Metagenomic Analysis of Serum Microbe-Derived Extracellular Vesicles and Diagnostic Models to Differentiate Ovarian Cancer and Benign Ovarian Tumor

 , , , , and
, , , , and 
Abstract
:
1. Introduction
2. Results
2.1. Characteristics of the Study Population
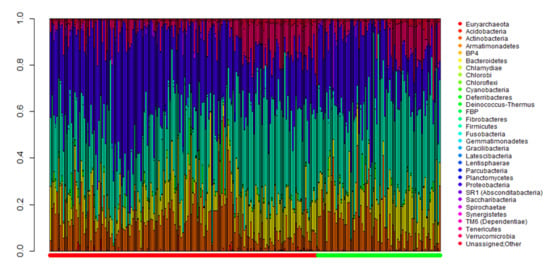
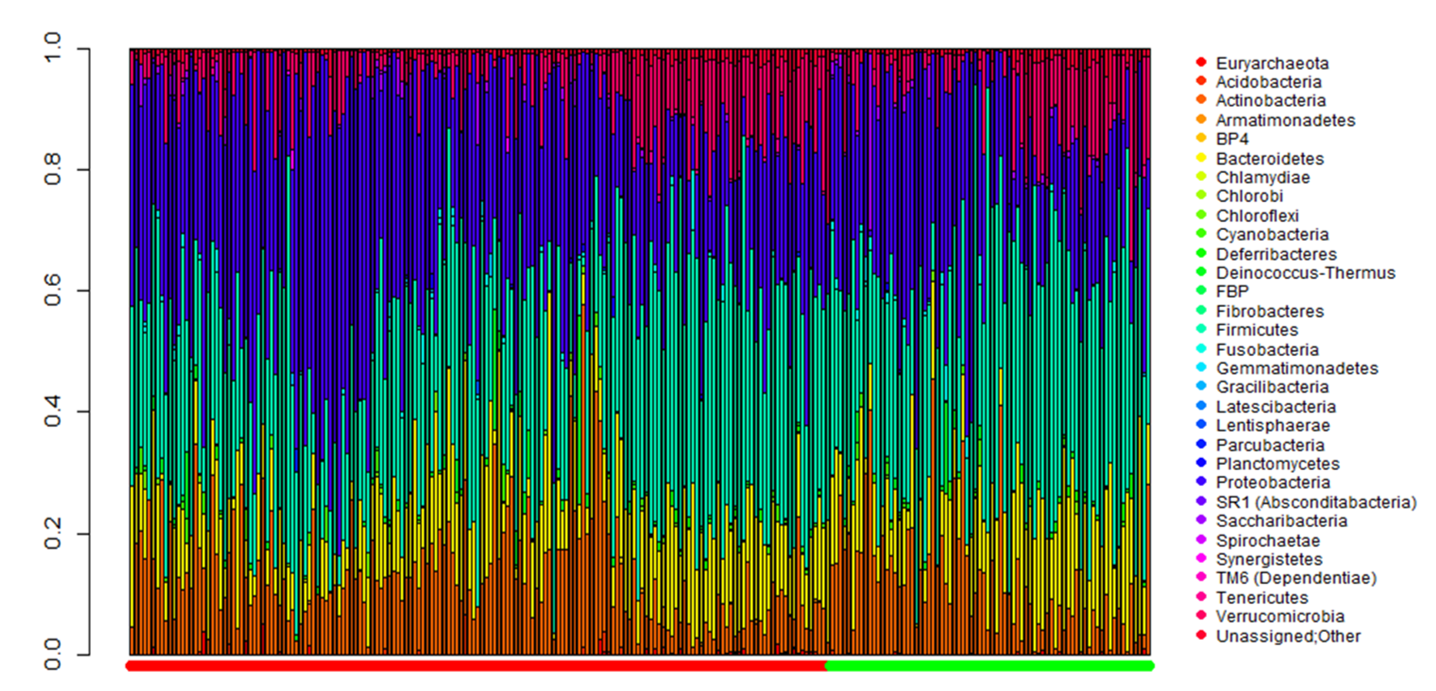
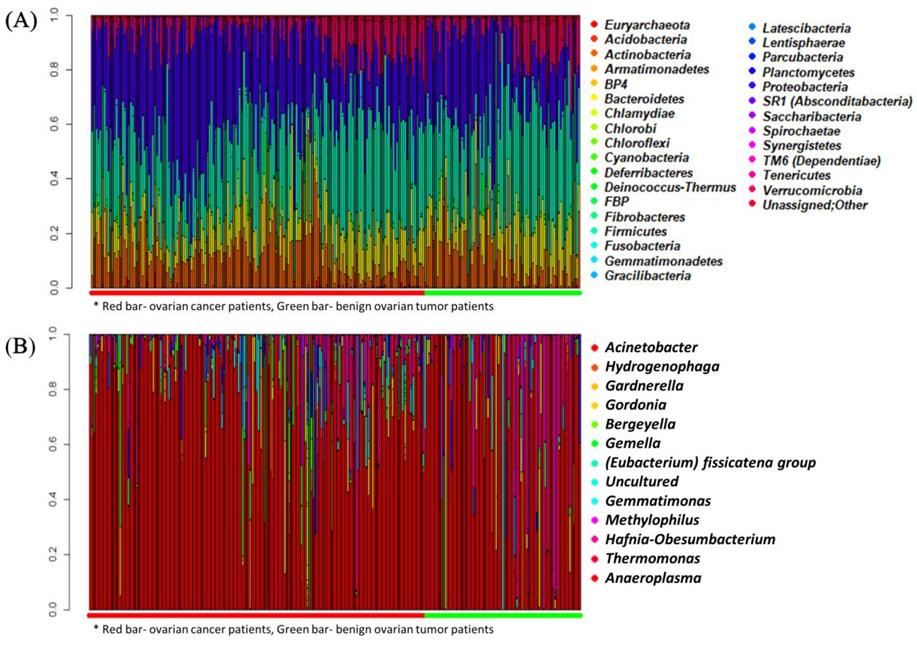
2.2. Comparison of Metagenomic Profiles between the Two Groups
2.3. Development of Diagnostic Models for Ovarian Cancer in the Training Set
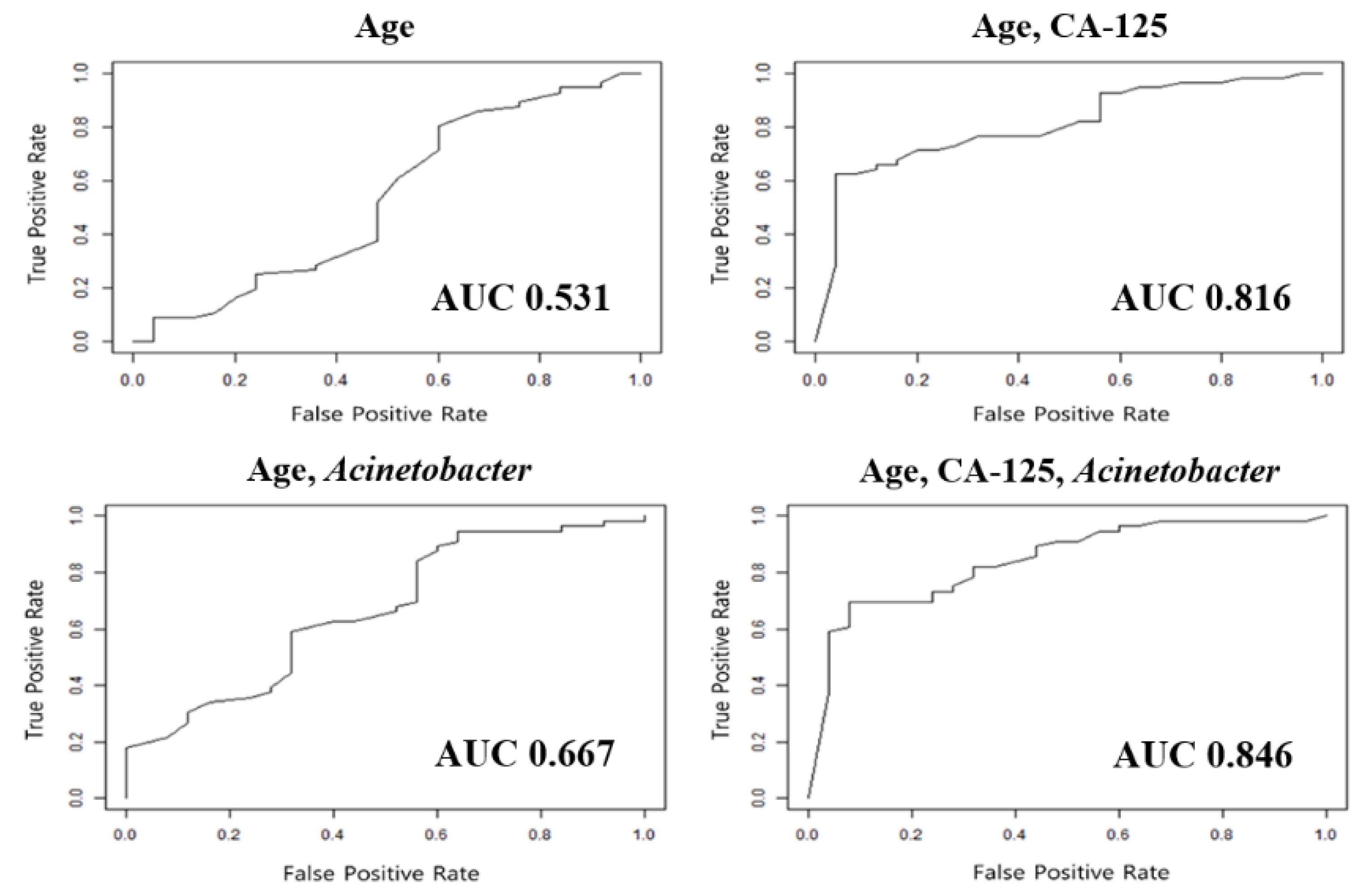
2.4. Validation of Diagnostic Models for Ovarian Cancer
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. EV Isolation and DNA Extraction from Serum Samples
4.3. Microbial Metagenomic Analysis
4.4. Analysis of Microbial Composition in the Microbiota
4.5. Development of Diagnostic Models for Ovarian Cancer
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, M.C.; Won, Y.-J.; Ko, M.J.; Kim, M.; Shim, S.-H.; Suh, D.H.; Kim, J.-W. Incidence of cervical, endometrial, and ovarian cancer in Korea during 1999–2015. J. Gynecol. Oncol. 2019, 30. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, L.A.; Huang, B.; Miller, R.W.; Tucker, T.; Goodrich, S.T.; Podzielinski, I.; DeSimone, C.P.; Ueland, F.R.; Van Nagell, J.R.; Seamon, L.G. Ten-Year Relative Survival for Epithelial Ovarian Cancer. Obstet. Gynecol. 2012, 120, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Myers, E.R.; Bastian, L.A.; Havrilesky, L.J.; Kulasingam, S.L.; Terplan, M.S.; Cline, K.E.; Gray, R.N.; McCrory, D.C. Management of adnexal mass. Evid. Rep. Assess. 2006, 2006, 1–145. [Google Scholar]
- Jacobs, I.; Oram, D.; Fairbanks, J.; Turner, J.; Frost, C.; Grudzinskas, J.G. A risk of malignancy index incorporating CA 125, ultrasound and menopausal status for the accurate preoperative diagnosis of ovarian cancer. BJOG: Int. J. Obstet. Gynaecol. 1990, 97, 922–929. [Google Scholar] [CrossRef]
