcutaneous Panc-1 xenografts models, when used in combination with 5-FU, with the increase in E-Cadherin expression and the decrease in Vimentin and CD133 expression | [38] | ||
| Efavirenz | HIV infection | In vitro | Impairment of clonogenicity and induction of apoptosis in distinct PC cell lines | [39] |
| In vitro | Inhibition of PC cell proliferation through a mechanism involving ROS production and mitochondrial membrane depolarization, along with phosphorylation of both ERK1/2 and p38 MAPK stress pathways, when used in combination with radiation | [40] | ||
| Nelfinavir | HIV infection | In vitro/ animal models | Inhibition of Akt phosphorylation in PC cells leading to enhanced radiosensitization of both wild-type and K-ras mutant cell lines; synergistic slowdown of tumor growth in Capan-2-bearing xenografts, when used in combination with radiation | [41] |
| In vitro | Impairment of clonogenicity and viability in distinct PC cells by affecting cell cycle and promoting apoptosis; synergistic antitumor effects in PC cells when used in combination with nitroxoline | [42] | ||
| Ritonavir | HIV infection | In vitro | Inhibition of PC cell viability by triggering the intrinsic apoptotic pathway and interfering with cell cycle machinery through the suppression of Akt and Rb phosphorylation in cells; impairment of PC cell motility and invasiveness; enhancement of antiproliferative effects in PC cells, when used in combination with gemcitabine | [43] |
| (Hydroxy)-Chloroquine | Malaria Systemic Lupus Erythematosus Rheumatoid arthritis | In vitro/ animal models | Inhibition of proliferation in several PC cell lines; improvement of survival in murine xenograft models of PC | [44] |
| In vitro/ animal models | Inhibition of autophagy in PSCs through reduced IL-6 expression and ECM protein production, leading to a quiescent state of PSCs; attenuation of invasion properties and liver metastasis formation in an orthotopic PC mouse model | [45] | ||
| In vitro | Inhibition of viability in metastatic PC cell lines, with enhanced effects in hypoxia | [46] | ||
| In vitro | Enhancement of antiproliferative effects of 5-fluorouracil, or gemcitabine by reversing autophagy-mediated cytoprotective mechanisms induced by chemotherapy drugs | [47] | ||
| In vitro/ animal models | Inhibition of autophagy leading to increased gemcitabine-induced cytotoxicity through ROS release, lysosomal membrane permeabilization and subsequent apoptosis in PC cells; reduction in tumor growth in xenograft PC models, when used in combination with gemcitabine | [48] | ||
| In vitro/ animal models | Enhancement of antitumor effects in both organoid and PDX models of PC, when used in combination with ERK/MAPK inhibitors, which induce a state of autophagy-dependence in PC cells due to impaired mitochondrial functions and glycolytic activity | [49] | ||
| In vitro/ animal models | Inhibition of autophagy leading to restoring MHC class I expression at the PC cell surface and reverting one of the PC immune escape mechanisms; slowdown of tumor growth in mouse model of PC, when used in combination with dual immune checkpoint blockade inhibitors (anti-CTL4 and anti-PD-1) | [50] | ||
| In vitro/ animal models | Inhibition of CXCL12/CXCR4 signaling, reduced phosphorylation of ERK and STAT3, downregulation of Hedgehog signaling, leading to elimination of Pa-CSCs; reduction in tumorigenicity and invasiveness in pancreatic cancer PDX models; improved outcomes of mice bearing primary xenografts, when used in combination with gemcitabine | [51] | ||
| Itraconazole | Fungal infections | In vitro/ animal models | Inhibition of PC cell proliferation by induction of apoptosis through ROS release, mitochondrial membrane depolarization and Bak-1 activation; inhibition of PC xenograft tumor growth | [52] |
| In vitro/ animal models | Inhibition of PC cell viability by apoptosis induction, together with impairment of migration, invasion and EMT of PC cells through TGF-β/SMAD2/3 signaling suppression; inhibition of tumor growth in GEMM of PC | [53] | ||
