Metabolic Reprogramming of Fibroblasts as Therapeutic Target in Rheumatoid Arthritis and Cancer: Deciphering Key Mechanisms Using Computational Systems Biology Approaches




Abstract
:Simple Summary
Abstract
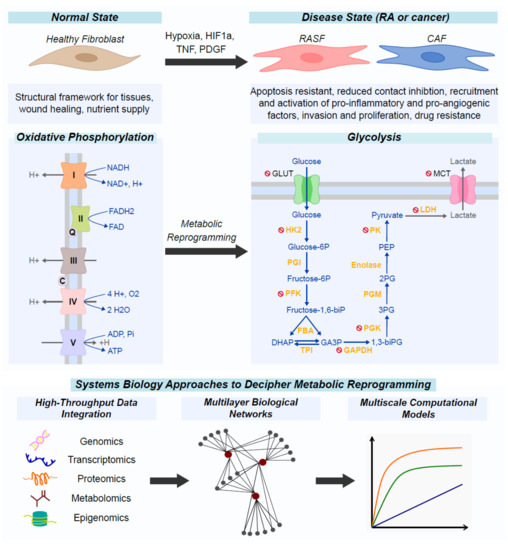
1. Introduction
2. The Role of Fibroblasts in Rheumatoid Arthritis
2.1. Origin of Rheumatoid Arthritis Synovial Fibroblasts
2.2. Population Heterogeneity in Rheumatoid Arthritis Synovial Fibroblasts
2.3. Epigenetic Modifications in Rheumatoid Arthritis Synovial Fibroblasts
3. The Role of Fibroblasts in Cancer
3.1. Origin of Cancer-Associated Fibroblasts
3.2. Population Heterogeneity in Cancer-Associated Fibroblasts
3.3. Epigenetic Alterations of Cancer-Associated Fibroblasts
4. Metabolic Reprogramming as an Alternative Survival Pathway in Rheumatoid Arthritis Synovial Fibroblasts and Cancer-Associated Fibroblasts
4.1. Metabolic Reprogramming of Fibroblasts in the Rheumatic Joint
4.2. Metabolic Reprogramming of Fibroblasts in Cancer
5. Metabolic Pathways as Therapeutic Targets in Rheumatoid Arthritis Synovial Fibroblasts and Cancer-Associated Fibroblasts
6. Immunometabolism as a Key Factor in Elucidating Signaling and Metabolic Crosstalks between Cancer-Associated Fibroblasts, Rheumatoid Arthritis Synovial Fibroblasts, and Immune Cells
6.1. High Throughput Technologies for Measurements and Analyses of Metabolic Components
6.2. Metabolomics and Integrative Analysis in RA
6.3. Metabolomics and Integrative Analysis in Cancer
7. Computational Systems Biology Approaches
7.1. Graphical Representations of Molecular Pathways in Rheumatoid Arthritis Synovial Fibroblasts and Cancer-Associated Fibroblasts
7.2. Computational Approaches for Metabolic Modeling and Various Mathematical Models Available in Rheumatoid Arthritis and Cancer
7.3. Toward Integrative Multi-Scale Cellular Models
8. Perspectives
8.1. Single-Cell Metabolomics, Transcriptomics, Proteomics
8.2. Integration Methods
8.3. Immunometabolism
8.4. Hybrid Modeling Approaches
8.5. HP Computing
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Virchow, R. Die Cellularpathologie in ihrer Begrundung auf Physiologische und Pathologische Gewebelehre; Hirschwald, A.: Berlin, Germany, 1862. [Google Scholar]
- Duval, M. Atlas D’embryologie; Masson G.: Paris, France, 1889. [Google Scholar]
- Huber, L.C.; Distler, O.; Tarner, I.; Gay, R.E.; Gay, S.; Pap, T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatology 2006, 45, 669–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegner, N.; Lundberg, K.; Kinloch, A.; Fisher, B.; Malmström, V.; Feldmann, M.; Venables, P.J. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol. Rev. 2010, 233, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Croft, C.B.; Tarin, D. Ultrastructural studies of wound healing in mouse skin. I. Epithelial behaviour. J. Anat. 1970, 106, 63–77. [Google Scholar] [PubMed]
- Bainbridge, P. Wound healing and the role of fibroblasts. J. Wound Care 2013, 22, 407–412. [Google Scholar] [CrossRef]
- Pinheiro, C.R.; Coelho, A.L.; de Oliveira, C.E.; Gasparoto, T.H.; Garlet, G.P.; Silva, J.S.; Santos, C.F.; Cavassani, K.A.; Hogaboam, C.M.; Campanelli, A.P. Recognition of Candida albicans by gingival fibroblasts: The role of TLR2, TLR4/CD14, and MyD88. Cytokine 2018, 106, 67–75. [Google Scholar] [CrossRef]
- Bellei, B.; Caputo, S.; Carbone, A.; Silipo, V.; Papaccio, F.; Picardo, M.; Eibenschutz, L. The Role of Dermal Fibroblasts in Nevoid Basal Cell Carcinoma Syndrome Patients: An Overview. Int. J. Mol. Sci. 2020, 21, 720. [Google Scholar] [CrossRef] [Green Version]
- Para, R.; Romero, F.; George, G.; Summer, R. Metabolic Reprogramming as a Driver of Fibroblast Activation in PulmonaryFibrosis. Am. J. Med. Sci. 2019, 357, 394–398. [Google Scholar] [CrossRef]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef]
- Turner, J.D.; Filer, A. The role of the synovial fibroblast in rheumatoid arthritis pathogenesis. Curr. Opin. Rheumatol. 2015, 27, 175–182. [Google Scholar] [CrossRef]
- Ospelt, C. Synovial fibroblasts in 2017. RMD Open 2017, 3, e000471. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for Fibrotic Diseases: Nearing the Starting Line. Sci. Transl. Med. 2013, 5, 167sr1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nihlberg, K. Fibroblasts as Matrix Modulating Cells in Asthma and COPD; Department of Experimental Medical Sciences: Lund University, Sweden, 2009; ISBN 9789186253509. [Google Scholar]
- Kiener, H.P.; Watts, G.F.M.; Cui, Y.; Wright, J.; Thornhill, T.S.; Sköld, M.; Behar, S.M.; Niederreiter, B.; Lu, J.; Cernadas, M.; et al. Synovial fibroblasts self-direct multicellular lining architecture and synthetic function in three-dimensional organ culture. Arthritis Rheum. 2010, 62, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef]
- Filer, A. The fibroblast as a therapeutic target in rheumatoid arthritis. Curr. Opin. Pharmacol. 2013, 13, 413–419. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [Green Version]
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Juarez, M.; Filer, A.; Buckley, C. Fibroblasts as therapeutic targets in rheumatoid arthritis and cancer. Swiss Med. Wkly. 2012, 142, 1–9. [Google Scholar] [CrossRef]
- Yoshitomi, H. Regulation of Immune Responses and Chronic Inflammation by Fibroblast-Like Synoviocytes. Front. Immunol. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Hirohata, S.; Yanagida, T.; Nagai, T.; Sawada, T.; Nakamura, H.; Yoshino, S.; Tomita, T.; Ochi, T. Induction of fibroblast-like cells from CD34+ progenitor cells of the bone marrow in rheumatoid arthritis. J. Leukoc. Biol. 2001, 70, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, A.J.; Zupan, J.; Riemen, A.H.K.; Kania, K.; Ansboro, S.; White, N.; Clark, S.M.; De Bari, C. Joint morphogenetic cells in the adult mammalian synovium. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, R.S.; Hülso, C.; Liu, Y.; Gasparini, S.J.; Fong-Yee, C.; Tu, J.; Stoner, S.; Stewart, P.M.; Raza, K.; Cooper, M.S.; et al. Characterisation of fibroblast-like synoviocytes from a murine model of joint inflammation. Arthritis Res. Ther. 2013, 15, R24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, J.; Hong, W.; Zhang, P.; Wang, X.; Körner, H.; Wei, W. Ontology and Function of Fibroblast-Like and Macrophage-Like Synoviocytes: How Do They Talk to Each Other and Can They Be Targeted for Rheumatoid Arthritis Therapy? Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat. Commun. 2018, 9, 789. [Google Scholar] [CrossRef] [Green Version]
