mTOR-Dependent Role of Sestrin2 in Regulating Tumor Progression of Human Endometrial Cancer

 ,
,  ,
, 
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
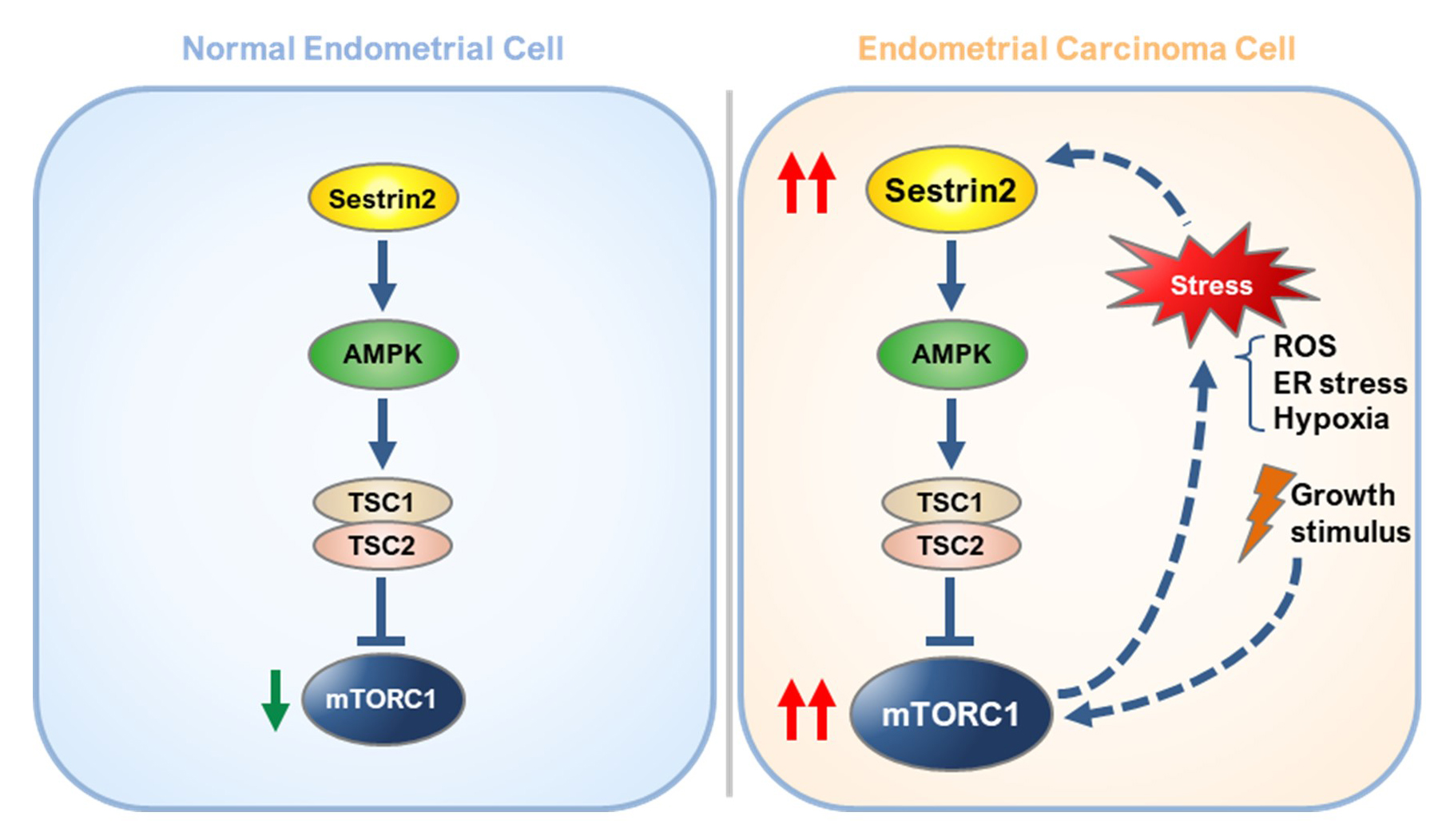
1. Introduction
2. Results
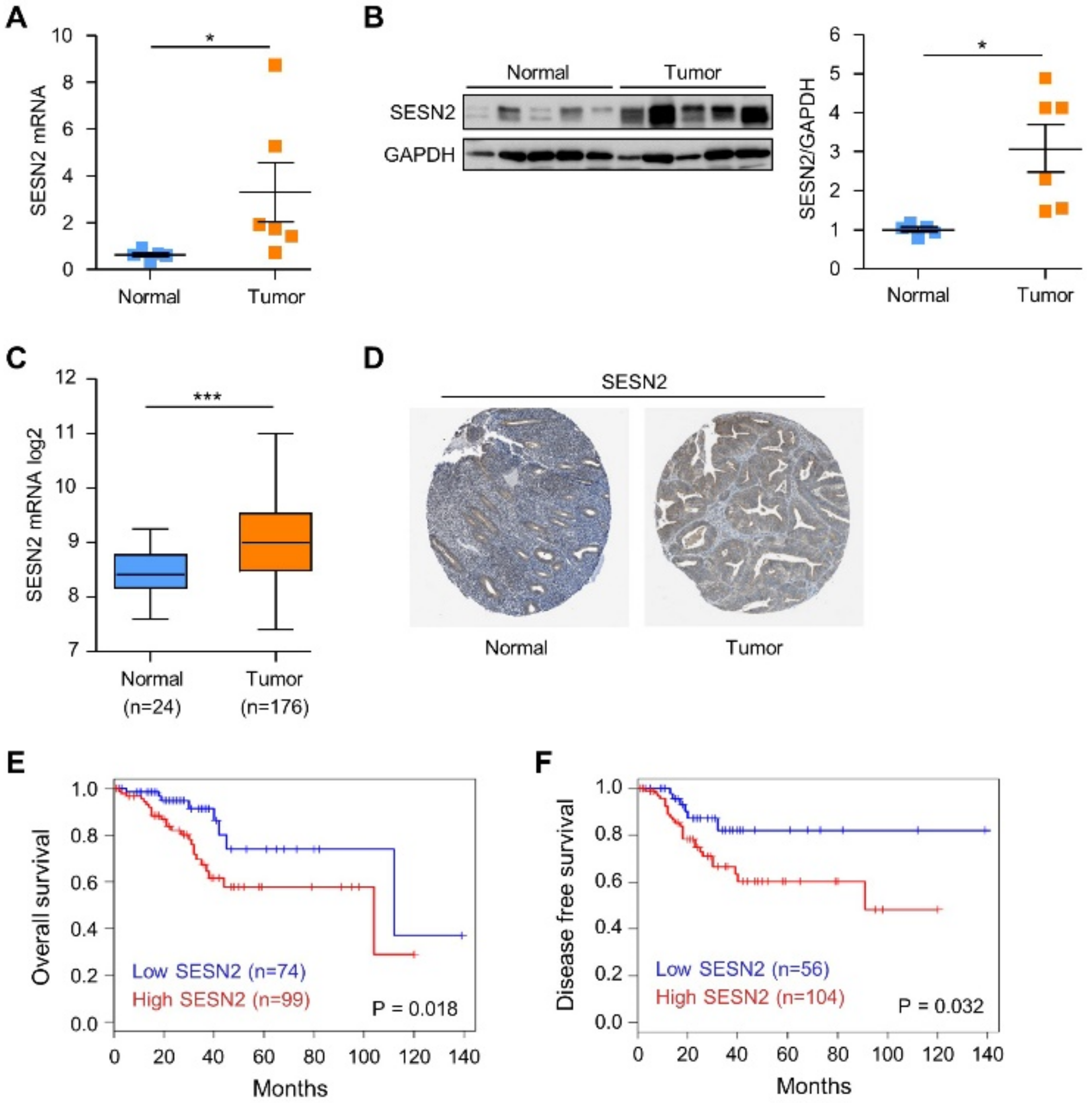
2.1. SESN2 Expression and Its Clinical Significance in Endometrial Cancer
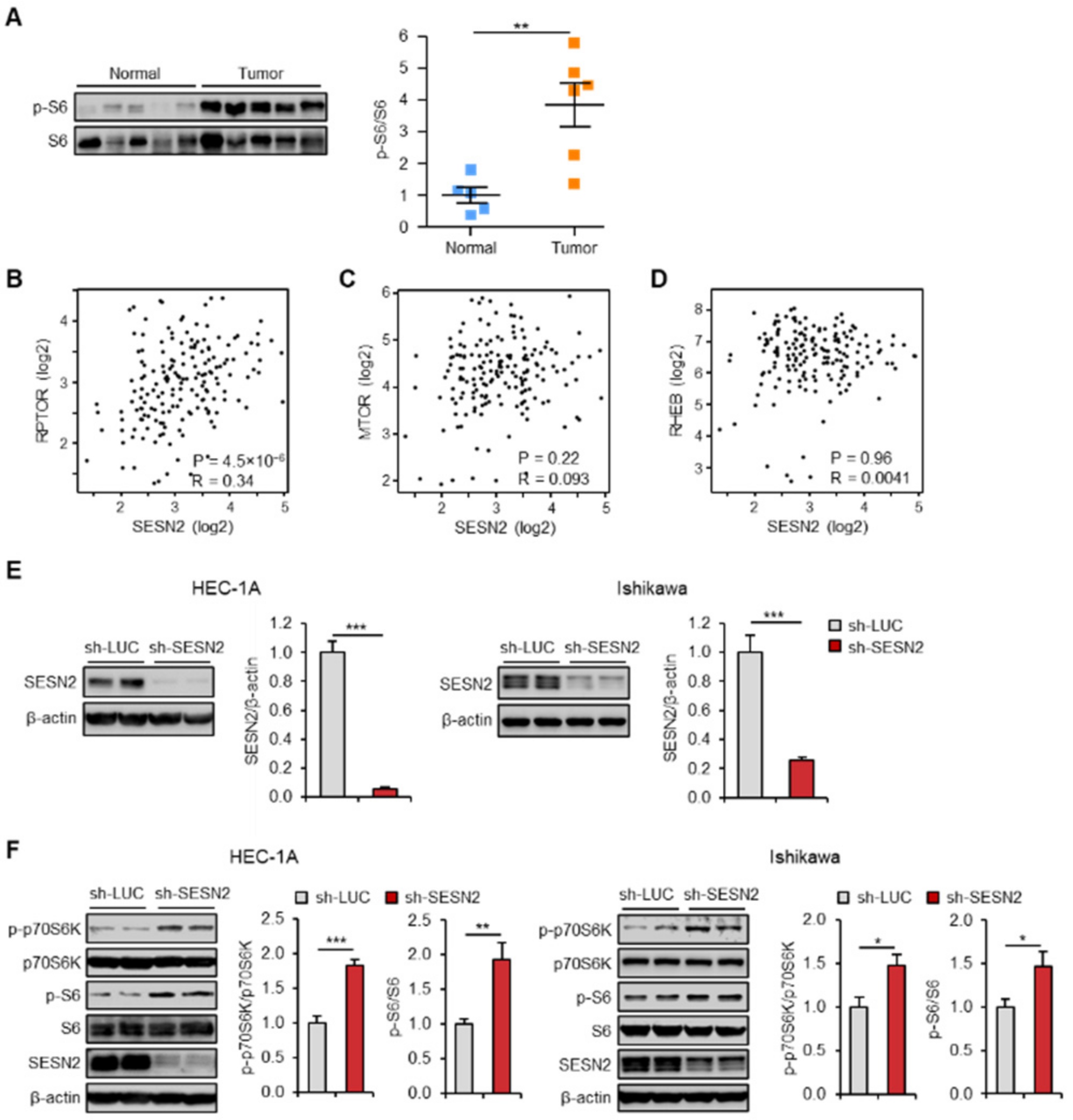
2.2. Expression of SESN2 Correlates with mTOR Pathway Activity in Endometrial Cancer
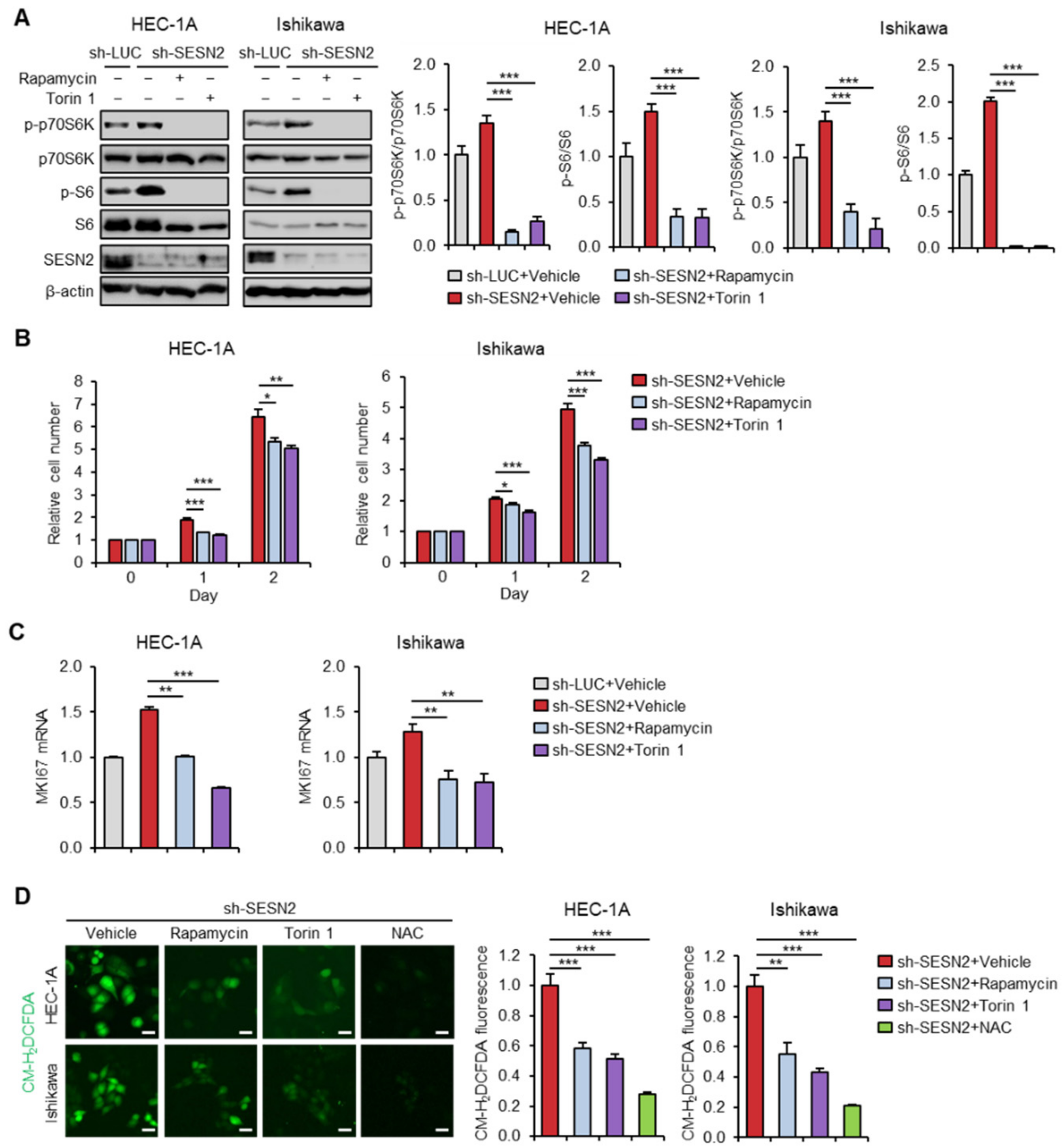
2.3. Knockdown of SESN2 Promotes Endometrial Cancer Cell Proliferation and ROS Production
2.4. SESN2 Regulates EMT and Migration in Endometrial Cancer Cells
2.5. SESN2 Regulates Endometrial Cancer Cell Proliferation and ROS Production in a mTOR-Dependent Manner
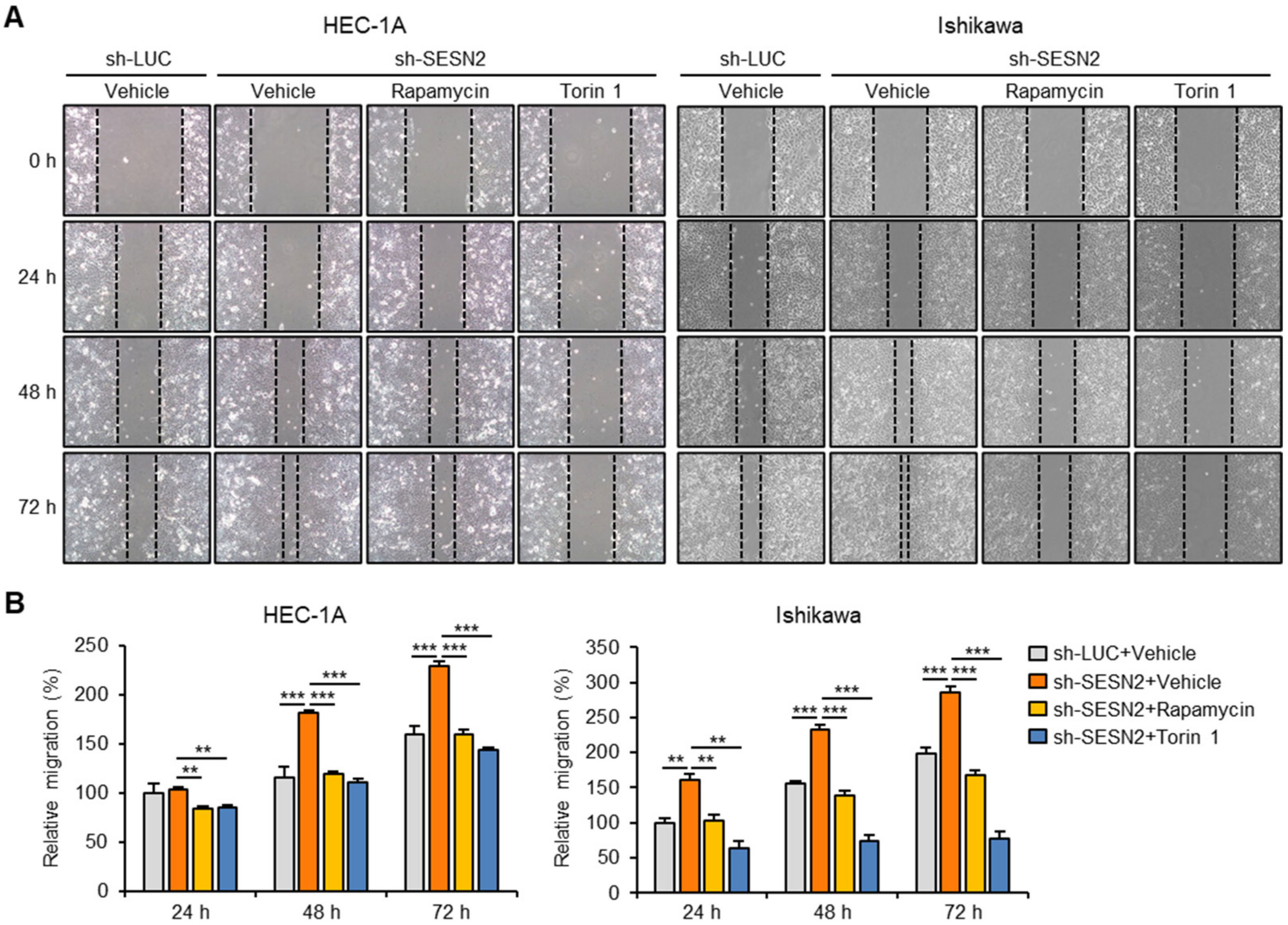
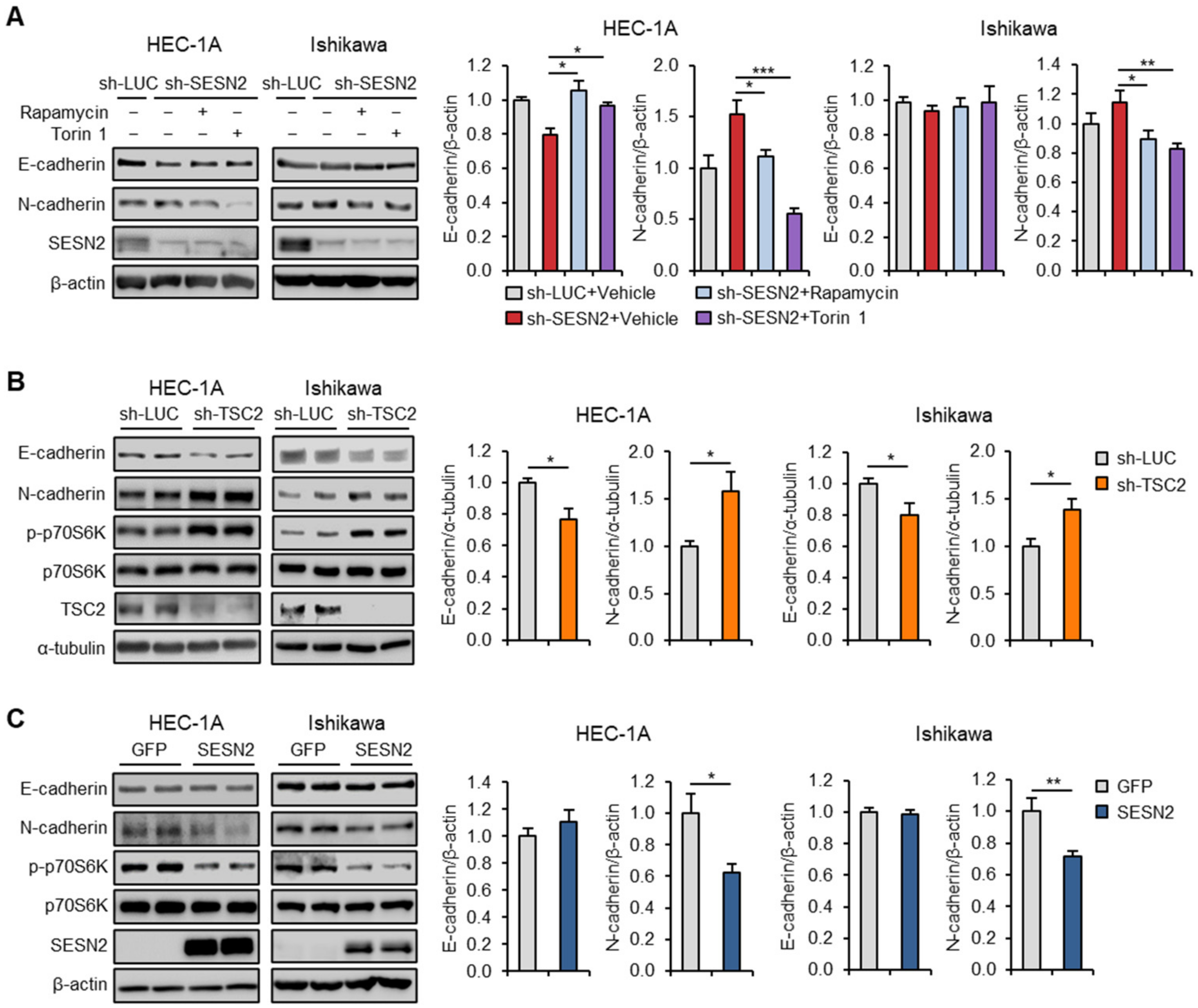
2.6. SESN2 Regulates EMT and Migration in Endometrial Cancer Cells via the mTORC1 Pathway
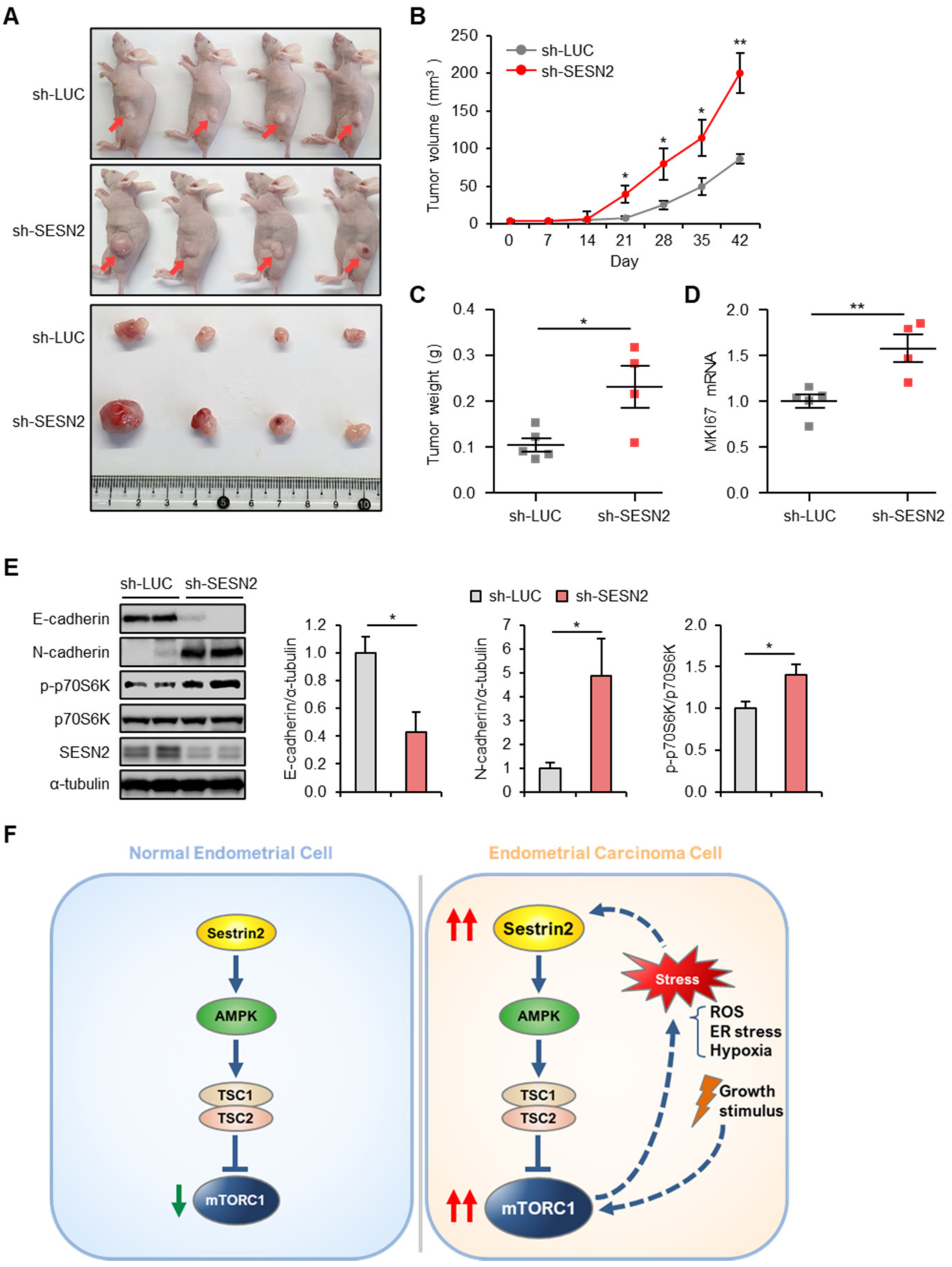
2.7. SESN2 Regulates Tumor Growth in a Xenograft Nude Mice Model
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Patient Tissue Samples
4.3. GEPIA Database Analysis
4.4. UCSC Cancer Genomics Browser Analysis
4.5. Cell Culture and Treatments
4.6. Viral Transduction
4.7. Quantitative Real-Time PCR
4.8. Immunoblotting
4.9. Cell Proliferation Assay
4.10. Detection of Reactive Oxygen Species (ROS)
4.11. Wound-Healing Assay
4.12. In Vivo Tumorigenesis