- Moore, R.G.; McMeekin, D.S.; Brown, A.K.; DiSilvestro, P.; Miller, M.C.; Allard, W.J.; Gajewski, W.; Kurman, R.; Bast, R.C.; Skates, S.J. A novel multiple marker bioassay utilizing HE4 and CA125 for the prediction of ovarian cancer in patients with a pelvic mass. Gynecol. Oncol. 2008, 112, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Karlsen, M.A.; Sandhu, N.; Høgdall, C.; Christensen, I.J.; Nedergaard, L.; Lundvall, L.; Engelholm, S.A.; Pedersen, A.T.; Hartwell, R.; Lydolph, M.; et al. Evaluation of HE4, CA125, risk of ovarian malignancy algorithm (ROMA) and risk of malignancy index (RMI) as diagnostic tools of epithelial ovarian cancer in patients with a pelvic mass. Gynecol. Oncol. 2012, 127, 379–383. [Google Scholar] [CrossRef]
- Liest, A.-L.; Omran, A.S.; Mikiver, R.; Rosenberg, P.; Uppugunduri, S. RMI and ROMA are equally effective in discriminating between benign and malignant gynecological tumors: A prospective population-based study. Acta Obstet. Gynecol. Scand. 2018, 98, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Van Gorp, T.; Cadron, I.; Despierre, E.; Daemen, A.; Leunen, K.; Amant, F.; Timmerman, D.; De Moor, B.; Vergote, I. HE4 and CA125 as a diagnostic test in ovarian cancer: Prospective validation of the Risk of Ovarian Malignancy Algorithm. Br. J. Cancer 2011, 104, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Lycke, M.; Kristjansdottir, B.; Sundfelt, K. A multicenter clinical trial validating the performance of HE4, CA125, risk of ovarian malignancy algorithm and risk of malignancy index. Gynecol. Oncol. 