- Croft, A.P.; Campos, J.; Jansen, K.; Turner, J.D.; Marshall, J.; Attar, M.; Savary, L.; Wehmeyer, C.; Naylor, A.J.; Kemble, S.; et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019, 570, 246–251. [Google Scholar] [CrossRef]
- Zerrouk, N.; Miagoux, Q.; Dispot, A.; Elati, M.; Niarakis, A. Identification of putative master regulators in rheumatoid arthritis synovial fibroblasts using gene expression data and network inference. Sci. Rep. 2020, 10, 16236. [Google Scholar] [CrossRef]
- Karami, J.; Aslani, S.; Jamshidi, A.; Garshasbi, M.; Mahmoudi, M. Genetic implications in the pathogenesis of rheumatoid arthritis; an updated review. Gene 2019, 702, 8–16. [Google Scholar] [CrossRef]
- Karouzakis, E.; Gay, R.E.; Gay, S.; Neidhart, M. Epigenetic control in rheumatoid arthritis synovial fibroblasts. Nat. Rev. Rheumatol. 2009, 5, 266–272. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Bera, S. CAF cellular glycolysis: Linking cancer cells with the microenvironment. Tumor Biol. 2016, 37, 8503–8514. [Google Scholar] [CrossRef]
- Pietras, K.; Östman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Micke, P.; Tman, A. Tumour-stroma interaction: Cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 2004, 45, S163–S175. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Mareel, M. Role of tissue stroma in cancer cell invasion. J. Pathol. 2003, 200, 429–447. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Räsänen, K.; Vaheri, A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 2010, 316, 2713–2722. [Google Scholar] [CrossRef]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.C.; Singer, C.; Hornby, A.; Rasmussen, A.; Cullen, K.J. Insulin-like growth factor mediated stromal-epithelial interactions in human breast cancer. Breast Cancer Res. Treat. 1994, 31, 249–261. [Google Scholar] [CrossRef]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. Biomed Res. Int. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Piek, E.; Heldin, C.-H.; Dijke, P. Ten Specificity, diversity, and regulation in TGF-β superfamily signaling. FASEB J. 1999, 13, 2105–2124. [Google Scholar] [CrossRef]
- Moustakas, A.; Souchelnytskyi, S.; Heldin, C.H. Smad regulation in TGF-beta signal transduction. J. Cell Sci. 2001, 114, 4359–4369. [Google Scholar]
- De Wever, O.; Nguyen, Q.; Van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent proinvasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- Grugan, K.D.; Miller, C.G.; Yao, Y.; Michaylira, C.Z.; Ohashi, S.; Klein-Szanto, A.J.; Diehl, J.A.; Herlyn, M.; Han, M.; Nakagawa, H.; et al. Fibroblast-secreted hepatocyte growth factor plays a functional role in esophageal squamous cell carcinoma invasion. Proc. Natl. Acad. Sci. USA 2010, 107, 11026–11031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.-L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551–7556. [Google Scholar] [CrossRef] [Green Version]
- Petersen, O.W.; Nielsen, H.L.; Gudjonsson, T.; Villadsen, R.; Rank, F.; Niebuhr, E.; Bissell, M.J.; Rønnov-Jessen, L. Epithelial to Mesenchymal Transition in Human Breast Cancer Can Provide a Nonmalignant Stroma. Am. J. Pathol. 2003, 162, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [Green Version]
- Cuiffo, B.G.; Karnoub, A.E. Mesenchymal stem cells in tumor development: Emerging roles and concepts. Cell Adhes. Migr. 2012, 6, 220–230. [Google Scholar] [CrossRef]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.W.; Takashi, S.; Baik, G.H.; Shibata, W.; DiPrete, B.; Betz, K.S.; et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [Green Version]
- Direkze, N.C.; Hodivala-Dilke, K.; Jeffery, R.; Hunt, T.; Poulsom, R.; Oukrif, D.; Alison, M.R.; Wright, N.A. Bone Marrow Contribution to Tumor-Associated Myofibroblasts and Fibroblasts. Cancer Res. 2004, 64, 8492–8495. [Google Scholar] [CrossRef] [Green Version]
- Wikström, P.; Marusic, J.; Stattin, P.; Bergh, A. Low stroma androgen receptor level in normal and tumor prostate tissue is related to poor outcome in prostate cancer patients. Prostate 2009, 69, 799–809. [Google Scholar] [CrossRef]
- Kanzaki, R.; Pietras, K. Heterogeneity of cancer-associated fibroblasts: Opportunities for precision medicine. Cancer Sci. 2020, 111, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, A.; Hum, N.R.; Martin, K.A.; Gilmore, S.F.; Peran, I.; Byers, S.W.; Wheeler, E.K.; Coleman, M.A.; Loots, G.G. Single-Cell Transcriptomic Analysis of Tumor-Derived Fibroblasts and Normal Tissue-Resident Fibroblasts Reveals Fibroblast Heterogeneity in Breast Cancer. Cancers 2020, 12, 1307. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef]
- Du, H.; Che, G. Genetic alterations and epigenetic alterations of cancer-associated fibroblasts. Oncol. Lett. 2017, 13, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Sun, Y.; Hou, Y.; Peng, Q.; Wang, L.; Luo, H.; Tang, X.; Zeng, Z.; Liu, M. MiRNA expression analysis of cancer-associated fibroblasts and normal fibroblasts in breast cancer. Int. J. Biochem. Cell Biol. 2012, 44, 2051–2059. [Google Scholar] [CrossRef]
- Enkelmann, A.; Heinzelmann, J.; von Eggeling, F.; Walter, M.; Berndt, A.; Wunderlich, H.; Junker, K. Specific protein and miRNA patterns characterise tumour-associated fibroblasts in bladder cancer. J. Cancer Res. Clin. Oncol. 2011, 137, 751–759. [Google Scholar] [CrossRef]
- Kekeeva, T.V.; Popova, O.P.; Shegai, P.V.; Alekseev, B.Y.; Andreeva, Y.Y.; Zaletaev, D.V.; Nemtsova, M.V. Aberrant methylation of p16, HIC1, N33, and GSTP1 in tumor epithelium and tumor-associated cells in prostate cancer. Mol. Biol. 2007, 41, 70–76. [Google Scholar] [CrossRef]