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; He, X.-Y.; Wang, R.; Wang, Z.; Wang, Y.-G. High leptin level is an independent risk factor of endometrial cancer: A meta-analysis. Cell. Physiol. Biochem. 2014, 34, 1477–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, F.; Shi, J.; Long, Y.; Tian, H.; Li, X.; Zhao, A.Z.; Li, R.F.; Chen, T. Adiponectin and endometrial cancer: A systematic review and meta-analysis. Cell. Physiol. Biochem. 2015, 36, 1670–1678. [Google Scholar] [CrossRef] [PubMed]
- Bradford, L.S.; Rauh-Hain, J.A.; Schorge, J.; Birrer, M.J.; Dizon, D.S. Advances in the management of recurrent endometrial cancer. Am. J. Clin. Oncol. 2015, 38, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.S. Improving survival after endometrial cancer: The big picture. J. Gynecol. Oncol. 2015, 26, 227–231. [Google Scholar] [CrossRef]
- Cao, C.; Zhou, J.-Y.; Xie, S.; Guo, X.-J.; Li, G.-T.; Gong, Y.-J.; Yang, W.-J.; Li, Z.; Zhong, R.-H.; Shao, H.-H.; et al. Metformin enhances nomegestrol acetate suppressing growth of endometrial cancer cells and may correlate to downregulating mTOR activity In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 3308. [Google Scholar] [CrossRef] [Green Version]
- Traen, K.; Holund, B.; Mogensen, O. Accuracy of preoperative tumor grade and intraoperative gross examination of myometrial invasion in patients with endometrial cancer. Acta Obstet. Gynecol. Scand. 2007, 86, 739–741. [Google Scholar] [CrossRef]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef] [Green Version]
- Thiem, S.; Pierce, T.P.; Palmieri, M.; Putoczki, T.L.; Buchert, M.; Preaudet, A.; Farid, R.O.; Love, C.; Catimel, B.; Lei, Z.; et al. mTORC1 inhibition restricts inflammation-associated gastrointestinal tumorigenesis in mice. J. Clin. Investig. 2013, 123, 767–781. [Google Scholar] [CrossRef] [Green Version]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Park, H.