Molecular Basis of Mismatch Repair Protein Deficiency in Tumors from Lynch Suspected Cases with Negative Germline Test Results

 ,
, 
Abstract
:1. Introduction
2. Results
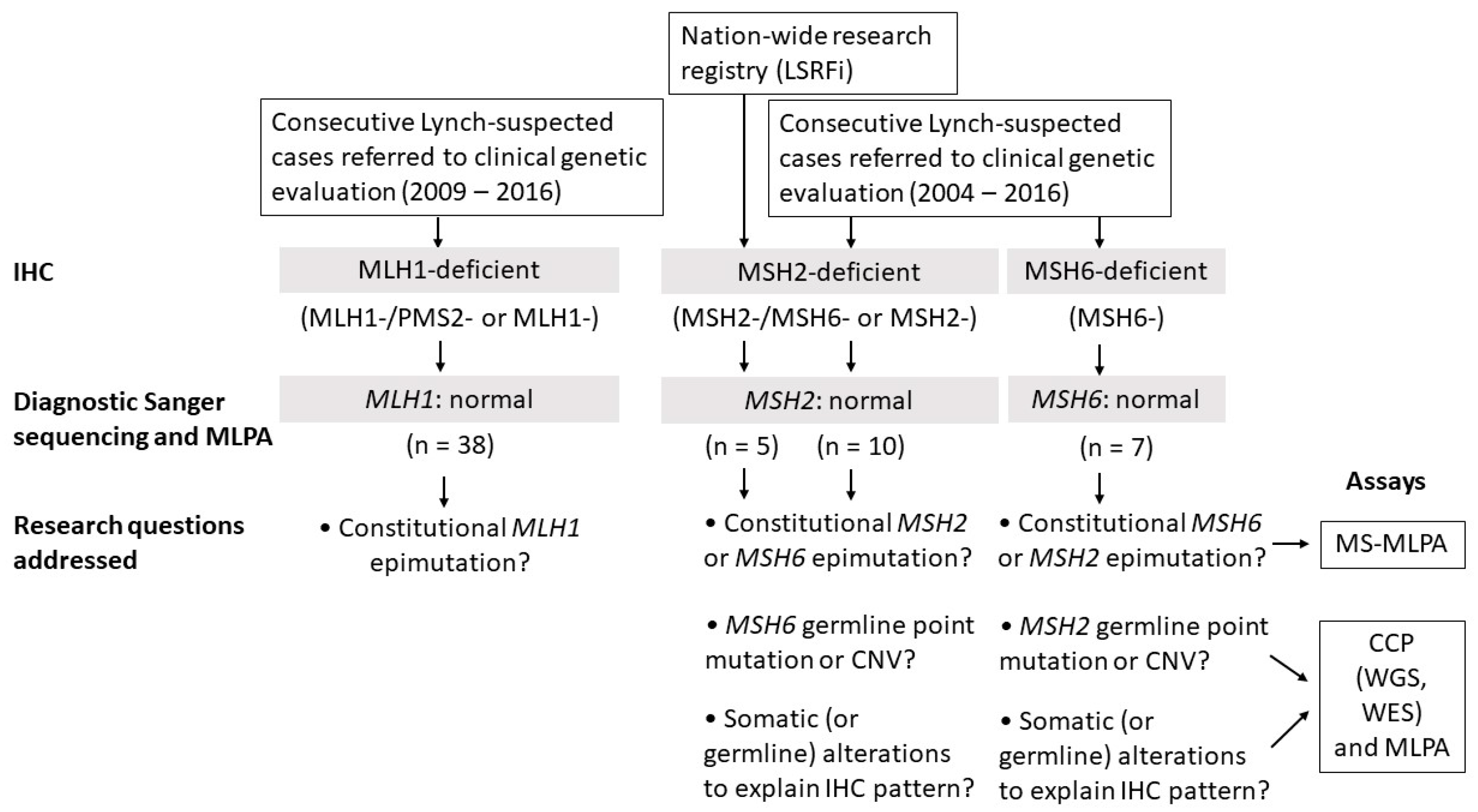
2.1. Clinicopathological Characteristics and Study Design
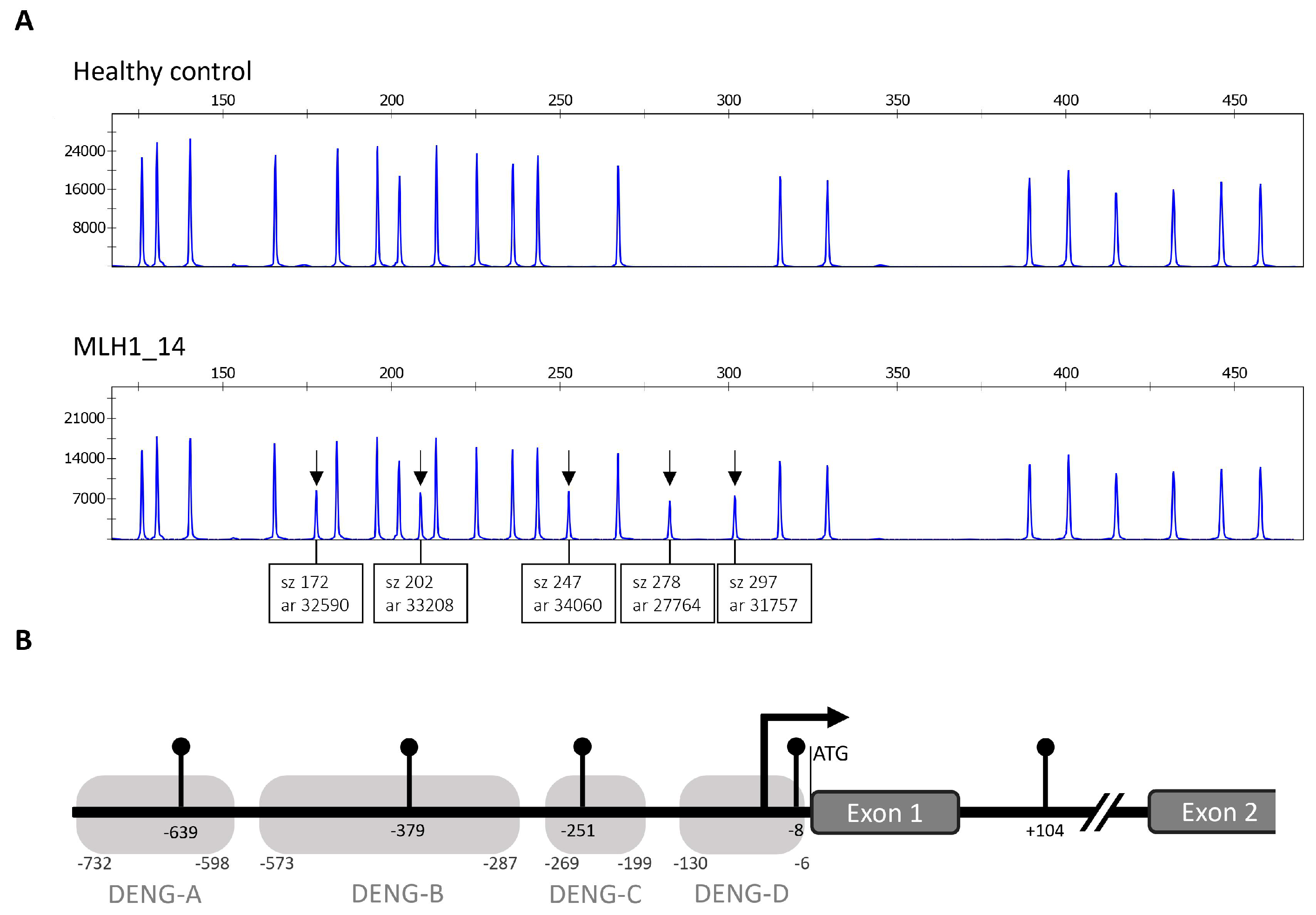
2.2. MLH1 Cohort
2.3. MSH2 Cohort
2.3.1. Consecutive MSH2-Deficient Cases from Diagnostics
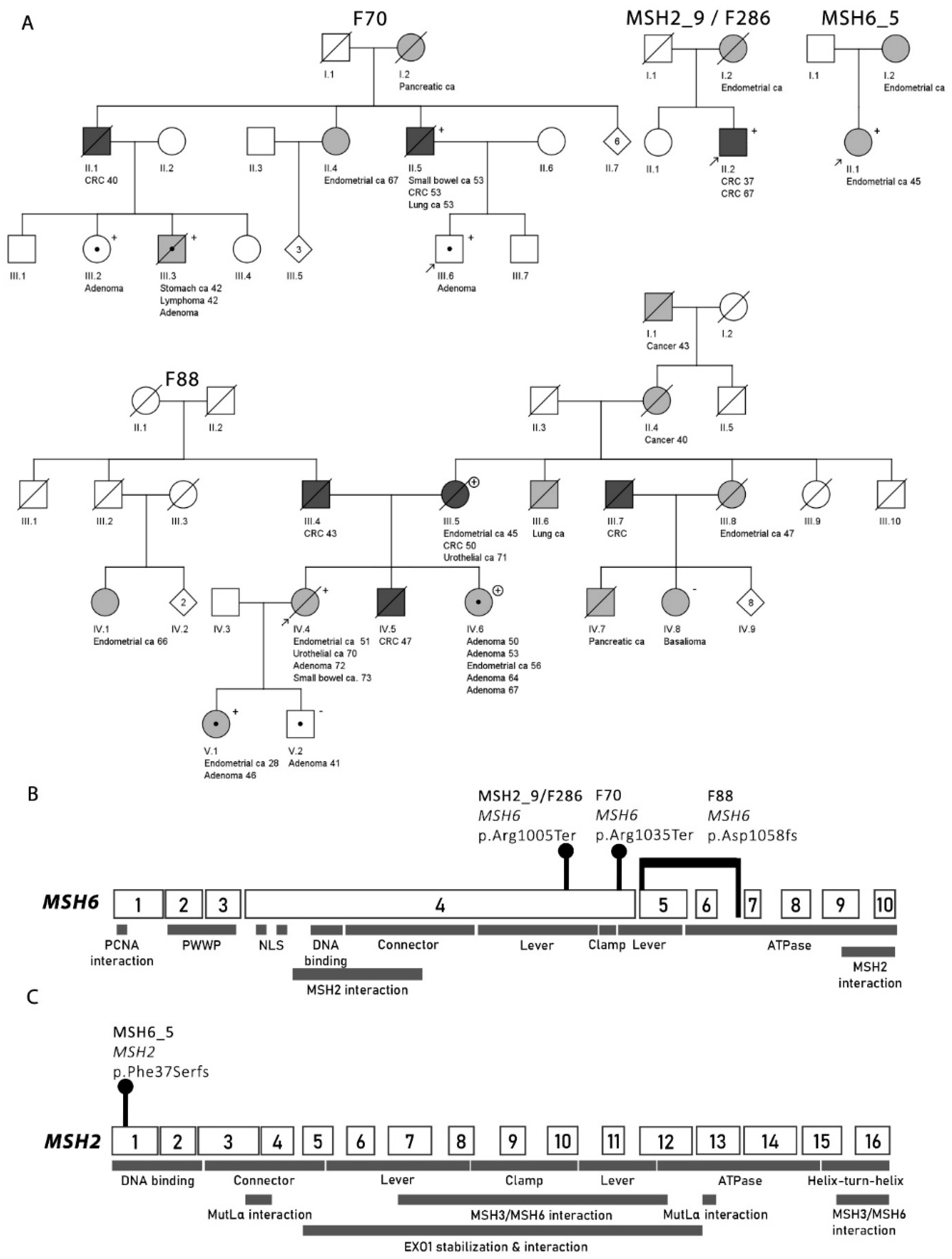
2.3.2. Families from the Hereditary Colorectal Cancer Registry of Finland (LSRFi)
F70
F88
2.4. MSH6 Cohort
2.5. Polymerase δ and Ɛ Mutation Analysis
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Immunohistochemical Staining (IHC)
4.3. Microsatellite Instability Analysis (MSI)
4.4. MMR Gene Methylation Status
4.5. MMR Gene Copy Number Status
4.6. Sanger Sequencing
4.7. Deep Sequencing and Variant Analysis
4.8. Somatic Mutation Profiling
4.9. Loss of Heterozygosity (LOH) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Patterns and Trends in Colorectal Cancer Incidence and Mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch Syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capellá, G.; Den Dunnen, J.T.; et al. Application of a Five-Tiered Scheme for Standardized Classification of 2,360 Unique Mismatch Repair Gene Variants Lodged on the InSiGHT Locus-Specific Database. Nat. Genet. 2013, 46, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, R.; Spier, I.; Zhao, B.; Kloth, M.; Marquez, J.; Hinrichsen, I.; Kirfel, J.; Tafazzoli, A.; Horpaopan, S.; Uhlhaas, S.; et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am. J. Hum. Genet. 2016, 99, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Olkinuora, A.; Nieminen, T.T.; Mårtensson, E.; Rohlin, A.; Ristimäki, A.; Koskenvuo, L.; Lepistö, A.; Gebre-Medhin, S.; Nordling, M.; Peltomäki, P. Biallelic Germline Nonsense Variant of MLH3 Underlies Polyposis Predisposition. Genet. Med. 2018, 21, 1868–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltomäki, P. Update