| Losartan | Hypertension | In vitro/ animal models | Blockade of AT1R leading to inhibition of VEGF synthesis and suppression of PC cell proliferation; improved survival benefit in rat orthotopic PC models, when used in combination with gemcitabine | [54] |
| In vitro/ animal models | Inhibition of PC growth in vivo by sequential administration of losartan- followed by gemcitabine-loaded magnetic mesoporous organosilica spheres leading to reduced stromal type I collagen and hyaluronic acid components in the ECM of the tumor | [55] | ||
| In vitro/ animal models | Blockade of AT1R leading to impaired stromal collagen and hyaluronan production by CAFs; improved overall survival in orthotopic PC mouse models through enhanced tumor perfusion, oxygen, chemo- and nanotherapeutics delivery | [56,57] | ||
| Animal models/ in patients | Blockade of AT1R leading to attenuated obesity-induced fibrosis, tumor progression and improved response to chemotherapy by preventing PSC activation, increasing CD8+ T cells, decreasing IL-1β, TANs and Tregs | [58] | ||
| Animal models | Slowdown of PC progression and improved survival in orthotopic models of PC by inhibiting aberrant TGF-β activity, collagen deposition and accumulation of Tregs | [59] | ||
| Metformin | Type 2 diabetes | In vitro | Inhibition of respiratory complex I leading to reduced ATP production, increased AMP/ATP ratio, activation of AMPK signaling, followed by downregulation of mTOR, which inhibits PC cell proliferation | [60] |
| In vitro/ animal models | Inhibition of PC cell proliferation through the suppression of insulin/IGF-I receptor activation and downstream signaling mediators IRS-1 and Akt; inhibition of tumor growth in PC xenografts mouse models | [61,62] | ||
| Nitroxoline | Urinary tract infections | In vitro | Inhibition of viability and clonogenicity, induction of cell cycle arrest and apoptosis, impairment of migration and invasion in PC cells through deregulation of Na/KATPase pump, β-catenin pathway, cytosolic iron homeostasis, together with ROS production and mitochondrial depolarization; synergistic antitumor effects in PC cells when used in combination with nelfinavir | [42,63] |
| Pimavanserin | Parkinson disease psychosis | In vitro/ animal models | Inhibition of viability, promotion of apoptosis and suppression of tumorsphere formation in PC cells through the abrogation of Akt/Gli1 signaling cascade leading to the downregulation of Oct-4, SOX2 and NANOG cancer stem cell markers; reduction in tumor growth in both subcutaneous and orthotopic xenografts models of PC | [64] |
| Pirfenidone | Idiopathic pulmonary fibrosis | In vitro | Inhibition of proliferation and promotion of cell cycle arrest in both PSCs and PC cells | [65] |
| In vitro/ animal models | Inhibition of proliferation, invasion and migration of PSCs through the suppression of PDGF-A, HGF, periostin, collagen type I and fibronectin; suppression of PC growth and metastasis in mice co-implanted with PC cells and PSCs by disrupting tumor-stromal interactions | [66] | ||
| Animal models | Inhibition of desmoplastic reactions and tumor growth in HapT1-derived orthotopic hamster PC models by reducing αSMA-positive cells and collagen deposition | [67] | ||
| In vitro | Inhibition of CHI3L1 and FN1, leading to reversion of gemcitabine resistance in PC cells | [68] | ||
| In vitro/ animal models | A liposome-based nanomedicine integrating pirfenidone and gemcitabine induces downregulation of collagen I and TGF-β in PSCs; slowdown of tumor growth in mice co-implanted with PC cells and PSCs through decreased stromal fibrosis and increased drug perfusion | [69,70] | ||
| In vitro/ animal models | Organosilica nanoparticles integrating pirfenidone and gemcitabine induce inhibition of PC cell viability by apoptosis induction, together with reduction in collagen I and fibronectin ECM components; reduction in tumor growth in mice co-implanted with PC cells and PSCs through decreased collagen I and fibronectin and enhanced endovascular osmotic pressure | [71] | ||