- Nielsen, B.S.; Jørgensen, S.; Fog, J.U.; Søkilde, R.; Christensen, I.J.; Hansen, U.; Brünner, N.; Baker, A.; Møller, S.; Nielsen, H.J. High levels of microRNA-21 in the stroma of colorectal cancers predict short disease-free survival in stage II colon cancer patients. Clin. Exp. Metastasis 2011, 28, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Luczak, M.W.; Jagodziński, P.P. The role of DNA methylation in cancer development. Folia Histochem. Cytobiol. 2006, 44, 143–154. [Google Scholar] [CrossRef]
- Mishra, P.; Kiebish, M.A.; Cullen, J.; Srinivasan, A.; Patterson, A.; Sarangarajan, R.; Narain, N.R.; Dobi, A. Genomic alterations of Tenascin C in highly aggressive prostate cancer: A meta-analysis. Genes Cancer 2019, 10, 150–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefèvre, S.; Knedla, A.; Tennie, C.; Kampmann, A.; Dinser, R.; Korb, A.; Schnäker, E.; Tarner, I.H.; Paul, D.; Evans, C.H.; et al. Synovial fibroblasts spread RA to unaffected joints. Nat. Med. 2013, 15, 1414–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-Associated Fibroblasts Drive the Progression of Metastasis through both Paracrine and Mechanical Pressure on Cancer Tissue. Mol. Cancer Res. 2012, 10, 1403–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Falconer, J.; Murphy, A.N.; Young, S.P.; Clark, A.R.; Tiziani, S.; Guma, M.; Buckley, C.D. Review: Synovial Cell Metabolism and Chronic Inflammation in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 984–999. [Google Scholar] [CrossRef]
- Tirone, T.A.; Brunicardi, F.C. Overview of Glucose Regulation. World J. Surg. 2001, 25, 461–467. [Google Scholar] [CrossRef]
- Marelli-Berg, F.M.; Fu, H.; Mauro, C. Molecular mechanisms of metabolic reprogramming in proliferating cells: Implications for T-cell-mediated immunity. Immunology 2012, 136, 363–369. [Google Scholar] [CrossRef]
- Fearon, U.; Hanlon, M.M.; Wade, S.M.; Fletcher, J.M. Altered metabolic pathways regulate synovial inflammation in rheumatoid arthritis. Clin. Exp. Immunol. 2019, 197, 170–180. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, P.G.; Farinon, M.; Sanchez-Lopez, E.; Miyamoto, S.; Guma, M. Fibroblast-Like Synoviocytes Glucose Metabolism as a Therapeutic Target in Rheumatoid Arthritis. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Barabási, A.-L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Saegusa, J.; Naka, I.; Tsuda, K.; Okano, T.; Akashi, K.; Sendo, S.; Ueda, Y.; Onishi, A.; Kogata, Y.; et al. Glutamine metabolism plays a crucial role in the pathogenesis of rheumatoid arthritis. Arthritis Rheumatol. 2016, 68, 1812–1813. [Google Scholar]
- Guma, M.; Sanchez-Lopez, E.; Lodi, A.; Garcia-Carbonell, R.; Tiziani, S.; Karin, M.; Lacal, J.C.; Firestein, G.S. Choline kinase inhibition in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Wang, J.; Xiao, Y.; Wang, C.; Qiu, Q.; Lao, M.; Yu, Y.; Li, Z.; Zhang, H.; Ye, Y.; et al. Glycogen Metabolism and Rheumatoid Arthritis: The Role of Glycogen Synthase 1 in Regulation of Synovial Inflammation via Blocking AMP-Activated Protein Kinase Activation. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Szanto, S.; Koreny, T.; Szekanecz, Z.; Mikecz, K.; Glant, T.T.; Varga, J. Inhibition of indoleamine 2,3-dioxygenase-mediated tryptophan catabolism accelerates collagen-induced arthritis in mice. Arthritis Res. Ther. 2007, 9, R50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante, M.F.; Garcia-Carbonell, R.; Whisenant, K.D.; Guma, M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 110. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, C. Sphingosine-1-Phosphate and Rheumatoid Arthritis: Pathological Implications and Potential Therapeutic Targets. In Innovative Rheumatology; InTech: Rijeka, Croatia, 2013; p. 13. [Google Scholar]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The Glycolytic Switch in Tumors: How Many Players Are Involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef]
- Becker, L.M.; O’Connell, J.T.; Vo, A.P.; Cain, M.P.; Tampe, D.; Bizarro, L.; Sugimoto, H.; McGow, A.K.; Asara, J.M.; Lovisa, S.; et al. Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020, 31. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Locasale, J.W.; Cantley, L.C. Altered metabolism in cancer. BMC Biol. 2010, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanaka, R.B.; Chandel, N.S. Targeting glucose metabolism for cancer therapy. J. Exp. Med. 2012, 209, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niebler, S.; Angele, P.; Kujat, R.; Bosserhoff, A.K. Hypoxia-inducible factor 1 Is an inductor of transcription factor activating protein 2 epsilon expression during chondrogenic differentiation. Biomed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bárdos, J.I.; Ashcroft, M. Hypoxia-inducible factor-1 and oncogenic signalling. BioEssays 2004, 26, 262–269. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Scott Budigner, G.R.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 2014, 1–18. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Waste Not, Want Not: Lactate Oxidation Fuels the TCA Cycle. Cell Metab. 2017, 26, 803–804. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Lin, Y.; Zhang, H.; Liu, C.; Cheng, Z.; Yang, X.; Zhang, J.; Xiao, Y.; Sang, N.; Qian, X.; et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020, 11, 267. [Google Scholar] [CrossRef]
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer Targets in the Glycolytic Metabolism of Tumors: A Comprehensive Review. Front. Pharmacol. 2011, 2, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Abboud, G.; Choi, S.-C.; Kanda, N.; Zeumer-Spataro, L.; Roopenian, D.C.; Morel, L. Inhibition of Glycolysis Reduces Disease Severity in an Autoimmune Model of Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Lu, Q.; Fan, H.; Zhang, X.; Ge, L.; Tian, R.; Wang, S.; Feng, T.; Pan, J.; Feng, J.; et al. Inhibition of hexokinases holds potential as treatment strategy for rheumatoid arthritis. Arthritis Res. Ther. 2019, 21, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante, M.F.; Oliveira, P.G.; Garcia-Carbonell, R.; Croft, A.P.; Smith, J.M.; Serrano, R.L.; Sanchez-Lopez, E.; Liu, X.; Kisseleva, T.; Hay, N.; et al. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann. Rheum. Dis. 2018, 77, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yan, X.; Li, X.; Zheng, Y.; Li, S.; Chang, X. PGK1, a glucose metabolism enzyme, may play an important role in rheumatoid arthritis. Inflamm. Res. 2016, 65, 815–825. [Google Scholar] [CrossRef]
