
Mutational Landscape of Virus- and UV-Associated Merkel Cell Carcinoma Cell Lines Is Comparable to Tumor Tissue

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
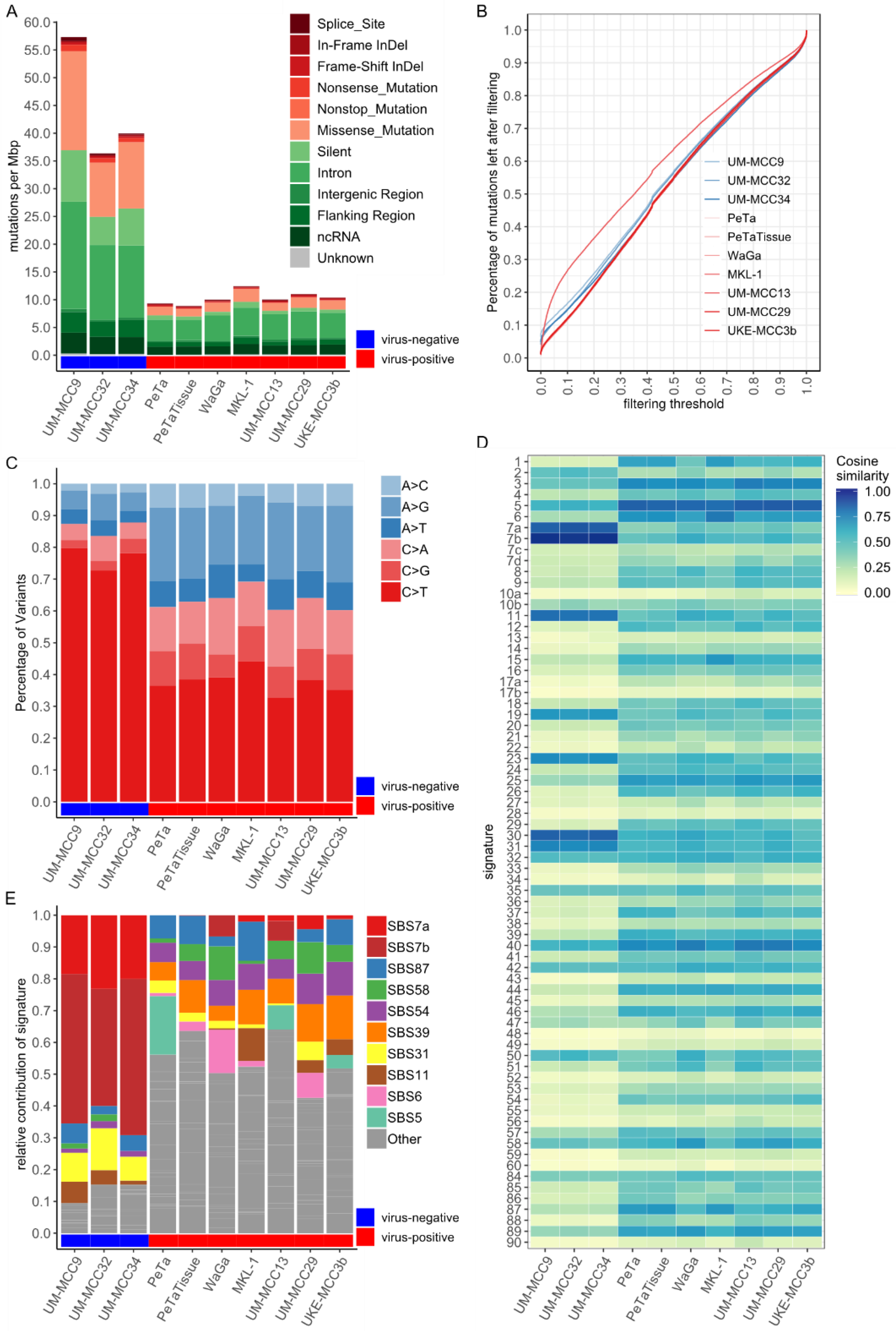
2.1. Mutational Burden and Signatures of MCC Cell Lines Are in Accordance with MCC Tissue Characteristics
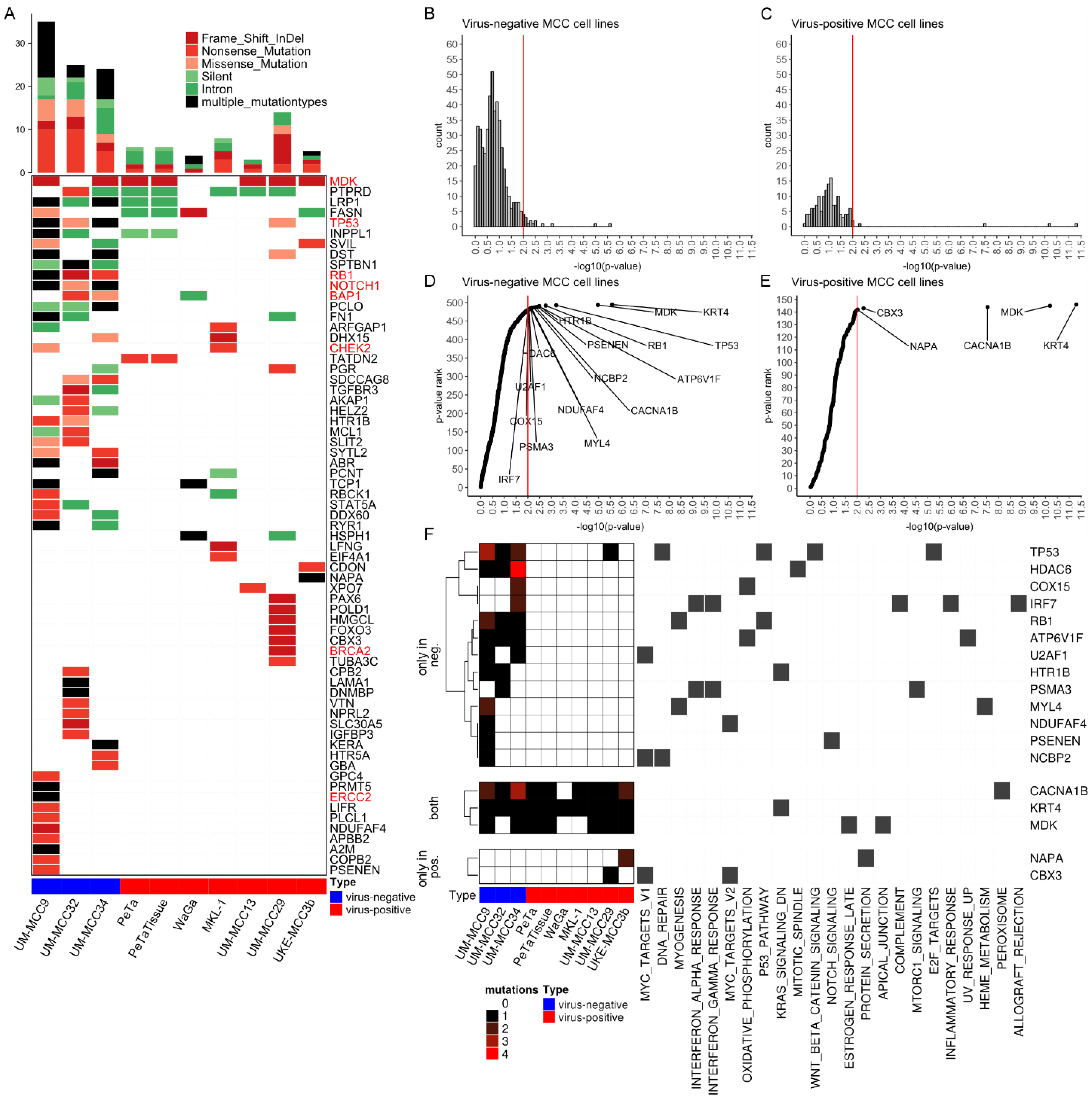
2.2. Mutations Altering Protein Structure
2.3. Significantly Mutated Genes
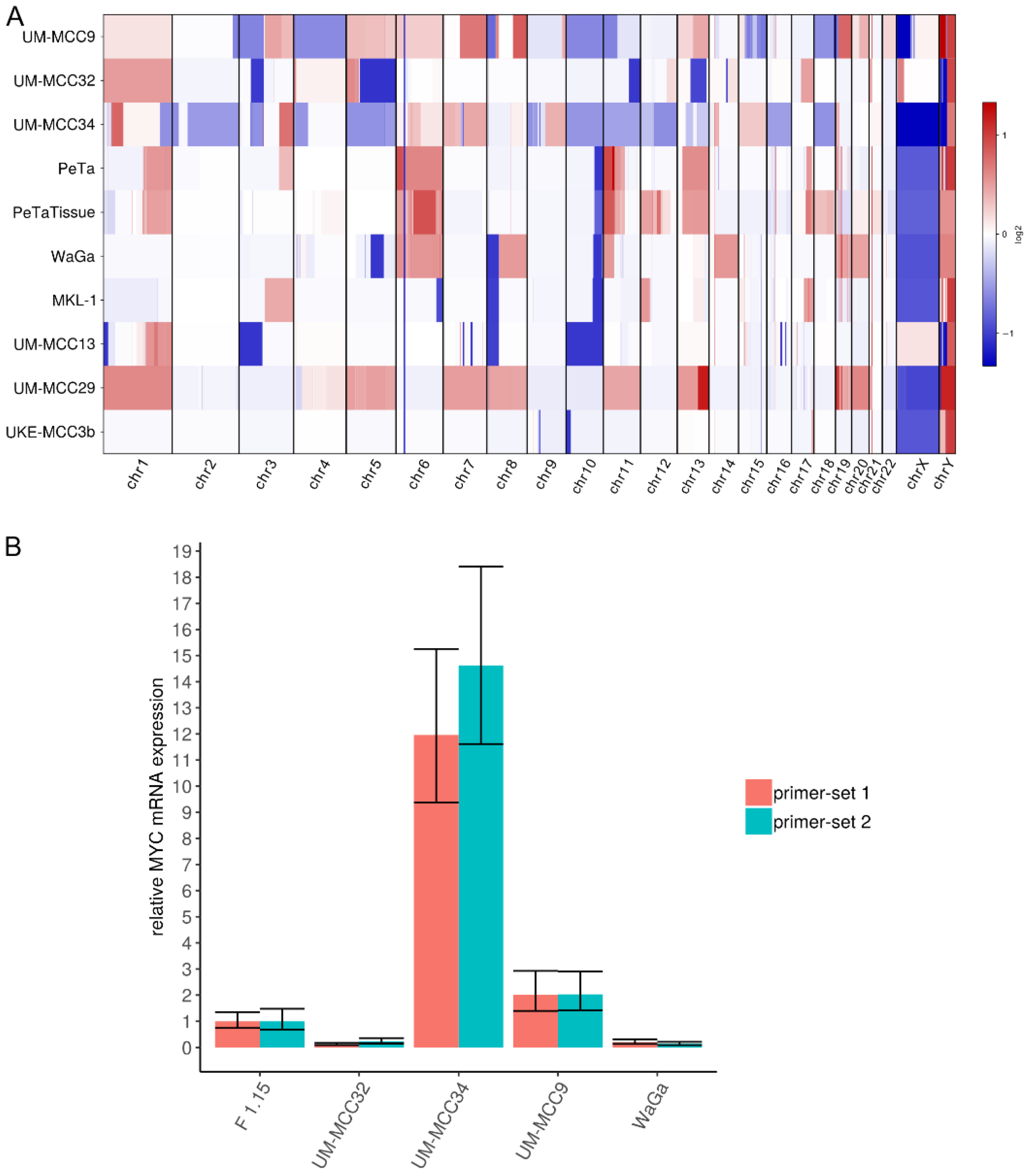
2.4. Characteristic Copy Number Variation Patterns in Virus-Positive MCC Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Tissues
4.2. Library Preparation and Sequencing
4.3. Alignment and Variant Calling
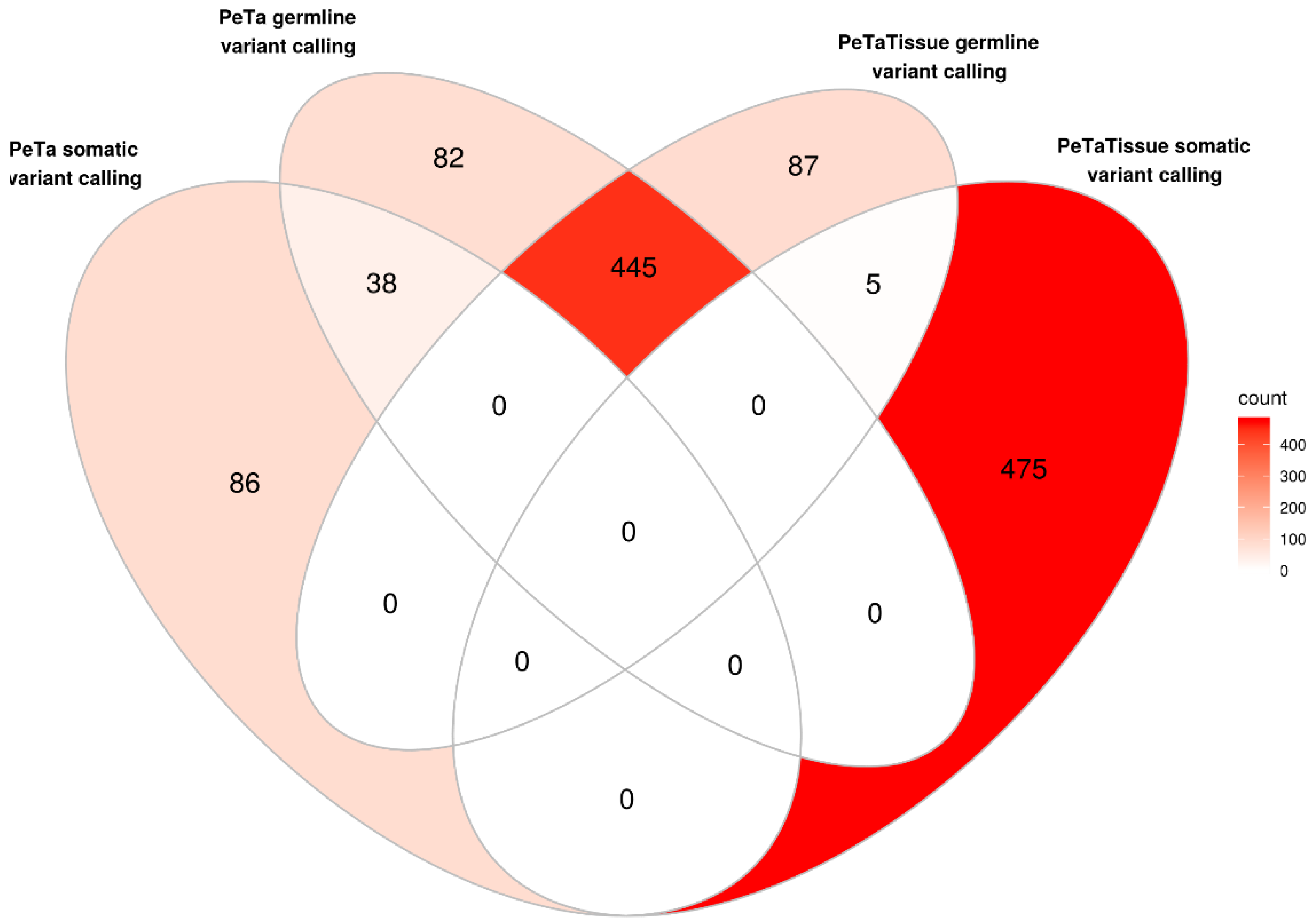
4.4. Variant Filtering
4.5. Quantitative Real-Time PCR (qRT-PCR)
4.6. Bioinformatic Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17077. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garneski, K.M.; Warcola, A.H.; Feng, Q.; Kiviat, N.B.; Helen Leonard, J.; Nghiem, P. Merkel Cell Polyomavirus Is More Frequently Present in North American than Australian Merkel Cell Carcinoma Tumors. J. Investig. Dermatol. 2009, 129, 246–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starrett, G.J.; Thakuria, M.; Chen, T.; Marcelus, C.; Cheng, J.; Nomburg, J.; Thorner, A.R.; Slevin, M.K.; Powers, W.; Burns, R.T.; et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020, 12, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knepper, T.C.; Montesion, M.; Russell, J.S.; Sokol, E.S.; Frampton, G.M.; Miller, V.A.; Albacker, L.A.; McLeod, H.L.; Eroglu, Z.; Khushalani, N.I.; et al. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5961. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.Q.; Waldeck, K.; Vergara, I.A.; Schröder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.J.