| Propranolol | Hypertension | In vitro/ animal models/ in patients | Inhibition of viability in primary human PC organoids, as a single agent and in combination with gemcitabine; abrogation of tumor growth, invasiveness and cancer-related immunosuppression, together with prolonged survival in KPC mice through ADRB2 blockade; improved clinical outcome in patients with stage II/III pancreatic adenocarcinoma undergoing surgery and receiving non-selective BBs, showing reduced nerve density, lower perineural invasion and decreased staining for BDNF (retrospective data) | [72] |
| In vitro/ animal models | Blockade of B1/B2-adrenoceptors leading to suppression of PC cell proliferation and invasiveness by inducing apoptosis and by inhibiting the expression of NF-kB, AP-1 and CREB, as well as the expression of MMP-9, MMP-2 and VEGF target genes | [73,74] | ||
| Animal models | Blockade of B1/B2-adrenoceptors leading to inhibition of tumor growth and prolonged survival in chronically stressed immunocompetent orthotopic syngeneic murine model of PC | [75] | ||
| Animal models | Blockade of B1/B2-adrenoceptors leading to inhibition of stress-induced tumor growth in PC xenograft animal models by decreasing Fz1, Wnt-1 and vimentin expression | [76] | ||
| Animal models | Prevention of PC development in hamsters with ethanol-induced pancreatitis by NNK, through the block of cAMP-dependent release of EGF and VEGF, together with the down-regulation of α7nAChR, ERK1/2 and p-CREB | [77] |
| Repurposed Drug | Register Trial Code/ Study Reference | Phase | Interventions | n. Patients | Status/Results |
|---|---|---|---|---|---|
| Chloroquine/ Hydroxychloroquine | NCT01777477 [78] | I | GEM + Chloroquine in mPC | 9 | Safe; 1 PR; 2 SD mTTP: 4 mo mOS: 7.6 mo |
| NCT01128296 [79] | I/II | Neoadjuvant GEM + Hydroxychloroquine in rPC | 35 | Safe; 94% resection rate 77% RO resection rate OS and DFS correlated with LC3-II expression in circulating PBMC | |
| NCT01978184 [80] | Randomized II | Neoadjuvant GEM/nabP ± hydroxychloroquine in rPC/brPC | 64 | Evans II grade response rate: 55.9% vs. 10% | |
| NCT01506973 [81] | Randomized II | GEM/nabP ± hydroxychloroquine in untreated laPC/mPC | 112 | 12-month OS: 41% vs. 49% ORR:38.2% vs. 21.1% mOS: 11.1 vs. 12.1 mo | |
| NCT04132505 | I | Binimetinib + hydroxychloroquine in mPC | 39 | Recruiting | |
| NCT03825289 | I | Trametinib + Hydroxychloroquine in aPC/mPC | 33 | Recruiting | |
| NCT04386057 | Randomized II | LY3214996 ± hydroxychloroquine in pretreated laPC/mPC | 52 | Recruiting | |
| Disulfiram | NCT02671890 | Partially randomized I | Disulfiram in refractory/ GEM-pretreated mPC | 74 | Recruiting |
| NCT03714555 | II | Disulfiram + copper gluconate in mPC treated with FOLFIRINOX, GEM/nabP or GEM and with rising CA19.9 | 42 | Recruiting | |
| Nelfinavir | [82] | I | Nelfinavir + Chemoradiation in locally laPC | 12 | Safe |
| NCT01068327 [83] | I | Nelfinavir + SBRT in brPC/laPC | 46 | Safe | |
| [84] | II | Nelfinavir + chemoradiation in laPC | 23 (prematurely closed due to drug unavailability) | 1yr-OS: 73.4% mOS:17.4 mo 1-yr PFS:21.8% mPFS:5.5 mo | |
| [85] | I/II | Oregovomab + SBRT + Nelfinavir in laPC | 11 (prematurely closed due to changed standard of treatment) | Safe | |
| NCT02024009 [86] | Randomized II | GEM/nab followed by chemoradiation ± nelfinavir in laPC | 289 | Recruiting | |
| Losartan | NCT01821729 [87] | II | Losartan + FOLFIRINOX + chemoradiation in laPC | 49 | R0 resection rate: 65% |
| NCT04539808 | II | Neoadjuvant FOLFIRINOX/GEM-nabP + chemoradiation + losartan in rPC/brPC | 40 | Recruiting | |
| Metformin | NCT01971034 [88] | II | Metformin + paclitaxel in laPC/mPC refractory to GEM | 20 | Terminated for futility |
| NCT01210911 [89] | Randomized II | GEM/erlotinib ± metformin in laPC/mPC | 121 | mOS: 56.7% vs. 63.9% (n.s.) | |
| NCT01167738 [90] | Randomized II | Cisplatin/epirubicin/capecitabine/GEM ± metformin in mPC | 60 | Terminated for futility | |
| NCT02336087 | I | GEM/nabP + dietary supplements + metformin in laPC | 21 | Active, not recruiting | |
| NCT01666730 | II | FOLFOX6 + metformin in mPC | 50 | Completed, results not published | |
| NCT02153450 | I | SBRT + metformin in rPC/laPC | 8 | Completed, results not published | |