-W.; Park, H.; Ro, S.-H.; Jang, I.; Semple, I.A.; Kim, D.N.; Kim, M.; Nam, M.; Zhang, D.; Yin, L.; et al. Hepatoprotective role of Sestrin2 against chronic ER stress. Nat. Commun. 2014, 5, 4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, S.S.; Case, S. Everolimus (RAD001) in the treatment of advanced renal cell carcinoma: A review. Oncologist 2010, 15, 236–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faivre, S.; Kroemer, G.; Raymond, E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 671–688. [Google Scholar] [CrossRef]
- Budanov, A.V. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid. Redox Signal. 2011, 15, 1679–1690. [Google Scholar] [CrossRef]
- Lee, J.H.; Budanov, A.V.; Karin, M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 2013, 18, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Budanov, A.V.; Park, E.J.; Birse, R.; Kim, T.E.; Perkins, G.A.; Ocorr, K.; Ellisman, M.H.; Bodmer, R.; Bier, E.; et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 2010, 327, 1223–1228. [Google Scholar] [CrossRef] [Green Version]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Shin, J.; Hong, Y.; Shin, S.M.; Shin, H.W.; Shin, J.; Lee, S.K.; Park, H.-W. Sestrin2 alleviates palmitate-induced endoplasmic reticulum stress, apoptosis, and defective invasion of human trophoblast cells. Am. J. Reprod. Immunol. 2020, 83, 13222. [Google Scholar] [CrossRef]