- Garcia-Carbonell, R.; Divakaruni, A.S.; Lodi, A.; Vicente-Suarez, I.; Saha, A.; Cheroutre, H.; Boss, G.R.; Tiziani, S.; Murphy, A.N.; Guma, M. Critical Role of Glucose Metabolism in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Arthritis Rheumatol. 2016, 68, 1614–1626. [Google Scholar] [CrossRef] [Green Version]
- Okano, T.; Saegusa, J.; Nishimura, K.; Takahashi, S.; Sendo, S.; Ueda, Y.; Morinobu, A. 3-bromopyruvate ameliorate autoimmune arthritis by modulating Th17/Treg cell differentiation and suppressing dendritic cell activation. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- McGarry, T.; Orr, C.; Wade, S.; Biniecka, M.; Wade, S.; Gallagher, L.; Low, C.; Veale, D.J.; Fearon, U. JAK/STAT Blockade Alters Synovial Bioenergetics, Mitochondrial Function, and Proinflammatory Mediators in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 1959–1970. [Google Scholar] [CrossRef] [Green Version]
- Phan, L.M.; Yeung, S.C.J.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Glycolysis in cancer: A potential target for therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1358–1366. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, J.; Xu, J.; Leong, W.S.; Liu, G.; Ji, T.; Cheng, Z.; Wang, J.; Lang, J.; Zhao, Y.; et al. Suppression of Tumor Energy Supply by Liposomal Nanoparticle-Mediated Inhibition of Aerobic Glycolysis. ACS Appl. Mater. Interfaces 2018, 10, 2347–2353. [Google Scholar] [CrossRef]
- Cardaci, S.; Desideri, E.; Ciriolo, M.R. Targeting aerobic glycolysis: 3-Bromopyruvate as a promising anticancer drug. J. Bioenerg. Biomembr. 2012, 44, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwarakarnath, B.S.; Jain, V. Targeting glucose metabolism with 2-deoxy-d-glucose for improving cancer therapy. Future Oncol. 2009, 5, 581–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, G.S.; Page, R.L.; Riviere, J.E.; Cline, J.M.; Thrall, D.E. Pharmacokinetics and toxicity of oral and intravenous lonidamine in dogs. Cancer Chemother. Pharmacol. 1996, 38, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Pålsson-McDermott, E.M.; O’Neill, L.A.J. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020, 30, 300–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biniecka, M.; Canavan, M.; McGarry, T.; Gao, W.; McCormick, J.; Cregan, S.; Gallagher, L.; Smith, T.; Phelan, J.J.; Ryan, J.; et al. Dysregulated bioenergetics: A key regulator of joint inflammation. Ann. Rheum. Dis. 2016, 75, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef] [Green Version]
- Wood, T.E.; Dalili, S.; Simpson, C.D.; Hurren, R.; Mao, X.; Saiz, F.S.; Gronda, M.; Eberhard, Y.; Minden, M.D.; Bilan, P.J.; et al. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol. Cancer Ther. 2008, 7, 3546–3555. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.H.; Ho, C.T.; Chen, Z.F.; Chen, L.C.; Whang-Peng, J.; Lin, T.N.; Ho, Y.S. The apple polyphenol phloretin inhibits breast cancer cell migration and proliferation via inhibition of signals by type 2 glucose transporter. J. Food Drug Anal. 2018, 26, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zheng, J.Y.; Liu, J.Q.; Yang, J.; Liu, Y.; Wang, C.; Ma, X.N.; Liu, B.L.; Xin, G.Z.; Liu, L.F. Succinate/NLRP3 inflammasome induces synovial fibroblast activation: Therapeutical effects of clematichinenoside AR on arthritis. Front. Immunol. 2016, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Ho, Y.S.; Tsai, C.Y.; Wang, Y.J.; Tseng, H.; Wei, P.L.; Lee, C.H.; Liu, R.S.; Lin, S.Y. In vitro and in vivo study of phloretin-induced apoptosis in human liver cancer cells involving inhibition of type II glucose transporter. Int. J. Cancer 2009, 124, 2210–2219. [Google Scholar] [CrossRef] [PubMed]
- Son, H.J.; Lee, J.; Lee, S.Y.; Kim, E.K.; Park, M.J.; Kim, K.W.; Park, S.H.; Cho, M. La Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediat. Inflamm. 2014, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, W.; Kawahito, Y.; Nagahara, H.; Kukida, Y.; Seno, T.; Yamamoto, A.; Kohno, M.; Oda, R.; Taniguchi, D.; Fujiwara, H.; et al. Monocarboxylate Transporter 4, Associated With the Acidification of Synovial Fluid, Is a Novel Therapeutic Target for Inflammatory Arthritis. Arthritis Rheumatol. 2015, 67, 2888–2896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Romero, I.L.; Litchfield, L.M.; Lengyel, E.; Locasale, J.W. Metformin targets central carbon metabolism and reveals mitochondrial requirements in human cancers. Cell Metab. 2018, 176, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Gurrapu, S.; Jonnalagadda, S.K.; Alam, M.A.; Nelson, G.L.; Sneve, M.G.; Drewes, L.R.; Mereddy, V.R. Monocarboxylate transporter 1 inhibitors as potential anticancer agents. ACS Med. Chem. Lett. 2015, 6, 558–561. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef] [Green Version]
- Crawford, S. Anti-inflammatory/antioxidant use in long-term maintenance cancer therapy: A new therapeutic approach to disease progression and recurrence. Ther. Adv. Med. Oncol. 2014, 6, 52–68. [Google Scholar] [CrossRef] [Green Version]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [Green Version]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-outschoorn, U. Metabolic Couplind and the Reverse Warburg Effect in Cancer, implications for novel biomarker and anticancer agent development. Semin. Oncol. 2018, 44, 198–203. [Google Scholar] [CrossRef]