2018, 151, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Dayyani, F.; Uhlig, S.; Colson, B.; Simon, K.; Rolny, V.; Morgenstern, D.; Schlumbrecht, M. Diagnostic Performance of Risk of Ovarian Malignancy Algorithm Against CA125 and HE4 in Connection With Ovarian Cancer: A Meta-analysis. Int. J. Gynecol. Cancer 2016, 26, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Fan, H.; Tang, X.; Zhai, H.; Zhang, Z. Diversified pattern of the human colorectal cancer microbiome. Gut Pathogens 2013, 5, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [Green Version]
- Bultman, S.J. Emerging roles of the microbiome in cancer. Carcinog 2013, 35, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Sinha, R.; Pei, Z.; Dominianni, C.; Wu, J.; Shi, J.; Goedert, J.J.; Hayes, R.B.; Yang, L. Human gut microbiome and risk for colorectal cancer. J. Natl. Cancer Inst. 2013, 105, 1907–1911. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.P.; Redinbo, M.R.; Bultman, S.J. The role of the microbiome in cancer development and therapy. CA Cancer J. Clin. 2017, 67, 326–344. [Google Scholar] [CrossRef] [Green Version]
- Raza, M.H.; Gul, K.; Arshad, A.; Riaz, N.; Waheed, U.; Rauf, A.; Aldakheel, F.; Alduraywish, S.; Rehman, M.U.; Abdullah, M.; et al. Microbiota in cancer development and treatment. J. Cancer Res. Clin. Oncol. 2018, 145, 49–63. [Google Scholar] [CrossRef]
- Kuehn, M. Bacterial outer membrane vesicles and the host-pathogen interaction. Genome Res. 2005, 19, 2645–2655. [Google Scholar] [CrossRef] [Green Version]
- Roier, S.; Zingl, F.G.; Cakar, F.; Schild, S. Bacterial outer membrane vesicle biogenesis: A new mechanism and its implications. Microb. Cell 2016, 3, 257–259. [Google Scholar] [CrossRef]
- Zhou, B.; Sun, C.; Huang, J.; Xia, M.; Guo, E.; Li, N.; Lu, H.; Shan, W.; Wu, Y.; Li, Y.; et al. The biodiversity Composition of Microbiome in Ovarian Carcinoma Patients. Sci. Rep. 2019, 9, 1691. [Google Scholar] [CrossRef] [PubMed]
- Pepper, I.L.; Gerba, C.P.; Gentry, T.J. Environmental Microbiology, 3rd ed.; Academic press: San Diego, CA, USA, 2014. [Google Scholar]