- Budanov, A.V.; Sablina, A.A.; Feinstein, E.; Koonin, E.V.; Chumakov, P.M.; Damsté, J.S.S.; Muyzer, G.; Abbas, B.; Rampen, S.W.; Massé, G.; et al. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004, 304, 596–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budanov, A.V.; Shoshani, T.; Faerman, A.; Zelin, E.; Kamer, I.; Kalinski, H.; Gorodin, S.; Fishman, A.; Chajut, A.; Einat, P.; et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 2002, 21, 6017–6031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, S.H.; D’Alessio, D.; Thomas, G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006, 3, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Budanov, A.V.; Talukdar, S.; Park, E.J.; Park, H.L.; Park, H.-W.; Bandyopadhyay, G.; Li, N.; Aghajan, M.; Jang, I.; et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012, 16, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Ro, S.H.; Kim, M.; Park, H.W.; Semple, I.A.; Park, H.; Cho, U.S.; Wang, W.; Guan, K.L.; Karin, M.; et al. Sestrin2 inhibits Mtorc1 through modulation of gator complexes. Sci. Rep. 2015, 5, 9502. [Google Scholar] [CrossRef]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.-L.; Karin, M.; Budanov, A.V. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014, 9, 1281–1291. [Google Scholar] [CrossRef] [Green Version]
- Ro, S.-H.; Xue, X.; Ramakrishnan, S.K.; Cho, C.-S.; Namkoong, S.; Jang, I.; Semple, I.A.; Ho, A.; Park, H.-W.; Shah, Y.M.; et al. Tumor suppressive role of sestrin2 during colitis and colon carcinogenesis. eLife 2016, 5, e12204. [Google Scholar] [CrossRef]
- Chen, S.; Yan, W.; Lang, W.; Yu, J.; Xu, L.; Xu, X.; Liu, Y.; Bao, H. SESN2 correlates with advantageous prognosis in hepatocellular carcinoma. Diagn. Pathol. 2017, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-B.; Xuan, Y.; Shi, W.-J.; Chi, F.; Xing, R.; Zeng, Y.-C. Sestrin2 expression is a favorable prognostic factor in patients with non-small cell lung cancer. Am. J. Transl. Res. 2016, 8, 1903–1909. [Google Scholar]