on Lynch Syndrome Genomics. Fam. Cancer 2016, 15, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, Y.M.C.; de Jong, A.E.; Morreau, H.; Tops, C.M.J.; Vasen, H.F.; Wijnen, J.T.; Breuning, M.H.; Brocker-Vriends, A.H.J.T. Diagnostic Approach and Management of Lynch Syndrome (Hereditary Nonpolyposis Colorectal Carcinoma): A Guide for Clinicians. CA Cancer J. Clin. 2006, 56, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.H.; Enns, R.; Heidelbaugh, J.; Barkun, A. American Gastroenterological Association Institute Guideline on the Diagnosis and Management of Lynch Syndrome. Gastroenterology 2015, 149, 777–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stjepanovic, N.; Moreira, L.; Carneiro, F.; Balaguer, F.; Cervantes, A.; Balmaña, J.; Martinelli, E. Hereditary Gastrointestinal Cancers: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2019, 30, 1558–1571. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Clendenning, M.; Sotamaa, K.; Prior, T.; Westman, J.A.; et al. Feasibility of Screening for Lynch Syndrome among Patients with Colorectal Cancer. J. Clin. Oncol. 2008, 26, 5783–5788. [Google Scholar] [CrossRef]
- Moreira, L.; Balaguer, F.; Lindor, N.; de la Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of Lynch Syndrome Among Patients With Colorectal Cancer. JAMA 2012, 308, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Loughrey, M.; Waring, P.; Tan, A.; Trivett, M.; Kovalenko, S.; Beshay, V.; Young, M.A.; McArthur, G.; Boussioutas, A.; Dobrovic, A. Incorporation of Somatic BRAF Mutation Testing into an Algorithm for the Investigation of Hereditary Non-Polyposis Colorectal Cancer. Fam. Cancer 2007, 6, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.M.; Wezel, T.; Van Den Akker, B.E.; Ventayol Garcia, M.; Ruano, D.; Tops, C.; Wagner, A.; Letteboer, T.; Gómez García, E.; Devilee, P.; et al. Combined Mismatch Repair and POLE/POLD1 Defects Explain Unresolved Suspected Lynch Syndrome Cancers. Eur. J. Hum. Genet. 