- Kinross, J.M.; Darzi, A.W.; Nicholson, J. Gut microbiome-host interactions in health and disease. Genome Med. 2011, 3, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walther-Antonio, M.; Chen, J.; Multinu, F.; Hokenstad, A.; Distad, T.J.; Cheek, E.H.; Keeney, G.L.; Creedon, D.J.; Nelson, H.; Mariani, A.; et al. Potential contribution of the uterine microbiome in the development of endometrial cancer. Genome Med. 2016, 8, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peleg, A.; Seifert, H.; Paterson, D.L. Acinetobacter baumannii: Emergence of a Successful Pathogen. Clin. Microbiol. Rev. 2008, 21, 538–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhash, K.; Medhekar, A.; Ghadyalpatil, N.; Noronha, V.; Biswas, S.; Kurkure, P.; Nair, R.; Kelkar, R. Blood stream infections in cancer patients: A single center experience of isolates and sensitivity pattern. Indian J. Cancer 2010, 47, 184. [Google Scholar] [CrossRef]
- Chiang, M.-C.; Kuo, S.-C.; Chen, S.-J.; Yang, S.-P.; Lee, Y.-T.; Chen, T.-L.; Fung, C.-P. Clinical characteristics and outcomes of bacteremia due to different genomic species of Acinetobacter baumannii complex in patients with solid tumors. Infection 2011, 40, 19–26. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Toll-like receptors; their physiological role and signal transduction system. Int. Immunopharmacol. 2001, 1, 625–635. [Google Scholar] [CrossRef]
- Knapp, S.; Wieland, C.W.; Florquin, S.; Pantophlet, R.; Dijkshoorn, L.; Tshimbalanga, N.; Akira, S.; Van Der Poll, T. Differential Roles of CD14 and Toll-like Receptors 4and 2 in MurineAcinetobacterPneumonia. Am. J. Respir. Crit. Care Med. 2006, 173, 122–129. [Google Scholar] [CrossRef]
- Erridge, C.; Moncayo-Nieto, O.L.; Morgan, R.; Young, M.; Poxton, I.R. Acinetobacter baumannii lipopolysaccharides are potent stimulators of human monocyte activation via Toll-like receptor 4 signalling. J. Med. Microbiol. 2007, 56, 165–171. [Google Scholar] [CrossRef]