- Liang, Y.; Zhu, J.; Huang, H.; Xiang, D.; Li, Y.; Zhang, D.; Li, J.; Wang, Y.; Jin, H.; Jiang, G.; et al. Sesn2/Sestrin 2 induction-mediated autophagy and inhibitory effect of Isorhapontigenin (Iso) on human bladder cancers. Autophagy 2016, 12, 1229–1239. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Huang, Q.; Niu, K.; Wang, B.; Li, Y.; Dai, C.; Chen, Z.-N.; Tao, K.; Dai, J.-Y. Sestrin 2 confers primary resistance to sorafenib by simultaneously activating AKT and AMPK in hepatocellular carcinoma. Cancer Med. 2018, 7, 5691–5703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR pathways in cancer and autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Nakashima, A.; Guo, L.; Coffman, K.; Tamanoi, F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene 2010, 29, 2746–2752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Guan, K.-L. mTOR as a central hub of nutrient signalling and cell growth. Nature 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Hecht, F.; Pessoa, C.F.; Gentile, L.B.; Rosenthal, D.; Carvalho, D.P.; Fortunato, R.S. The role of oxidative stress on breast cancer development and therapy. Tumor Biol. 2016, 37, 4281–4291. [Google Scholar] [CrossRef]
- Morry, J.; Ngamcherdtrakul, W.; Yantasee, W. Oxidative stress in cancer and fibrosis: Opportunity for therapeutic intervention with antioxidant compounds, enzymes, and nanoparticles. Redox Biol. 2017, 11, 240–253. [Google Scholar] [CrossRef]