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-Associated Mutations Underlie the Etiology of MCV-Negative Merkel Cell Carcinomas. Cancer Res. 2015, 75, 5228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, P.W.; Collie, A.M.B.; Hovelson, D.H.; Cani, A.K.; Verhaegen, M.E.; Patel, R.M.; Fullen, D.R.; Omata, K.; Dlugosz, A.A.; Tomlins, S.A.; et al. Next generation sequencing of Cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. Mod. Pathol. 2016, 29, 240–248. [Google Scholar] [CrossRef]
- Veija, T.; Sarhadi, V.K.; Koljonen, V.; Bohling, T.; Knuutila, S. Hotspot mutations in polyomavirus positive and negative Merkel cell carcinomas. Cancer Genet. 2016, 209, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.R.; Tomson, B.N.; Elkin, S.K.; Marchlik, E.; Carter, J.L.; Kurzrock, R. Genomic portfolio of Merkel cell carcinoma as determined by comprehensive genomic profiling: Implications for targeted therapeutics. Oncotarget 2016, 7, 23454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, C.A.; Jones, A.; Reynolds, J.; Stuart, J.; Pirisi, L.; Botrous, P.; Wells, J. Neuroendocrine Merkel Cell Carcinoma Is Associated with Mutations in Key DNA Repair, Epigenetic and Apoptosis Pathways: A Case-Based Study Using Targeted Massively Parallel Sequencing. Neuroendocrinology 2015, 101, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Harms, P.W.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.-M.; Dhanasekaran, S.M.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res. 2015, 75, 3720–3727. [Google Scholar] [CrossRef] [Green Version]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2015, 7, 3403. [Google Scholar] [CrossRef] [PubMed]