2016, 24, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Mensenkamp, A.R.; Vogelaar, I.P.; van Zelst–Stams, W.A.G.; Goossens, M.; Ouchene, H.; Hendriks–Cornelissen, S.J.B.; Kwint, M.P.; Hoogerbrugge, N.; Nagtegaal, I.D.; Ligtenberg, M.J.L. Somatic Mutations in MLH1 and MSH2 are a Frequent Cause of Mismatch-Repair Deficiency in Lynch Syndrome-Like Tumors. Gastroenterology 2014, 146, 643–646.e8. [Google Scholar] [CrossRef]
- Deng, G.; Chen, A.; Hong, J.; Chae, H.S.; Kim, Y.S. Methylation of CpG in a Small Region of the hMLH1 Promoter Invariably Correlates with the Absence of Gene Expression. Cancer Res. 1999, 59, 2029. [Google Scholar] [PubMed]
- Deng, G.; Chen, A.; Pong, E.; Kim, Y.S. Methylation in hMLH1 Promoter Interferes With Its Binding to Transcription Factor CBF and Inhibits Gene Expression. Oncogene 2001, 20, 7120–7127. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Pearlman, R.; Markow, M.; Knight, D.; Chen, W.; Arnold, C.A.; Pritchard, C.C.; Hampel, H.; Frankel, W.L. Two-Stain Immunohistochemical Screening for Lynch Syndrome in Colorectal Cancer may Fail to Detect Mismatch Repair Deficiency. Mod. Pathol. 2018, 31, 1891–1900. [Google Scholar] [CrossRef]
- Morak, M.; Käsbauer, S.; Kerscher, M.; Laner, A.; Nissen, A.; Benet-Pagès, A.; Schackert, H.; Keller, G.; Massdorf, T.; Holinski-Feder, E. Loss of MSH2 and MSH6 due to Heterozygous Germline Defects in MSH3 and MSH6. Fam. Cancer 2017, 16, 491–500. [Google Scholar] [CrossRef]
- Mäki-Nevala, S.; Valo, S.; Ristimäki, A.; Sarhadi, V.; Knuutila, S.; Nyström, M.; Renkonen-Sinisalo, L.; Lepistö, A.; Mecklin, J.; Peltomäki, P. DNA Methylation Changes and Somatic Mutations as Tumorigenic Events in Lynch Syndrome-Associated Adenomas Retaining Mismatch Repair Protein Expression. EBioMedicine 2019, 39, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Gylling, A.; Ridanpää, M.; Vierimaa, O.; Aittomäki, K.; Avela, K.; Kääriäinen, H.; Laivuori, H.; Pöyhönen, M.; Sallinen, S.; Wallgren-Pettersson, C.; et al. Large Genomic