- Bae, S.-H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Mittal, V. Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Pavlidou, A.; Vlahos, N.F. Molecular alterations of PI3K/Akt/mTOR pathway: A therapeutic target in endometrial cancer. Sci. World J. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Korets, S.B.; Czok, S.; Blank, S.V.; Curtin, J.P.; Schneider, R.J. Targeting the mTOR/4E-BP pathway in endometrial cancer. Clin. Cancer Res. 2011, 17, 7518–7528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR Interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. p53 strikes mTORC1 by employing sestrins. Cell Metab. 2008, 8, 184–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Fu, Z.; Fang, M.; Guo, J.; Zhao, Q.; Lu, W.; Zhou, Q. Decreased expression of sestrin 2 predicts unfavorable outcome in colorectal cancer. Oncol. Rep. 2014, 33, 1349–1357. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The interplay of reactive oxygen species, hypoxia, inflammation, and sirtuins in cancer initiation and progression. Oxidative Med. Cell. Longev. 2015, 2016, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Heberle, A.M.; Prentzell, M.T.; Van Eunen, K.; Bakker, B.M.; Grellscheid, S.N.; Thedieck, K. Molecular mechanisms of mTOR regulation by stress. Mol. Cell. Oncol. 2014, 2, e970489. [Google Scholar] [CrossRef] [Green Version]
- D’Orazi, G.; Cirone, M. Mutant p53 and cellular stress pathways: A criminal alliance that promotes cancer progression. Cancers 2019, 11, 614. [Google Scholar] [CrossRef] [Green Version]