- González-Vela, M.d.C.; Curiel-Olmo, S.; Derdak, S.; Beltran, S.; Santibañez, M.; Martínez, N.; Castillo-Trujillo, A.; Gut, M.; Sánchez-Pacheco, R.; Almaraz, C.; et al. Shared Oncogenic Pathways Implicated in Both Virus-Positive and UV-Induced Merkel Cell Carcinomas. J. Investig. Dermatol. 2017, 137, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Cimino, P.J.; Robirds, D.H.; Tripp, S.R.; Pfeifer, J.D.; Abel, H.J.; Duncavage, E.J. Retinoblastoma gene mutations detected by whole exome sequencing of Merkel cell carcinoma. Mod. Pathol. 2014, 27, 1073–1087. [Google Scholar] [CrossRef] [Green Version]
- Starrett, G.J.; Marcelus, C.; Cantalupo, P.G.; Katz, J.P.; Cheng, J.; Akagi, K.; Thakuria, M.; Rabinowits, G.; Wang, L.C.; Symer, D.E.; et al. Merkel Cell Polyomavirus Exhibits Dominant Control of the Tumor Genome and Transcriptome in Virus-Associated Merkel Cell Carcinoma. mBio 2017, 8, e02079-02016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, K.G.; Lemos, B.D.; Feng, B.; Jaimes, N.; Peñas, P.F.; Bi, X.; Maher, E.; Cohen, L.; Helen Leonard, J.; Granter, S.R.; et al. Array-CGH Reveals Recurrent Genomic Changes in Merkel Cell Carcinoma Including Amplification of L-Myc. J. Investig. Dermatol. 2009, 129, 1547–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrama, D.; Sarosi, E.-M.; Adam, C.; Ritter, C.; Kaemmerer, U.; Klopocki, E.; König, E.-M.; Utikal, J.; Becker, J.C.; Houben, R. Characterization of six Merkel cell polyomavirus-positive Merkel cell carcinoma cell lines: Integration pattern suggest that large T antigen truncating events occur before or during integration. Int. J. Cancer 2019, 145, 1020–1032. [Google Scholar] [CrossRef]
- Van Gele, M.; Leonard, J.H.; Van Roy, N.; Van Limbergen, H.; Van Belle, S.; Cocquyt, V.; Salwen, H.; De Paepe, A.; Speleman, F. Combined karyotyping, CGH and M-FISH analysis allows detailed characterization of unidentified chromosomal rearrangements in Merkel cell carcinoma. Int. J. Cancer 2002, 101, 137–145. [Google Scholar] [CrossRef]