- Monti, D.; Sotgia, F.; Whitaker-menezes, D.; Birbe, R.; Berger, A.; Lazar, M.; Cotzia, P.; Draganova-tacheva, R.; Lin, Z.; Domingo-vidal, M.; et al. Pilot study demonstrating metabolic and anti-proliferative effects of in vivo anti-oxidant supplementation with NAcetylcysteine in Breast Cancer. Semin. Oncol. 2018, 44, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Han, F.; Yang, S.; Wu, J.; Zhan, W. Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: Involvement of the Akt-mTOR signaling pathway. Cancer Lett. 2015, 358, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Colen, C.B.; Shen, Y.; Ghoddoussi, F.; Yu, P.; Francis, T.B.; Koch, B.J.; Monterey, M.D.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic Targeting of Lactate Efflux by Malignant Glioma Inhibits Invasiveness and Induces Necrosis: An In Vivo Study. Neoplasia 2011, 13, 620–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathupala, S.P.; Colen, C.B.; Parajuli, P.; Sloan, A.E. Lactate and malignant tumors: A therapeutic target at the end stage of glycolysis. J. Bioenerg. Biomembr. 2007, 39, 73–77. [Google Scholar] [CrossRef] [Green Version]
- Kohlmann, A.; Zech, S.G.; Li, F.; Zhou, T.; Squillace, R.M.; Commodore, L.; Greenfield, M.T.; Lu, X.; Miller, D.P.; Huang, W.S.; et al. Fragment growing and linking lead to novel nanomolar lactate dehydrogenase inhibitors. J. Med. Chem. 2013, 56, 1023–1040. [Google Scholar] [CrossRef]
- John, P.; Sidney, P.C. The Role of Glycolysis in the Growth of Tumor Cells. J. Biol. Chem. 1961, 236, 2786–2790. [Google Scholar]
- Billiard, J.; Dennison, J.B.; Briand, J.; Annan, R.S.; Chai, D.; Colón, M.; Dodson, C.S.; Gilbert, S.A.; Greshock, J.; Jing, J.; et al. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 2013, 1, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Rani, R.; Kumar, V. Recent Update on Human Lactate Dehydrogenase Enzyme 5 (hLDH5) Inhibitors: A Promising Approach for Cancer Chemotherapy. J. Med. Chem. 2016, 59, 487–496. [Google Scholar] [CrossRef]
- Shelley, M.D.; Hartley, L.; Fish, R.G.; Groundwater, P.; Morgan, J.J.G.; Mort, D.; Mason, M.; Evans, A. Stereo-specific cytotoxic effects of gossypol enantiomers and gossypolone in tumour cell lines. Cancer Lett. 1999, 135, 171–180. [Google Scholar] [CrossRef]
- Flack, M.R.; Pyle, R.G.; Mullen, N.M.; Lorenzo, B.; Wu, Y.W.; Knazek, R.A.; Nisula, B.C.; Reidenberg, M.M. Oral Gossypol in the Treatment of Metastatic Adrenal Cancer. J. Clin. Endocrinol. Metab. 2014, 76, 1019–1024. [Google Scholar]
- Van Poznak, C.; Seidman, A.D.; Reidenberg, M.M.; Moasser, M.M.; Sklarin, N.; Van Zee, K.; Borgen, P.; Gollub, M.; Bacotti, D.; Yao, T.J.; et al. Oral gossypol in the treatment of patients with refractory metastatic breast cancer: A phase I/II clinical trial. Breast Cancer Res. Treat. 2001, 66, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Paterni, I.; Rani, R.; Minutolo, F. Small-molecule inhibitors of human LDH5. Future Med. Chem. 2013, 5, 1967–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manerba, M.; Vettraino, M.; Fiume, L.; di Stefano, G.; Sartini, A.; Giacomini, E.; Buonfiglio, R.; Roberti, M.; Recanatini, M. Galloflavin (CAS 568-80-9): A Novel Inhibitor of Lactate Dehydrogenase. ChemMedChem 2012, 7, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stefano, G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef]
- Granchi, C.; Roy, S.; Giacomelli, C.; MacChia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef]
- Fiume, L.; Manerba, M.; Vettraino, M.; Di Stefano, G. Inhibition of lactate dehydrogenase activity as an approach to cancer therapy. Future Med. Chem. 2014, 6, 429–445. [Google Scholar] [CrossRef]
- Maftouh, M.; Avan, A.; Sciarrillo, R.; Granchi, C.; Leon, L.G.; Rani, R.; Funel, N.; Smid, K.; Honeywell, R.; Boggi, U.; et al. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br. J. Cancer 2014, 110, 172–182. [Google Scholar] [CrossRef]
- Schneider, C.C.; Archid, R.; Fischer, N.; Bühler, S.; Venturelli, S.; Berger, A.; Burkard, M.; Kirschniak, A.; Bachmann, R.; Königsrainer, A.; et al. Metabolic alteration—Overcoming therapy resistance in gastric cancer via PGK-1 inhibition in a combined therapy with standard chemotherapeutics. Int. J. Surg. 2015, 22, 92–98. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.S.; Sharp, P.A. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J. Exp. Med. 2012, 209, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Zeng, S.; Huang, M.; Qiu, Q.; Xiao, Y.; Shi, M.; Zhan, Z.; Liang, L.; Yang, X.; Xu, H. Inhibition of 6-phosphofructo-2-kinase suppresses fibroblast-like synoviocytes-mediated synovial inflammation and joint destruction in rheumatoid arthritis. Br. J. Pharmacol. 2017, 174, 893–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trefely, S.; Khoo, P.-S.; Krycer, J.R.; Chaudhuri, R.; Fazakerley, D.J.; Parker, B.L.; Sultani, G.; Lee, J.; Stephan, J.-P.; Torres, E.; et al. Kinome Screen Identifies PFKFB3 and Glucose Metabolism as Important Regulators of the Insulin/Insulin-like Growth Factor (IGF)-1 Signaling Pathway. J. Biol. Chem. 2015, 290, 25834–25846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clem, B.; Telang, S.; Clem, A.; Yalcin, A.; Meier, J.; Simmons, A.; Rasku, M.A.; Arumugam, S.; Dean, W.L.; Eaton, J.; et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 2008, 7, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, Y.; Wang, C.; Xia, W.R.; Zheng, J.Y.; Yang, J.; Liu, B.; Liu, J.Q.; Liu, L.F. Succinate induces synovial angiogenesis in rheumatoid arthritis through metabolic remodeling and HIF-1α/VEGF axis. Free Radic. Biol. Med. 2018, 126, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.T.; Tu, S.H.; Yang, P.S.; Hsu, S.P.; Lee, W.H.; Ho, C.T.; Wu, C.H.; Lai, Y.H.; Chen, M.Y.; Chen, L.C. Apple Polyphenol Phloretin Inhibits Colorectal Cancer Cell Growth via Inhibition of the Type 2 Glucose Transporter and Activation of p53-Mediated Signaling. J. Agric. Food Chem. 2016, 64, 6826–6837. [Google Scholar] [CrossRef] [PubMed]
- Makowski, L.; Chaib, M.; Rathmell, J.C. Immunometabolism: From basic mechanisms to translation. Immunol. Rev. 2020, 295, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.J.; Pearce, E.L. Driving immunity: All roads lead to metabolism. Nat. Rev. Immunol. 2018, 18, 81–82. [Google Scholar] [CrossRef]
- Viale, A.; Draetta, G.F. Metabolic Features of Cancer Treatment Resistance. In Recent Results in Cancer Research; Springer: New York, NY, USA, 2016; Volume 207, pp. 135–156. ISBN 9783319421186. [Google Scholar]
