Autophagy-Dependent Reactivation of Epstein-Barr Virus Lytic Cycle and Combinatorial Effects of Autophagy-Dependent and Independent Lytic Inducers in Nasopharyngeal Carcinoma

 , , and
, , and
Abstract
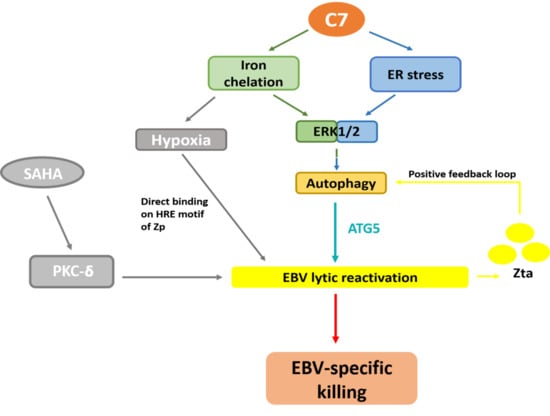
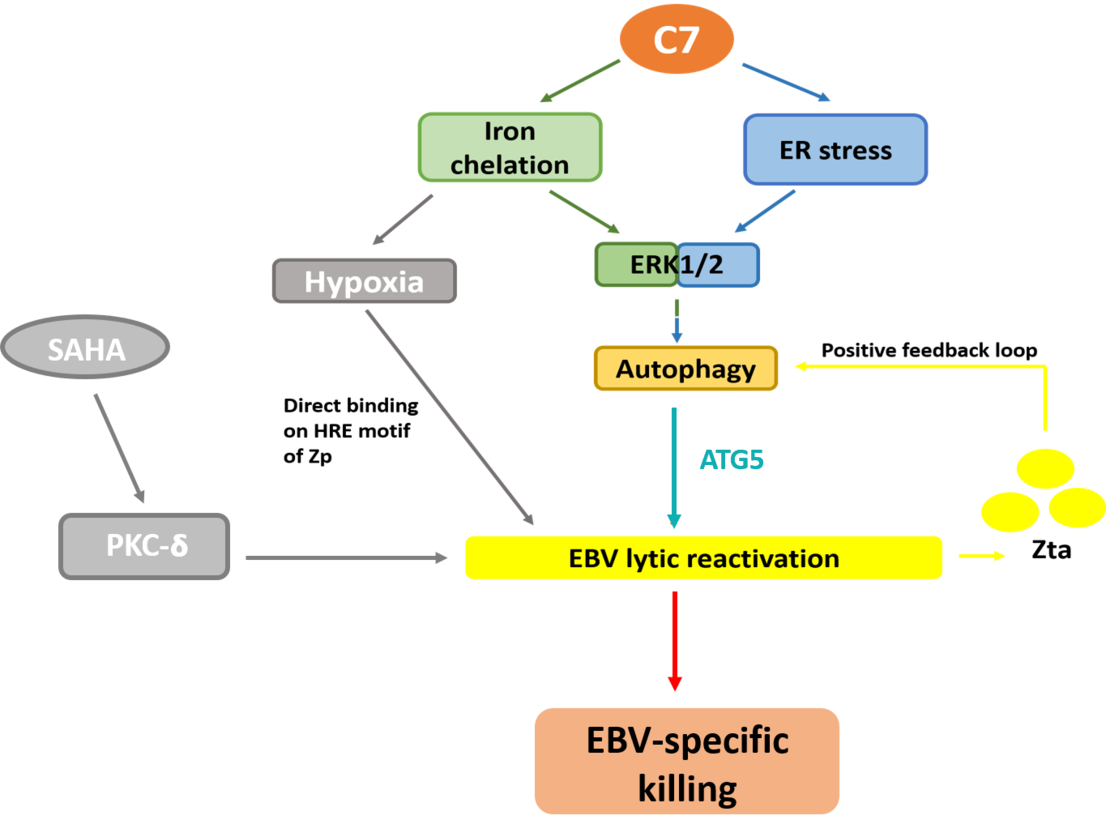