- Van Gele, M.; Speleman, F.; Vandesompele, J.; Van Roy, N.; Leonard, J.H. Characteristic pattern of chromosomal gains and losses in Merkel cell carcinoma detected by comparative genomic hybridization. Cancer Res. 1998, 58, 1503–1508. [Google Scholar]
- Larramendy, M.L.; Koljonen, V.; Böhling, T.; Tukiainen, E.; Knuutila, S. Recurrent DNA copy number changes revealed by comparative genomic hybridization in primary Merkel cell carcinomas. Mod. Pathol. 2004, 17, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Härle, M.; Arens, N.; Moll, I.; Back, W.; Schulz, T.; Scherthan, H. Comparative genomic hybridization (CGH) discloses chromosomal and subchromosomal copy number changes in Merkel cell carcinomas. J. Cutan. Pathol. 1996, 23, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Gele, M.V.; Boyle, G.M.; Cook, A.L.; Vandesompele, J.; Boonefaes, T.; Rottiers, P.; Roy, N.V.; De Paepe, A.; Parsons, P.G.; Leonard, J.H.; et al. Gene-expression profiling reveals distinct expression patterns for Classic versus Variant Merkel cell phenotypes and new classifier genes to distinguish Merkel cell from small-cell lung carcinoma. Oncogene 2004, 23, 2732–2742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daily, K.; Coxon, A.; Williams, J.S.; Lee, C.-C.R.; Coit, D.G.; Busam, K.J.; Brownell, I. Assessment of Cancer Cell Line Representativeness Using Microarrays for Merkel Cell Carcinoma. J. Investig. Dermatol. 2015, 135, 1138–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houben, R.; Dreher, C.; Angermeyer, S.; Borst, A.; Utikal, J.; Haferkamp, S.; Peitsch, W.K.; Schrama, D.; Hesbacher, S. Mechanisms of p53 restriction in Merkel cell carcinoma cells are independent of the Merkel cell polyoma virus T antigens. J. Investig. Dermatol. 2013, 133, 2453–2460. [Google Scholar] [CrossRef] [Green Version]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.-J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956. [Google Scholar] [CrossRef]