- Yecies, J.L.; Manning, B.D. mTOR links oncogenic signaling to tumor cell metabolism. J. Mol. Med. 2011, 89, 221–228. [Google Scholar] [CrossRef]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris-Hanon, O.; Furmento, V.A.; Rodríguez-Varela, M.S.; Mucci, S.; Espinosa, D.F.; Romorini, L.; Sevlever, G.E.; Scassa, M.E.; Videla-Richardson, G.A. The cell cycle inhibitors p21Cip1 and p27Kip1 control proliferation but enhance DNA damage resistance of glioma stem cells. Neoplasia 2017, 19, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Llanos, S.; Garcia-Pedrero, J.M. A new mechanism of regulation of P21 by the Mtorc1/4e-Bp1 pathway predicts clinical outcome of head and neck cancer. Mol. Cell Oncol. 2016, 3, e1159275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abukhdeir, A.M.; Park, B.H. p21 and p27: Roles in carcinogenesis and drug resistance. Expert Rev. Mol. Med. 2008, 10, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, K.; Seo, S.; Ki, S.H.; Shin, S.M. Sestrin2 inhibits hypoxia-inducible factor-1α accumulation via AMPK-mediated prolyl hydroxylase regulation. Free. Radic. Biol. Med. 2016, 101, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yin, K.; Falcon, D.M.; Xue, X. The interaction of Hemin and Sestrin2 modulates oxidative stress and colon tumor growth. Toxicol. Appl. Pharmacol. 2019, 374, 77–85. [Google Scholar] [CrossRef]
- Wei, J.-L.; Fang, M.; Fu, Z.-X.; Zhang, S.; Guo, J.-B.; Wang, R.; Lv, Z.-B.; Xiong, Y.-F. Sestrin 2 suppresses cells proliferation through AMPK/mTORC1 pathway activation in colorectal cancer. Oncotarget 2017, 8, 49318–49328. [Google Scholar] [CrossRef] [Green Version]
- Buitrago-Molina, L.E.; Marhenke, S.; Longerich, T.; Sharma, A.; Boukouris, A.E.; Geffers, R.; Guigas, B.; Manns, M.P.; Vogel, A. The degree of liver injury determines the role of p21 in liver regeneration and hepatocarcinogenesis in mice. Hepatology 2013, 58, 1143–1152. [Google Scholar] [CrossRef]