Rearrangements and Germline Epimutations in Lynch Syndrome. Int. J. Cancer 2009, 124, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Niessen, R.C.; Hofstra, R.M.; Westers, H.; Ligtenberg, M.J.L.; Kooi, K.; Jager, P.O.; de Groote, M.L.; Dijkhuizen, T.; Olderode-Berends, M.J.; Hollema, H.; et al. Germline Hypermethylation of MLH1 and EPCAM Deletions are a Frequent Cause of Lynch Syndrome. Genes Chromosomes Cancer 2009, 48, 737–744. [Google Scholar] [CrossRef]
- Hitchins, M.P. Constitutional epimutation as mechanism for cancer causality and heritability? Nat. Rev. Cancer 2015, 15, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chew, M.H.; Goh, X.W.; Tan, S.Y.; Loi, C.T.T.; Tan, Y.M.; Law, H.Y.; Koh, P.K.; Tang, C.L. Systematic Study on Genetic and Epimutational Profile of a Cohort of Amsterdam Criteria-Defined Lynch Syndrome in Singapore. PLoS ONE 2014, 9, e94170. [Google Scholar] [CrossRef] [PubMed]
- Morak, M.; Heidenreich, B.; Keller, G.; Hampel, H.; Laner, A.; de la Chapelle, A.; Holinski-Feder, E. Biallelic MUTYH Mutations can Mimic Lynch Syndrome. Eur. J. Hum. Genet. 2014, 22, 1334–1337. [Google Scholar] [CrossRef] [Green Version]
- Porkka, N.; Lahtinen, L.; Ahtiainen, M.; Böhm, J.P.; Kuopio, T.; Eldfors, S.; Mecklin, J.; Seppälä, T.T.; Peltomäki, P. Epidemiological, Clinical and Molecular Characterization of Lynch-like Syndrome: A Population-based Study. Int. J. Cancer 2019, 145, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Valeri, N.; Gasparini, P.; Fabbri, M.; Braconi, C.; Veronese, A.; Lovat, F.; Adair, B.; Vannini, I.; Fanini, F.; Bottoni, A.; et al. Modulation of Mismatch Repair and Genomic Stability by miR-155. Proc. Natl. Acad. Sci. USA 2010, 107, 6982–6987. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Pigeon, H.; Quillet-Mary, A.; Louat, T.; Schambourg, A.; Humbert, O.; Selves, J.; Salles, B.; Laurent, G.; Lautier, D. hMutSα is Protected from Ubiquitin-Proteasome-Dependent Degradation by Atypical Protein Kinase Cζ Phosphorylation. J. Mol. Biol. 2005, 348, 63–74. [Google Scholar] [CrossRef]