- Kim, C.; Jeong, Y.-J.; Lee, J.; Jeon, S.-J.; Park, S.-R.; Kang, M.-J.; Park, J.-H.; Park, J.-H. Essential role of toll-like receptor 4 in Acinetobacter baumannii-induced immune responses in immune cells. Microb. Pathog. 2013, 54, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.H.; Lee, J.H.; Kim, B.R.; Kim, S.I.; Park, T.I.; Lee, J.C.; Lee, Y.-C. Acinetobacter baumannii Outer Membrane Vesicles Elicit a Potent Innate Immune Response via Membrane Proteins. PLoS ONE 2013, 8, e71751. [Google Scholar] [CrossRef] [PubMed]
- Nho, J.S.; Jun, S.H.; Oh, M.H.; Park, T.I.; Choi, C.W.; Kim, S.I.; Choi, C.H.; Lee, J.C. Acinetobacter nosocomialis secretes outer membrane vesicles that induce epithelial cell death and host inflammatory responses. Microb. Pathog. 2015, 81, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; McFarland-Mancini, M.M.; Funk, H.M.; Husseinzadeh, N.; Mounajjed, T.; Drew, A. Toll-like receptor expression in normal ovary and ovarian tumors. Cancer Immunol. Immunother. 2009, 58, 1375–1385. [Google Scholar] [CrossRef]
- Kelly, M.G.; Alvero, A.; Chen, R.; Silasi, D.-A.; Abrahams, V.M.; Chan, S.; Visintin, I.; Rutherford, T.; Mor, G. TLR-4 Signaling Promotes Tumor Growth and Paclitaxel Chemoresistance in Ovarian Cancer. Cancer Res. 2006, 66, 3859–3868. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, T.; Okamoto, A.; Enomoto, T.; Hamano, T.; Aotani, E.; Terao, Y.; Suzuki, N.; Mikami, M.; Yaegashi, N.; Kato, K.; et al. Randomized Phase III Trial of Irinotecan Plus Cisplatin Compared With Paclitaxel Plus Carboplatin As First-Line Chemotherapy for Ovarian Clear Cell Carcinoma: JGOG3017/GCIG Trial. J. Clin. Oncol. 2016, 34, 2881–2887. [Google Scholar] [CrossRef]