- Wang, L.-X.; Zhu, X.-M.; Yao, Y.-M. Sestrin2: Its potential role and regulatory mechanism in host immune response in diseases. Front. Immunol. 2019, 10, 2797. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-Y.; Wu, X.-Q.; Deng, R.; Sun, T.; Feng, G.-K.; Zhu, X.-F. Upregulation of sestrin 2 expression via JNK pathway activation contributes to autophagy induction in cancer cells. Cell. Signal. 2013, 25, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeussler, M.; Zweig, A.S.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Hinrichs, A.S.; Gonzalez, J.N.; et al. The UCSC genome browser database: 2019 update. Nucleic Acids Res. 2019, 47, D853–D858. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, J.; Bae, J.; Park, S.; Kang, H.-G.; Shin, S.M.; Won, G.; Kim, J.-S.; Cho, S.-G.; Choi, Y.; Oh, S.-M.; et al. mTOR-Dependent Role of Sestrin2 in Regulating Tumor Progression of Human Endometrial Cancer. Cancers 2020, 12, 2515. https://doi.org/10.3390/cancers12092515
Shin J, Bae J, Park S, Kang H-G, Shin SM, Won G, Kim J-S, Cho S-G, Choi Y, Oh S-M, et al. mTOR-Dependent Role of Sestrin2 in Regulating Tumor Progression of Human Endometrial Cancer. Cancers. 2020; 12(9):2515. https://doi.org/10.3390/cancers12092515
Chicago/Turabian StyleShin, Jiha, Jeongyun Bae, Sumi Park, Hyun-Goo Kang, Seong Min Shin, Gunho Won, Jong-Seok Kim, Ssang-Goo Cho, Youngsok Choi, Sang-Muk Oh, and et al. 2020. "mTOR-Dependent Role of Sestrin2 in Regulating Tumor Progression of Human Endometrial Cancer" Cancers 12, no. 9: 2515. https://doi.org/10.3390/cancers12092515