- Renkonen, E.; Zhang, Y.; Lohi, H.; Salovaara, R.; Abdel-Rahman, W.; Nilbert, M.; Aittomaki, K.; Jarvinen, H.; Mecklin, J.; Lindblom, A.; et al. Altered Expression of MLH1, MSH2, and MSH6 in Predisposition to Hereditary Nonpolyposis Colorectal Cancer. J. Clin. Oncol. 2003, 21, 3629. [Google Scholar] [CrossRef]
- Isola, J.; DeVries, S.; Chu, L.; Ghazvini, S.; Waldman, F. Analysis of Changes in DNA Sequence Copy Number by Comparative Genomic Hybridization in Archival Paraffin-Embedded Tumor Samples. Am. J. Pathol. 2014, 145, 1301–1308. [Google Scholar]
- Loukola, A.; Eklin, K.; Laiho, P.; Salovaara, R.; Kristo, P.; Jarvinen, H.; Mecklin, J.; Launonen, V.; Aaltonen, L.A. Microsatellite Marker Analysis in Screening for Hereditary Nonpolyposis Colorectal Cancer (HNPCC). Cancer Res. 2001, 61, 4545. [Google Scholar] [PubMed]
- Church, D.N.; Stelloo, E.; Nout, R.A.; Valtcheva, N.; Depreeuw, J.; ter Haar, N.; Noske, A.; Amant, F.; Tomlinson, I.P.M.; Wild, P.J.; et al. Prognostic Significance of POLE Proofreading Mutations in Endometrial Cancer. J. Natl. Cancer Inst. 2015, 107, 1. [Google Scholar] [CrossRef] [PubMed]
- Valle, L.; Hernández-Illán, E.; Bellido, F.; Aiza, G.; Castillejo, A.; Castillejo, M.; Navarro, M.; Seguí, N.; Vargas, G.; Guarinos, C.; et al. New Insights into POLE and POLD1 Germline Mutations in Familial Colorectal Cancer and Polyposis. Hum. Mol. Genet. 2014, 23, 3506–3512. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Wood, R.D.; Mitchell, M.; Sgouros, J.; Lindahl, T. Human DNA Repair Genes. Science 2001, 291, 1284–1289. [Google Scholar] [CrossRef] [Green Version]
- Porkka, N.; Valo, S.; Nieminen, T.T.; Olkinuora, A.; Mäki-Nevala, S.; Eldfors, S.; Peltomäki, S. Sequencing of Lynch Syndrome Tumors Reveals the Importance of Epigenetic Alterations. Oncogene 2017, 8, 108020–108030. [Google Scholar] [CrossRef] [Green Version]