- RA, C.; IS, H.; TW, M. Re: Regulation of Cancer Cell Metabolism. J. Urol. Surg. 2017, 222. [Google Scholar] [CrossRef]
- Kareva, I.; Hahnfeldt, P. The Emerging “Hallmarks” of Metabolic Reprogramming and Immune Evasion: Distinct or Linked? Cancer Res. 2013, 73, 2737–2742. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Shoshan, J.; Maysel-Auslender, S.; Mor, A.; Keren, G.; George, J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1α. Eur. J. Immunol. 2008, 38, 2412–2418. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Lochner, M.; Berod, L.; Sparwasser, T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015, 36, 81–91. [Google Scholar] [CrossRef]
- Guo, C.; Chen, S.; Liu, W.; Ma, Y.; Li, J.; Fisher, P.B.; Fang, X.; Wang, X. Immunometabolism: A new target for improving cancer immunotherapy. Adv. Cancer Res. 2019, 143, 195–253. [Google Scholar] [CrossRef]
- Yu, Y.-R.; Ho, P.-C. Sculpting tumor microenvironment with immune system: From immunometabolism to immunoediting. Clin. Exp. Immunol. 2019, 197, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic Reprogramming of Cancer-Associated Fibroblasts by IDH3α Downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [Green Version]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8+ T Cells to protect tumour cells. Nat. Commun. 2018, 9, 948. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. Immunometabolism in early and late stages of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 291–301. [Google Scholar] [CrossRef]
- Pucino, V.; Certo, M.; Varricchi, G.; Marone, G.; Ursini, F.; Rossi, F.W.; De Paulis, A.; Mauro, C.; Raza, K.; Buckley, C.D. Metabolic Checkpoints in Rheumatoid Arthritis. Front. Physiol. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.; Liu, H.; Xu, L.; Li, Y.; Liu, X.; Shi, L.; Su, Y.; Qiu, X.; Zhang, X.; Yang, Y.; et al. Hypoxia-inducible factor-1α perpetuates synovial fibroblast interactions with T cells and B cells in rheumatoid arthritis. Eur. J. Immunol. 2016, 46, 742–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucino, V.; Bombardieri, M.; Pitzalis, C.; Mauro, C. Lactate at the crossroads of metabolism, inflammation, and autoimmunity. Eur. J. Immunol. 2017, 47, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Artyomov, M.N.; Van den Bossche, J. Immunometabolism in the Single-Cell Era. Cell Metab. 2020, 32, 710–725. [Google Scholar] [CrossRef]
- Han, J.; Datla, R.; Chan, S.; Borchers, C.H. Mass spectrometry-based technologies for high-throughput metabolomics. Bioanalysis 2009, 1, 1665–1684. [Google Scholar] [CrossRef]
- Li, C.; Chen, B.; Fang, Z.; Leng, Y.; Wang, D.; Chen, F.; Xu, X.; Sun, Z. Metabolomics in the development and progression of rheumatoid arthritis: A systematic review. Jt. Bone Spine 2020, 87, 425–430. [Google Scholar] [CrossRef]
- Sweeney, S.R.; Kavanaugh, A.; Lodi, A.; Wang, B.; Boyle, D.; Tiziani, S.; Guma, M. Metabolomic profiling predicts outcome of rituximab therapy in rheumatoid arthritis. RMD Open 2016, 2, e000289. [Google Scholar] [CrossRef] [Green Version]
- Funk, R.S.; Singh, R.K.; Becker, M.L. Metabolomic Profiling to Identify Molecular Biomarkers of Cellular Response to Methotrexate In Vitro. Clin. Transl. Sci. 2020, 13, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Teitsma, X.M.; Yang, W.; Jacobs, J.W.G.; Pethö-Schramm, A.; Borm, M.E.A.; Harms, A.C.; Hankemeier, T.; van Laar, J.M.; Bijlsma, J.W.J.; Lafeber, F.P.J.G. Baseline metabolic profiles of early rheumatoid arthritis patients achieving sustained drug-free remission after initiating treat-to-target tocilizumab, methotrexate, or the combination: Insights from systems biology. Arthritis Res. Ther. 2018, 20, 230. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.K.; Kim, S.; Hwang, J.; Kim, J.; Kim, K.H.; Cha, H.S. GC/TOF-MS-based metabolomic profiling in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Jt. Bone Spine 2016, 83, 707–713. [Google Scholar] [CrossRef]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 2019, 20, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Damiani, C.; Maspero, D.; Di Filippo, M.; Colombo, R.; Pescini, D.; Graudenzi, A.; Westerhoff, H.V.; Alberghina, L.; Vanoni, M.; Mauri, G. Integration of single-cell RNA-seq data into population models to characterize cancer metabolism. PLoS Comput. Biol. 2019, 15, e1006733. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.K.; DeBerardinis, R.J. Applications of metabolomics to study cancer metabolism. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Ortmayr, K.; Dubuis, S.; Zampieri, M. Metabolic profiling of cancer cells reveals genome-wide crosstalk between transcriptional regulators and metabolism. Nat. Commun. 2019, 10, 1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Zhao, H.; Zhang, X.; Zhao, X.; Song, Z.; Ouyang, J. Metabolic Discrimination of Breast Cancer Subtypes at the Single-Cell Level by Multiple Microextraction Coupled with Mass Spectrometry. Anal. Chem. 2019, 91, 3667–3674. [Google Scholar] [CrossRef] [PubMed]
- Le Novère, N.; Hucka, M.; Mi, H.; Moodie, S.; Schreiber, F.; Sorokin, A.; Demir, E.; Wegner, K.; Aladjem, M.I.; Wimalaratne, S.M.; et al. The Systems Biology Graphical Notation. Nat. Biotechnol. 2009, 27, 735–741. [Google Scholar] [CrossRef]
- Le Novere, N. Quantitative and logic modelling of molecular and gene networks. Nat. Rev. Genet. 2015, 16, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Ostaszewski, M.; Kalliolias, G.D.; Chiocchia, G.; Olaso, R.; Petit-Teixeira, E.; Helikar, T.; Niarakis, A. Computational Systems Biology Approach for the Study of Rheumatoid Arthritis: From a Molecular Map to a Dynamical Model. Genom. Comput. Biol. 2017, 4, 100050. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Kalliolias, G.D.; Ostaszewski, M.; Veyssiere, M.; Pilalis, E.; Gawron, P.; Mazein, A.; Bonnet, E.; Petit-Teixeira, E.; Niarakis, A. RA-map: Building a state-of-the-art interactive knowledge base for rheumatoid arthritis. Database 2020, 2020, 1–18. [Google Scholar] [CrossRef]
- Krogan, N.J.; Lippman, S.; Agard, D.A.; Ashworth, A.; Ideker, T. The Cancer Cell Map Initiative: Defining the Hallmark Networks of Cancer. Mol. Cell 2015, 58, 690–698. [Google Scholar] [CrossRef] [Green Version]