- Kim, S.I.; Lim, M.C.; Lim, J.; Won, Y.-J.; Seo, S.-S.; Kang, S.; Park, S.-Y. Incidence of epithelial ovarian cancer according to histologic subtypes in Korea, 1999 to 2012. J. Gynecol. Oncol. 2016, 27. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.J.; Kim, H.; Lee, Y.; Lee, H.J.; Park, C.H.K.; Yang, J.; Kim, Y.-K.; Kym, S.; Ahn, Y.M. Comparison of serum microbiome composition in bipolar and major depressive disorders. J. Psychiatr. Res. 2020, 123, 31–38. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Kwon, S.; Lee, B.; Yoon, S. CASPER: Context-aware scheme for paired-end reads from high-throughput amplicon sequencing. BMC Bioinform. 2014, 15, S10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | All (n = 242, %) | Training Set | Test Set | ||||
|---|---|---|---|---|---|---|---|
| Cancer (n = 110, %) | Benign (n = 51, %) | p | Cancer (n = 56, %) | Benign (n = 25, %) | p | ||
| Age, years | |||||||
| Mean ± SD | 52.3 ± 13.4 | 53.8 ± 12.3 | 48.2 ± 15.9 | 0.031 | 53.4 ± 11.5 | 51.8 ± 15.7 | 0.658 |
| BMI, kg/m2 | |||||||
| Mean ± SD | 23.0 ± 3.4 | 22.6 ± 3.1 | 23.1 ± 3.6 | 0.387 | 23.1 ± 3.6 | 23.9 ± 4.2 | 0.370 |
| Menopause | 141 (58.3) | 68 (61.8) | 26 (51.0) | 0.194 | 34 (60.7) | 13 (52.0) | 0.463 |
| Comorbidities | |||||||
| Hypertension | 55 (22.7) | 27 (24.5) | 8 (15.7) | 0.205 | 11 (19.6) | 9 (36.0) | 0.115 |
| Diabetes | 21 (8.7) | 11 (10.0) | 6 (11.8) | 0.735 | 1 (1.8) | 3 (12.0) | 0.085 |
| Dyslipidemia | 34 (14.0) | 18 (16.4) | 6 (11.8) | 0.446 | 6 (10.7) | 4 (16.0) | 0.489 |
| Serum CA-125, IU/mL | |||||||
| Median (range) | 126.3 (2.3–10000.0) | 331.1 (2.3–10000.0) | 22.3 (3.5–1821.0) | <0.001 | 432.3 (7.7–9909.0) | 20.6 (5.7–1710.0) | <0.001 |
| FIGO stage | |||||||
| I | 52 (21.5) | 33 (30.0) | 19 (33.9) | ||||
| II | 10 (4.1) | 6 (5.5) | 4 (7.1) | ||||
| III | 75 (31.0) | 53 (48.2) | 22 (39.3) | ||||
| IV | 29 (12.0) | 18 (16.4) | 11 (19.6) | ||||