- Ollikainen, M.; Abdel-Rahman, W.M.; Moisio, A.; Lindroos, A.; Kariola, R.; Järvelä, I.; Pöyhönen, M.; Butzow, R.; Peltomäki, P. Molecular Analysis of Familial Endometrial Carcinoma: A Manifestation of Hereditary Nonpolyposis Colorectal Cancer Or a Separate Syndrome? J. Clin. Oncol. 2005, 23, 4609–4616. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Case ID | IHC Pattern | Germline Mutation | Second Hit | Additional Somatic Alterations Relevant for IHC Pattern |
|---|---|---|---|---|
| MSH2_9/F286 II.2 | MSH2-, MSH6- | MSH6 c.3013C>T, | None identified | MSH2 c.2528delG, |
| p.(Arg1005Ter) * | p.(Cys843fs) | |||
| F70 III.6 (index) | MSH2-, MSH6- | MSH6 c.3103C>T, | MSH2-KCNK12-MSH6 deletion | (MSH2-KCNK12-MSH6 deletion) |
| p.(Arg1035Ter)* | ||||
| F70 II.5 | MSH6- | MSH6 c.3103C>T, | MSH6 c.2932C>T, | Not applicable |
| p.(Arg1035Ter)* | p.(Gln978Ter) | |||
| F88 IV.4 | MSH2-, MSH6- | MSH6 c.1052+2023_c.3557-179, | MSH6 c.3261dupC, | MSH3 c.1148delA, |
| p.(Asp1058Alafs) | p.(Phe1088Leufs) * | (pLys383Argfs) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olkinuora, A.; Gylling, A.; Almusa, H.; Eldfors, S.; Lepistö, A.; Mecklin, J.-P.; Nieminen, T.T.; Peltomäki, P. Molecular Basis of Mismatch Repair Protein Deficiency in Tumors from Lynch Suspected Cases with Negative Germline Test Results. Cancers 2020, 12, 1853. https://doi.org/10.3390/cancers12071853
Olkinuora A, Gylling A, Almusa H, Eldfors S, Lepistö A, Mecklin J-P, Nieminen TT, Peltomäki P. Molecular Basis of Mismatch Repair Protein Deficiency in Tumors from Lynch Suspected Cases with Negative Germline Test Results. Cancers. 2020; 12(7):1853. https://doi.org/10.3390/cancers12071853
Chicago/Turabian StyleOlkinuora, Alisa, Annette Gylling, Henrikki Almusa, Samuli Eldfors, Anna Lepistö, Jukka-Pekka Mecklin, Taina Tuulikki Nieminen, and Päivi Peltomäki. 2020. "Molecular Basis of Mismatch Repair Protein Deficiency in Tumors from Lynch Suspected Cases with Negative Germline Test Results" Cancers 12, no. 7: 1853. https://doi.org/10.3390/cancers12071853