- Rozenblatt-Rosen, O.; Regev, A.; Oberdoerffer, P.; Nawy, T.; Hupalowska, A.; Rood, J.E.; Ashenberg, O.; Cerami, E.; Coffey, R.J.; Demir, E.; et al. The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell 2020, 181, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Kuperstein, I.; Bonnet, E.; Nguyen, H.A.; Cohen, D.; Viara, E.; Grieco, L.; Fourquet, S.; Calzone, L.; Russo, C.; Kondratova, M.; et al. Atlas of Cancer Signalling Network: A systems biology resource for integrative analysis of cancer data with Google Maps. Oncogenesis 2015, 4, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noronha, A.; Daníelsdóttir, A.D.; Gawron, P.; Jóhannsson, F.; Jónsdóttir, S.; Jarlsson, S.; Gunnarsson, J.P.; Brynjólfsson, S.; Schneider, R.; Thiele, I.; et al. ReconMap: An interactive visualization of human metabolism. Bioinformatics 2017, 33, 605–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.L.; Kocabaş, P.; Wang, H.; Cholley, P.-E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An atlas of human metabolism. Sci. Signal. 2020, 13, eaaz1482. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, E.; Bucher, J.; Ryll, A.; Niklas, J.; Mauch, K.; Klamt, S.; Rocha, M.; Saez-Rodriguez, J. Bridging the layers: Towards integration of signal transduction, regulation and metabolism into mathematical models. Mol. Biosyst. 2013, 9, 1576. [Google Scholar] [CrossRef] [Green Version]
- Reed, J.L.; Senger, R.S.; Antoniewicz, M.R.; Young, J.D. Computational Approaches in Metabolic Engineering. J. Biomed. Biotechnol. 2010, 2010, 1–7. [Google Scholar] [CrossRef]
- Raman, K.; Chandra, N. Flux balance analysis of biological systems: Applications and challenges. Brief. Bioinform. 2009, 10, 435–449. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Cazzaniga, P.; Damiani, C.; Besozzi, D.; Colombo, R.; Nobile, M.; Gaglio, D.; Pescini, D.; Molinari, S.; Mauri, G.; Alberghina, L.; et al. Computational Strategies for a System-Level Understanding of Metabolism. Metabolites 2014, 4, 1034–1087. [Google Scholar] [CrossRef]
- Wagner, A.; Wang, C.; DeTomaso, D.; Avila-Pacheco, J.; Zaghouani, S.; Fessler, J.; Akama-Garren, E.; Pierce, K.; Ron-Harel, N.; Douglas, V.P.; et al. In silico modeling of metabolic state in single Th17 cells reveals novel regulators of inflammation and autoimmunity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Baker, M.; Denman-Johnson, S.; Brook, B.S.; Gaywood, I.; Owen, M.R. Mathematical modelling of cytokine-mediated inflammation in rheumatoid arthritis. Math. Med. Biol. 2013, 30, 311–337. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, F.R.; Chaplain, M.A.J.; Eftimie, R. Quantitative Predictive Modelling Approaches to Understanding Rheumatoid Arthritis: A Brief Review. Cells 2019, 9, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moise, N.; Friedman, A. Rheumatoid arthritis—A mathematical model. J. Theor. Biol. 2019, 461, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Rullmann, J.A.C.; Struemper, H.; Defranoux, N.A.; Ramanujan, S.; Meeuwisse, C.M.L.; van Elsas, A. Systems biology for battling rheumatoid arthritis: Application of the Entelos PhysioLab platform. IEE Proc. Syst. Biol. 2005, 152, 256. [Google Scholar] [CrossRef] [PubMed]
- Deisboeck, T.S.; Stamatakos, G.S. (Eds.) Multiscale Cancer Modeling; CRC Press: Boca Raton, FL, USA, 2010; ISBN 9780429075605. [Google Scholar]
- Metzcar, J.; Wang, Y.; Heiland, R.; Macklin, P. A Review of Cell-Based Computational Modeling in Cancer Biology. JCO Clin. Cancer Inform. 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bouhaddou, M.; Barrette, A.M.; Stern, A.D.; Koch, R.J.; DiStefano, M.S.; Riesel, E.A.; Santos, L.C.; Tan, A.L.; Mertz, A.E.; Birtwistle, M.R. A mechanistic pan-cancer pathway model informed by multi-omics data interprets stochastic cell fate responses to drugs and mitogens. PLoS Comput. Biol. 2018, 14, e1005985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, K.-A.; Gong, C.; Jamalian, S.; Popel, A. Multiscale Agent-Based and Hybrid Modeling of the Tumor Immune Microenvironment. Processes 2019, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Hartung, N.; Mollard, S.; Barbolosi, D.; Benabdallah, A.; Chapuisat, G.; Henry, G.; Giacometti, S.; Iliadis, A.; Ciccolini, J.; Faivre, C.; et al. Mathematical Modeling of Tumor Growth and Metastatic Spreading: Validation in Tumor-Bearing Mice. Cancer Res. 2014, 74, 6397–6407. [Google Scholar] [CrossRef] [Green Version]
- Shamsi, M.; Saghafian, M.; Dejam, M.; Sanati-Nezhad, A. Mathematical Modeling of the Function of Warburg Effect in Tumor Microenvironment. Sci. Rep. 2018, 8, 8903. [Google Scholar] [CrossRef]
- Gong, C.; Milberg, O.; Wang, B.; Vicini, P.; Narwal, R.; Roskos, L.; Popel, A.S. A computational multiscale agent-based model for simulating spatio-temporal tumour immune response to PD1 and PDL1 inhibition. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef] [Green Version]
- Smallbone, K.; Gatenby, R.A.; Gillies, R.J.; Maini, P.K.; Gavaghan, D.J. Metabolic changes during carcinogenesis: Potential impact on invasiveness. J. Theor. Biol. 2007, 244, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, M.; Finley, S.D. Computational Model Predicts the Effects of Targeting Cellular Metabolism in Pancreatic Cancer. Front. Physiol. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndt, N.; Egners, A.; Mastrobuoni, G.; Vvedenskaya, O.; Fragoulis, A.; Dugourd, A.; Bulik, S.; Pietzke, M.; Bielow, C.; van Gassel, R.; et al. Kinetic modelling of quantitative proteome data predicts metabolic reprogramming of liver cancer. Br. J. Cancer 2020, 122, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Yizhak, K.; Le Dévédec, S.E.; Rogkoti, V.M.; Baenke, F.; Boer, V.C.; Frezza, C.; Schulze, A.; Water, B.; Ruppin, E. A computational study of the Warburg effect identifies metabolic targets inhibiting cancer migration. Mol. Syst. Biol. 2014, 10, 744. [Google Scholar] [CrossRef]
- Vazquez, A.; Liu, J.; Zhou, Y.; Oltvai, Z.N. Catabolic efficiency of aerobic glycolysis: The Warburg effect revisited. BMC Syst. Biol. 2010, 4. [Google Scholar] [CrossRef] [Green Version]