| NCT02005419 | II | Metformin + GEM in resected PC | 300 | Completed, results not published | |
| Propranolol | EudraCT 2018-000415-25 [91] | Randomized II | Perioperative propranolol + etodolac vs. placebo in rPC | 80 | Recruiting |
| Repurposed Drug | Predicted Targets (Probability Score) |
|---|---|
| Auranofin | — |
| Haloperidol | 5-HT2b receptor (1), Dopamine D5 receptor (1), Alpha-2a adrenergic receptor (1), Adrenergic receptor alpha-2 (1), Histamine H2 receptor (1), Alpha-2b adrenergic receptor (1), Muscarinic acetylcholine receptor M5 (1), Dopamine D1 receptor (1), Muscarinic acetylcholine receptor M2 (1), Serotonin 1a (5-HT1a) receptor (1), Muscarinic acetylcholine receptor M1 (1), Dopamine D2 receptor (1), Dopamine D4 receptor (1), Norepinephrine transporter (1), Alpha-1d adrenergic receptor (1), Serotonin 2a (5-HT2a) receptor (1), Serotonin 2c (5-HT2c) receptor (1), Serotonin transporter (1), Alpha-1a adrenergic receptor (1), Histamine H1 receptor (1), Potassium channel subfamily K member 2 (1), Mu opioid receptor (1), Dopamine D3 receptor (1), HERG (1), Sigma opioid receptor (1), Serotonin 7 (5-HT7) receptor (1), Serotonin 6 (5-HT6) receptor (1), Histone H1.0 (1), P-glycoprotein 1 (1), Anti-estrogen binding site (AEBS) (1) |
| Penfluridol | — |
| Parbendazole | — |
| Disulfiram | Alpha-2a adrenergic receptor (1), Alpha-2b adrenergic receptor (1), Dopamine D1 receptor (1), Dopamine D2 receptor (1), Dopamine D4 receptor (1), Mu opioid receptor (1), Dopamine D3 receptor (1), Kappa Opioid receptor (1), Dopamine transporter (1), C-C chemokine receptor type 4 (1), Interleukin-8 receptor B (1), Adenosine A3 receptor (1), Cytochrome P450 1A2 (1), Serotonin 6 (5-HT6) receptor (1), MAP kinase ERK1 (1), C-C chemokine receptor type 2 (1), Monoglyceride lipase (1), Serotonin 1b (5-HT1b) receptor (1) |
| Doxycycline | — |
| Efavirenz | — |
| Nelfinavir | Neurokinin 2 receptor (0.97), Mu opioid receptor (0.97), Tyrosine-protein kinase FYN (0.74), Dopamine D1 receptor (0.74), Norepinephrine transporter (0.74), Adenosine A3 receptor (0.74), Cytochrome P450 3A4 (0.74) |
| Ritonavir | Cytochrome P450 3A4 (0.64), Neurokinin 2 receptor (0.62) |
| Chloroquine | HERG (1), Quinone reductase 2 (1), Prion protein (1), Histamine N-methyltransferase (0.67), Histamine H3 receptor (0.67) |
| Hydroxychloroquine | Muscarinic acetylcholine receptor M2 (1), Alpha-1d adrenergic receptor (1) |
| Itraconazole | — |
| Losartan | Type-1 angiotensin II receptor (1), Angiotensin converting enzyme (0.91), Cytochrome P450 2C9 (0.91) |
| Metformin | — |
| Nitroxoline | Cyclooxygenase-2 (1), Methionine aminopeptidase 2 (1) |
| Pimavanserin | — |
| Pirfenidone | — |
| Propranolol | Serotonin 2b (5-HT2b) receptor (1), Serotonin 1b (5-HT1b) receptor (1), Adrenergic receptor beta (1), Beta-1 adrenergic receptor (1), Serotonin 1a (5-HT1a) receptor (1), Serotonin 2a (5-HT2a) receptor (1), Serotonin 2c (5-HT2c) receptor (1), Serotonin transporter (1), Beta-3 adrenergic receptor (1), Sigma opioid receptor (1), Cytochrome P450 2D6 (1), Cytochrome P450 1A2 (1), Serotonin 6 (5-HT6) receptor (1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Lellis, L.; Veschi, S.; Tinari, N.; Mokini, Z.; Carradori, S.; Brocco, D.; Florio, R.; Grassadonia, A.; Cama, A. Drug Repurposing, an Attractive Strategy in Pancreatic Cancer Treatment: Preclinical and Clinical Updates. Cancers 2021, 13, 3946. https://doi.org/10.3390/cancers13163946
De Lellis L, Veschi S, Tinari N, Mokini Z, Carradori S, Brocco D, Florio R, Grassadonia A, Cama A. Drug Repurposing, an Attractive Strategy in Pancreatic Cancer Treatment: Preclinical and Clinical Updates. Cancers. 2021; 13(16):3946. https://doi.org/10.3390/cancers13163946
Chicago/Turabian StyleDe Lellis, Laura, Serena Veschi, Nicola Tinari, Zhirajr Mokini, Simone Carradori, Davide Brocco, Rosalba Florio, Antonino Grassadonia, and Alessandro Cama. 2021. "Drug Repurposing, an Attractive Strategy in Pancreatic Cancer Treatment: Preclinical and Clinical Updates" Cancers 13, no. 16: 3946. https://doi.org/10.3390/cancers13163946