- Verhaegen, M.E.; Mangelberger, D.; Weick, J.W.; Vozheiko, T.D.; Harms, P.W.; Nash, K.T.; Quintana, E.; Baciu, P.; Johnson, T.M.; Bichakjian, C.K.; et al. Merkel cell carcinoma dependence on bcl-2 family members for survival. J. Investig. Dermatol. 2014, 134, 2241–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, S.K.; Yoshida, K.; Machida, Y.; Khaira, P.; Chaudhuri, B.; Wohlschlegel, J.A.; Leffak, M.; Yates, J.; Dutta, A. Replication from oriP of Epstein-Barr Virus Requires Human ORC and Is Inhibited by Geminin. Cell 2001, 106, 287–296. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Carter, M.D.; Gaston, D.; Huang, W.-Y.; Greer, W.L.; Pasternak, S.; Ly, T.Y.; Walsh, N.M. Genetic profiles of different subsets of Merkel cell carcinoma show links between combined and pure MCPyV-negative tumors. Hum. Pathol. 2018, 71, 117–125. [Google Scholar] [CrossRef]
- Patel, S.A.; Rodrigues, P.; Wesolowski, L.; Vanharanta, S. Genomic control of metastasis. Br. J. Cancer 2021, 124, 3–12. [Google Scholar] [CrossRef]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, S.T.; Gould, V.E.; Salwen, H.R.; Herst, C.V.; Le Beau, M.M.; Lee, I.; Bauer, K.; Marder, R.J.; Andersen, R.; Kies, M.S.; et al. Establishment and characterization of a neuroendocrine skin carcinoma cell line. Lab. Investig. 1987, 56, 302–312. [Google Scholar]
- Rickman, D.S.; Beltran, H.; Demichelis, F.; Rubin, M.A. Biology and evolution of poorly differentiated neuroendocrine tumors. Nat. Med. 2017, 23, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Ireland, A.S.; Micinski, A.M.; Kastner, D.W.; Guo, B.; Wait, S.J.; Spainhower, K.B.; Conley, C.C.; Chen, O.S.; Guthrie, M.R.; Soltero, D.; et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2020, 38, 60–78.e12. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Kannan, A.; Lin, Z.; Stack, B.C.; Suen, J.Y.; Gao, L. BET Protein Inhibitor JQ1 Attenuates Myc-Amplified MCC Tumor Growth In Vivo. Cancer Res. 2014, 74, 7090. [Google Scholar] [CrossRef] [Green Version]