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
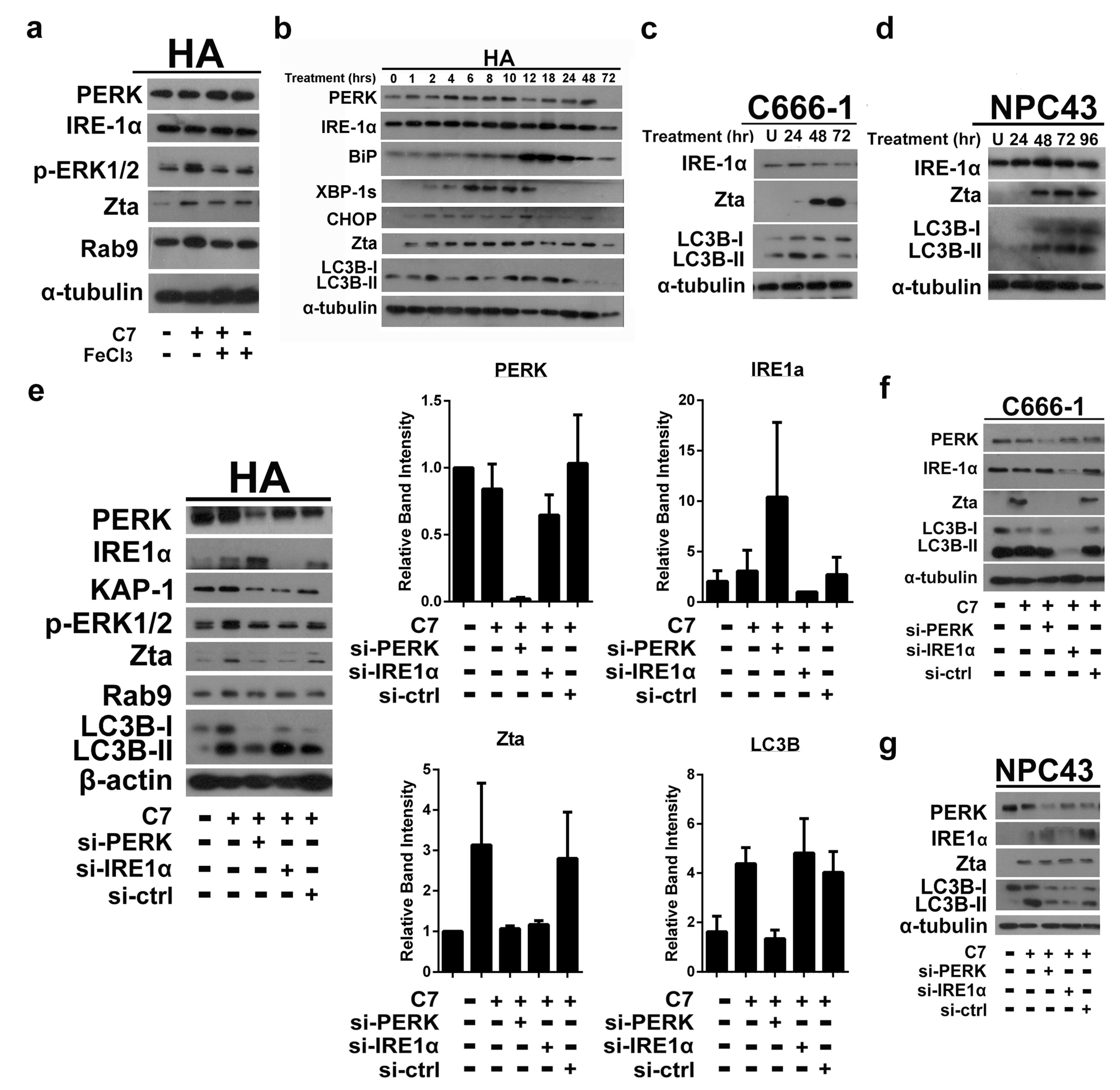
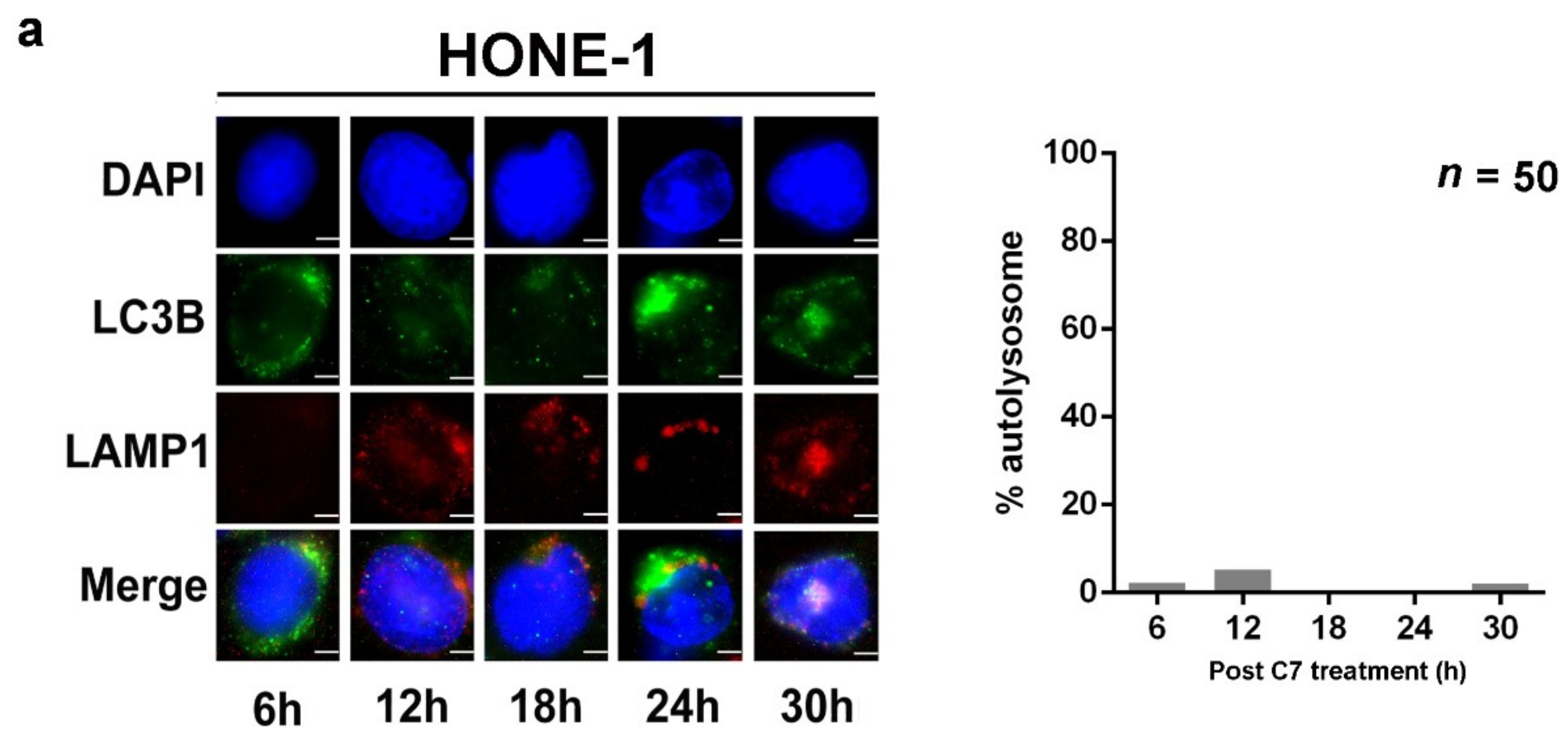