| Histologic type | |||||||
| Epithelial ovarian cancer | |||||||
| High-grade serous | 88 (36.4) | 60 (54.5) | 28 (50.0) | ||||
| Low-grade serous | 8 (3.3) | 6 (5.5) | 2 (3.6) | ||||
| Mucinous | 15 (6.2) | 10 (9.1) | 5 (8.9) | ||||
| Endometrioid | 16 (6.6) | 9 (8.2) | 7 (12.5) | ||||
| Clear cell | 29 (12.0) | 18 (16.4) | 11 (19.6) | ||||
| Mixed | 6 (2.5) | 3 (2.7) | 3 (5.4) | ||||
| Others | 4 (1.7) | 4 (3.6) | 0 | ||||
| Benign ovarian tumor | |||||||
| Mucinous cystadenoma | 28 (11.6) | 24 (47.1) | 4 (16.0) | ||||
| With fibroma | 5 (2.1) | 4 (7.8) | 1 (4.0) | ||||
| Without fibroma | 23 (9.5) | 20 (39.2) | 3 (12.0) | ||||
| Serous cystadenoma | 15 (6.2) | 8 (15.7) | 7 (28.0) | ||||
| With fibroma | 4 (1.7) | 3 (5.9) | 1 (4.0) | ||||
| Without fibroma | 11 (4.5) | 5 (9.8) | 6 (24.0) | ||||
| Seromucinous cystadenoma | 6 (2.5) | 4 (7.8) | 2 (8.0) | ||||
| With fibroma | 2 (0.8) | 1 (2.0) | 1 (4.0) | ||||
| Without fibroma | 4 (1.7) | 3 (5.9) | 1 (4.0) | ||||
| Endometriotic cyst | 8 (3.3) | 4 (7.8) | 4 (16.0) | ||||
| Mature cystic teratoma | 8 (3.3) | 6 (11.8) | 2 (8.0) | ||||
| Fibroma/fibrothecoma | 9 (3.7) | 3 (5.9) | 6 (24.0) | ||||
| Paratubal cyst | 2 (0.8) | 2 (3.9) | 0 | ||||
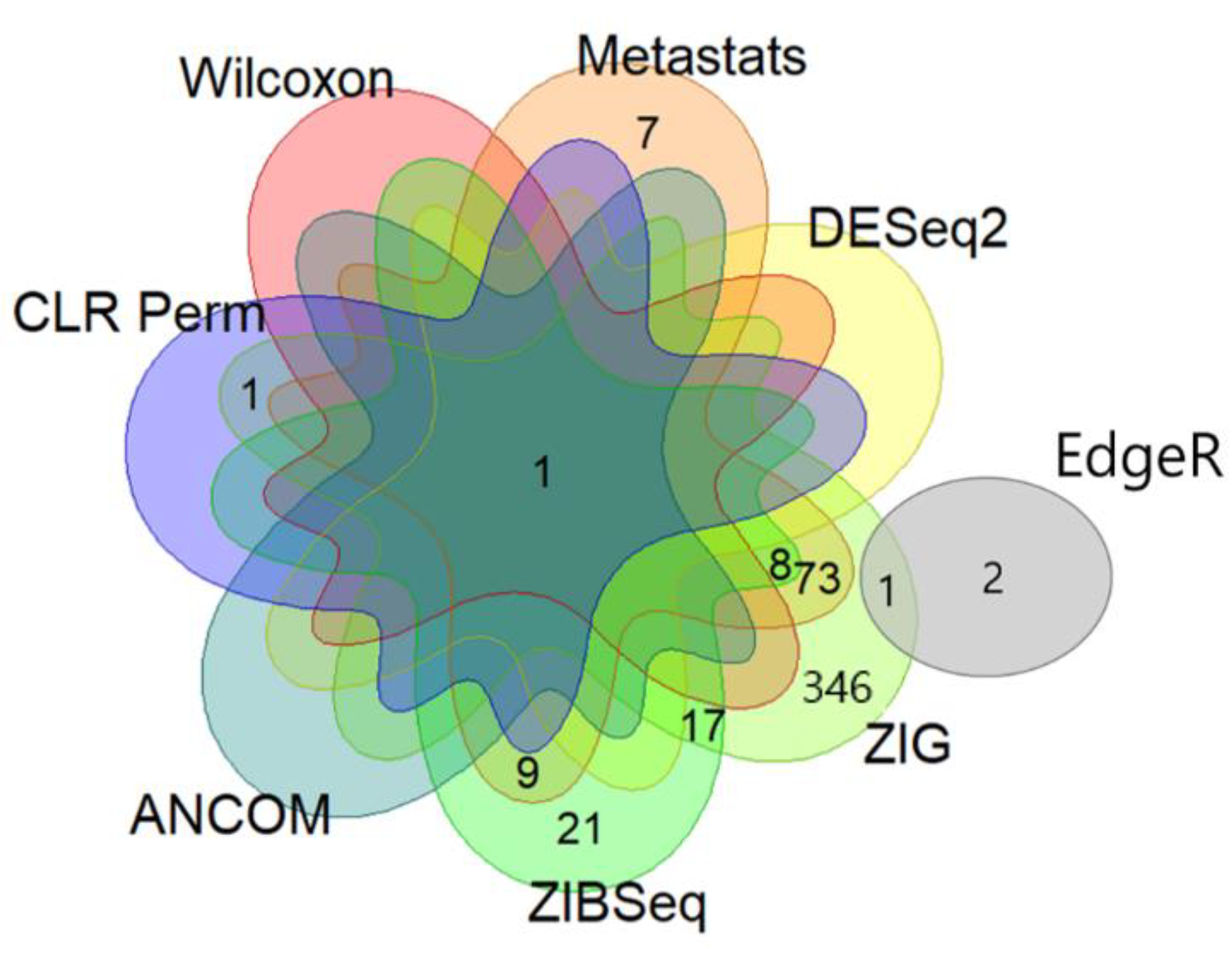
| Genus | Wilcoxon | Metastats | EdgeR | DESeq2 | ZIG | ZIBSeq | ANCOM | CLR Perm |
|---|---|---|---|---|---|---|---|---|
| Acinetobacter | <0.001 | 0.008 | 0.093 | 0.043 | <0.001 | <0.001 | Acinetobacter | <0.001 |
| Isoptericola | 0.841 | <0.001 | 0.487 | 1 | 0.046 | <0.001 | Not detected | 0.855 |
| Terrisporobacter | 0.841 | 0.023 | 0.600 | 1 | <0.001 | <0.001 | Not detected | 0.944 |
| SM1A02 | 0.989 | 0.008 | 0.528 | 1 | <0.001 | 0.002 | Not detected | 0.935 |
| Candidatus Alysiosphaera | 0.841 | <0.001 | 0.476 | 1 | 0.015 | <0.001 | Not detected | 0.901 |
| Ralstonia | 0.841 | <0.001 | 0.462 | 1 | <0.001 | 0.005 | Not detected | 0.913 |