- Kwun, H.J.; Wendzicki, J.A.; Shuda, Y.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen induces genome instability by E3 ubiquitin ligase targeting. Oncogene 2017, 36, 6784–6792. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Maura, F.; Degasperi, A.; Nadeu, F.; Leongamornlert, D.; Davies, H.; Moore, L.; Royo, R.; Ziccheddu, B.; Puente, X.S.; Avet-Loiseau, H.; et al. A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 2019, 10, 2969. [Google Scholar] [CrossRef]
- Molofsky, A.V.; He, S.; Bydon, M.; Morrison, S.J.; Pardal, R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 2005, 19, 1432–1437. [Google Scholar] [CrossRef] [Green Version]
- Stemple, D.L.; Anderson, D.J. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell 1992, 71, 973–985. [Google Scholar] [CrossRef]
- Fan, K.; Spassova, I.; Gravemeyer, J.; Ritter, C.; Horny, K.; Lange, A.; Gambichler, T.; Ødum, N.; Schrama, D.; Schadendorf, D.; et al. Merkel cell carcinoma-derived exosome-shuttle miR-375 induces fibroblast polarization by inhibition of RBPJ and p53. Oncogene 2020. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; Kim, D.R. pcr: An R package for quality assessment, analysis and testing of qPCR data. PeerJ 2018, 6, e4473. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MCC Cell Line Type | Virus-Negative | Virus-Positive |
|---|---|---|
| Tumor Mutational Burden | high (on average, 44.5 mut/Mbp) | low (on average, 10.5 mut/Mbp) |
| Mutagenic processes detected | UV-light-associated DNA damage (SBS7a, SBS7b) | flat mutation profile without prominent signatures. |