- Heidary, Z.; Ghaisari, J.; Moein, S.; Haghjooy Javanmard, S. The double-edged sword role of fibroblasts in the interaction with cancer cells; an agent-based modeling approach. PLoS ONE 2020, 15, e0232965. [Google Scholar] [CrossRef]
- Helikar, T.; Konvalina, J.; Heidel, J.; Rogers, J.A. Emergent decision-making in biological signal transduction networks. Proc. Natl. Acad. Sci. USA 2008, 105, 1913–1918. [Google Scholar] [CrossRef] [Green Version]
- Aghamiri, S.S.; Singh, V.; Naldi, A.; Helikar, T.; Soliman, S.; Niarakis, A. Automated inference of Boolean models from molecular interaction maps using CaSQ. Bioinformatics 2020, 1–13. [Google Scholar] [CrossRef]
- Marmiesse, L.; Peyraud, R.; Cottret, L. FlexFlux: Combining metabolic flux and regulatory network analyses. BMC Syst. Biol. 2015, 9, 93. [Google Scholar] [CrossRef] [Green Version]
- Ulfenborg, B. Vertical and horizontal integration of multi-omics data with miodin. BMC Bioinform. 2019, 20, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Palsson, B.; Zengler, K. The challenges of integrating multi-omic data sets. Nat. Chem. Biol. 2010, 6, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Szigeti, B.; Roth, Y.D.; Sekar, J.A.P.; Goldberg, A.P.; Pochiraju, S.C.; Karr, J.R. A blueprint for human whole-cell modeling. Curr. Opin. Syst. Biol. 2018, 7, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karr, J.R.; Sanghvi, J.C.; MacKlin, D.N.; Gutschow, M.V.; Jacobs, J.M.; Bolival, B.; Assad-Garcia, N.; Glass, J.I.; Covert, M.W. A whole-cell computational model predicts phenotype from genotype. Cell 2012, 150, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macklin, P. Key challenges facing data-driven multicellular systems biology. Gigascience 2019, 8, 1–8. [Google Scholar] [CrossRef]
- Evers, T.M.J.; Hochane, M.; Tans, S.J.; Heeren, R.M.A.; Semrau, S.; Nemes, P.; Mashaghi, A. Deciphering Metabolic Heterogeneity by Single-Cell Analysis. Anal. Chem. 2019, 91, 13314–13323. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Zhang, Y.; Liu, J. Advancing single-cell proteomics and metabolomics with microfluidic technologies. Analyst 2019, 144, 846–858. [Google Scholar] [CrossRef]
- Bock, C.; Farlik, M.; Sheffield, N.C. Multi-Omics of Single Cells: Strategies and Applications. Trends Biotechnol. 2016, 34, 605–608. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, R.; Rao, M.; Zhang, C.; Agrati, C.; Ippolito, G.; Wang, F.-S.; Zumla, A.; Maeurer, M. Immunometabolism: New insights and lessons from antigen-directed cellular immune responses. Semin. Immunopathol. 2020, 42, 279–313. [Google Scholar] [CrossRef]
- Galgani, M.; Bruzzaniti, S.; Matarese, G. Immunometabolism and autoimmunity. Curr. Opin. Immunol. 2020, 67, 10–17. [Google Scholar] [CrossRef]
- Bardini, R.; Politano, G.; Benso, A.; Di Carlo, S. Multi-level and hybrid modelling approaches for systems biology. Comput. Struct. Biotechnol. J. 2017, 15, 396–402. [Google Scholar] [CrossRef] [Green Version]
- Fromentin, J.; Eveillard, D.; Roux, O. Hybrid modeling of biological networks: Mixing temporal and qualitative biological properties. BMC Syst. Biol. 2010, 4. [Google Scholar] [CrossRef] [PubMed]
- Bersanelli, M.; Mosca, E.; Remondini, D.; Giampieri, E.; Sala, C.; Castellani, G.; Milanesi, L. Methods for the integration of multi-omics data: Mathematical aspects. BMC Bioinform. 2016, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiore, S.; Bakhouya, M.; Smari, W.W. On the road to exascale: Advances in High Performance Computing and Simulations—An overview and editorial. Future Gener. Comput. Syst. 2018, 82, 450–458. [Google Scholar] [CrossRef]
- HPC Wire. Available online: https://www.hpcwire.com/off-the-wire/hpc-exascale-centre-of-excellence-will-support-european-personalised-medicine/ (accessed on 27 October 2020).



{kind=link}
{kind=link}
{kind=link}
| Metabolic Target | Drug or Compound | |
|---|---|---|
| Rheumatoid Arthritis Synovial Fibroblast | Cancer-Associated Fibroblast | |
| HK2 | 3-Bromopyruvate * [95,96,97,98,99,100] 2-Deoxyglucose * [95] Lonidamine * [96] Tofacitinib [101] | 3-Bromopyruvate * [102,103,104,105,106] 2-Deoxyglucose * [102,107] Lonidamine * [102,108] T-Lipo-3-BP [104] |
| GLUT | WZB117* [109] Tumor Necrosis Factor-α inhibitor [110] | WZB117 * [111] Fasentin [112] Phloretin [102,113,114,115] |
| MCT | Metformin * [95,109,116] MCT4-siRNA [117] | Metformin * [91,102,118,119] Quercetin [120,121] NAC [121,122,123,124,125,126] α-Cyano-4-hydroxycinnamic [119,127,128] Acetylcysteine combined with Topotecan [66,123] |
| LDH | Tofacitinib [101] | FX11 [102,122,129] Oxamate [126,130] Quinoline 3-sulfonamides [131] Gossypol [132,133,134,135,136] Galloflavin [137,138] NHI [139,140,141] |
| PGK | PGK1-SiRNA [98] | Adenovirus-shPGK1 [142] |
| PK | TEPP-46 [109] Tumor Necrosis Factor-α inhibitor [110] | Shikonin and its analogs [143] Alkannin [94] PKM2-siRNA [144] |
| PFK | 3 PO * [110] PFK15 [145,146] PFKFB3-SiRNA [146] | 3 PO * [147] |
| GAPDH | Heptelidic Acid [109] | 3-Bromopyruvate * [105,106] |
| Tumor Necrosis Factor-α inhibitor [110] | ||
| SDH | Saponin [114] Dimethyl Malonate [148] | 3-Bromopyruvate * [105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aghakhani, S.; Zerrouk, N.; Niarakis, A. Metabolic Reprogramming of Fibroblasts as Therapeutic Target in Rheumatoid Arthritis and Cancer: Deciphering Key Mechanisms Using Computational Systems Biology Approaches. Cancers 2021, 13, 35. https://doi.org/10.3390/cancers13010035
Aghakhani S, Zerrouk N, Niarakis A. Metabolic Reprogramming of Fibroblasts as Therapeutic Target in Rheumatoid Arthritis and Cancer: Deciphering Key Mechanisms Using Computational Systems Biology Approaches. Cancers. 2021; 13(1):35. https://doi.org/10.3390/cancers13010035
Chicago/Turabian StyleAghakhani, Sahar, Naouel Zerrouk, and Anna Niarakis. 2021. "Metabolic Reprogramming of Fibroblasts as Therapeutic Target in Rheumatoid Arthritis and Cancer: Deciphering Key Mechanisms Using Computational Systems Biology Approaches" Cancers 13, no. 1: 35. https://doi.org/10.3390/cancers13010035