| Hydrogenophaga | 0.771 | <0.001 | 0.872 | 1 | <0.001 | 0.027 | Not detected | 0.809 |
| Pseudorhodoferax | 0.921 | 0.024 | 0.811 | 1 | <0.001 | 0.007 | Not detected | 0.779 |
| Bryobacter | 0.841 | 0.023 | 0.420 | 1 | 0.007 | 0.999 | Not detected | 0.849 |
| Varibaculum | 0.841 | 0.013 | 0.600 | 1 | 0.599 | <0.001 | Not detected | 0.416 |
| Model | Training Set | Test Set | ||||
|---|---|---|---|---|---|---|
| Sensitivity | Specificity | AUC | Sensitivity | Specificity | AUC | |
| Age | 0.554 | 0.490 | 0.589 | 0.518 | 0.520 | 0.531 |
| Age, CA-125 | 0.773 | 0.686 | 0.809 | 0.768 | 0.560 | 0.816 |
| Age, Acinetobacter | 0.827 | 0.529 | 0.770 | 0.839 | 0.440 | 0.667 |
| Age, CA-125, Acinetobacter | 0.864 | 0.784 | 0.898 | 0.821 | 0.680 | 0.846 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.I.; Kang, N.; Leem, S.; Yang, J.; Jo, H.; Lee, M.; Kim, H.S.; Dhanasekaran, D.N.; Kim, Y.-K.; Park, T.; et al. Metagenomic Analysis of Serum Microbe-Derived Extracellular Vesicles and Diagnostic Models to Differentiate Ovarian Cancer and Benign Ovarian Tumor. Cancers 2020, 12, 1309. https://doi.org/10.3390/cancers12051309
Kim SI, Kang N, Leem S, Yang J, Jo H, Lee M, Kim HS, Dhanasekaran DN, Kim Y-K, Park T, et al. Metagenomic Analysis of Serum Microbe-Derived Extracellular Vesicles and Diagnostic Models to Differentiate Ovarian Cancer and Benign Ovarian Tumor. Cancers. 2020; 12(5):1309. https://doi.org/10.3390/cancers12051309
Chicago/Turabian StyleKim, Se Ik, Nayeon Kang, Sangseob Leem, Jinho Yang, HyunA Jo, Maria Lee, Hee Seung Kim, Danny N. Dhanasekaran, Yoon-Keun Kim, Taesung Park, and et al. 2020. "Metagenomic Analysis of Serum Microbe-Derived Extracellular Vesicles and Diagnostic Models to Differentiate Ovarian Cancer and Benign Ovarian Tumor" Cancers 12, no. 5: 1309. https://doi.org/10.3390/cancers12051309