| Coding mutations | many mutations with potential functional effect; many mutated genes | few mutations with potential functional effect; few mutated genes |
| Copy Number Variations | many widespread CNVs | few, characteristic CNVs |
| Cell Line | MCPyV Status | Established From | Localization of Primary | Time in Culture | Reference |
|---|---|---|---|---|---|
| UM-MCC9 | negative | primary, scalp | scalp | >6 years | [28] |
| UM-MCC32 | negative | primary, scalp | scalp | >6 years | [28] |
| UM-MCC34 | negative | axillary lymph node metastasis | presumably arm | >6 years | [28] |
| PeTa | positive | primary, trunk | trunk | >7 years | [17,26] |
| WaGa | positive | malignant ascites | head | >10 years | [17,33] |
| MKL-1 | positive | nodal metastasis | unknown | >30 years | [17,34] |
| UM-MCC13 | positive | metastasis, leg | presumably leg | >6 years | [28] |
| UM-MCC29 | positive | inguinal lymph node metastasis | presumably leg | >6 years | [28] |
| UKE-MCC3b | positive | metastasis, trunk | trunk | >3 years | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horny, K.; Gerhardt, P.; Hebel-Cherouny, A.; Wülbeck, C.; Utikal, J.; Becker, J.C. Mutational Landscape of Virus- and UV-Associated Merkel Cell Carcinoma Cell Lines Is Comparable to Tumor Tissue. Cancers 2021, 13, 649. https://doi.org/10.3390/cancers13040649
Horny K, Gerhardt P, Hebel-Cherouny A, Wülbeck C, Utikal J, Becker JC. Mutational Landscape of Virus- and UV-Associated Merkel Cell Carcinoma Cell Lines Is Comparable to Tumor Tissue. Cancers. 2021; 13(4):649. https://doi.org/10.3390/cancers13040649
Chicago/Turabian StyleHorny, Kai, Patricia Gerhardt, Angela Hebel-Cherouny, Corinna Wülbeck, Jochen Utikal, and Jürgen C. Becker. 2021. "Mutational Landscape of Virus- and UV-Associated Merkel Cell Carcinoma Cell Lines Is Comparable to Tumor Tissue" Cancers 13, no. 4: 649. https://doi.org/10.3390/cancers13040649