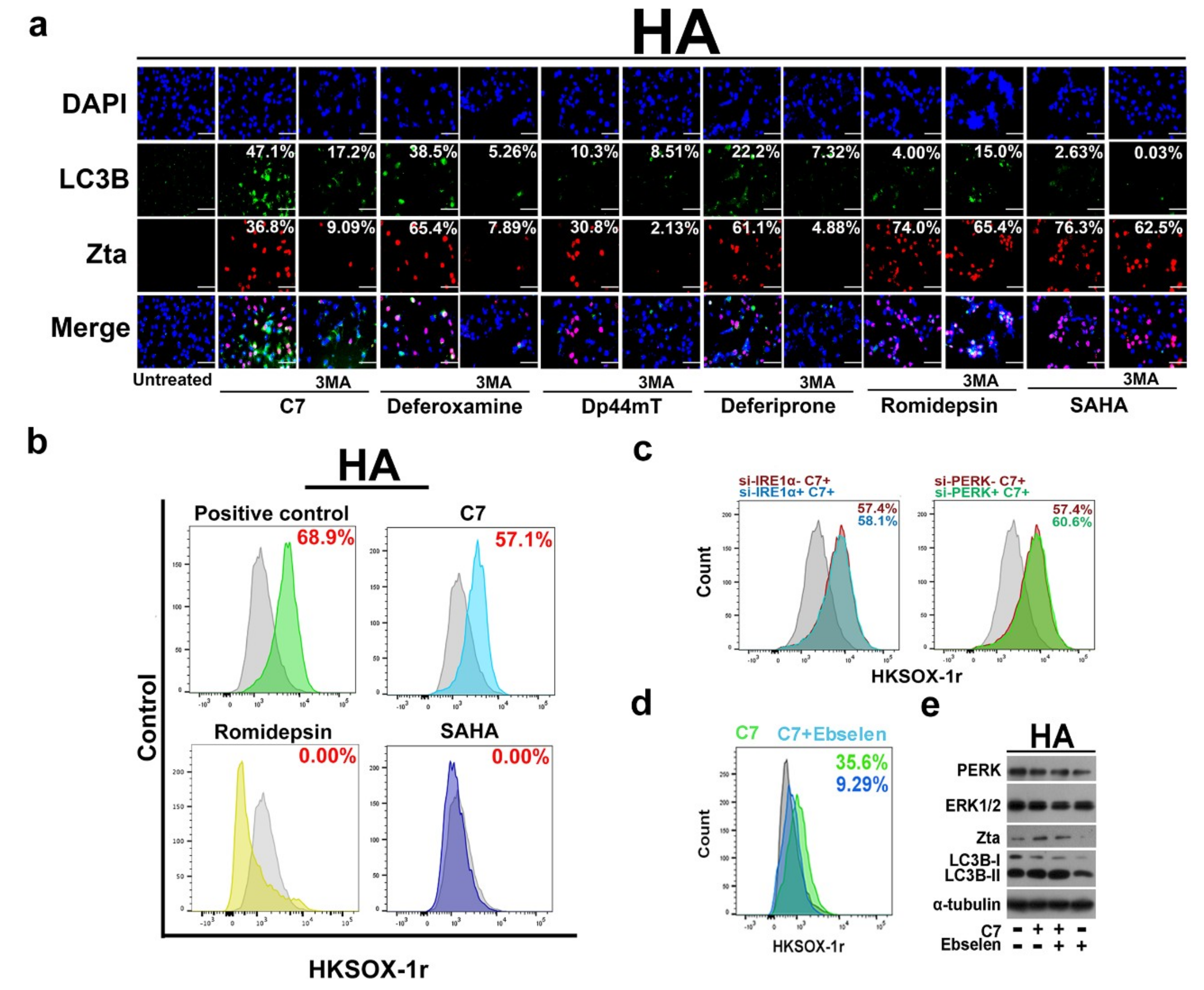
2.1. Endoplasmic Reticulum (ER) Stress Is Partially Required for Autophagy Initiation and EBV Lytic Reactivation
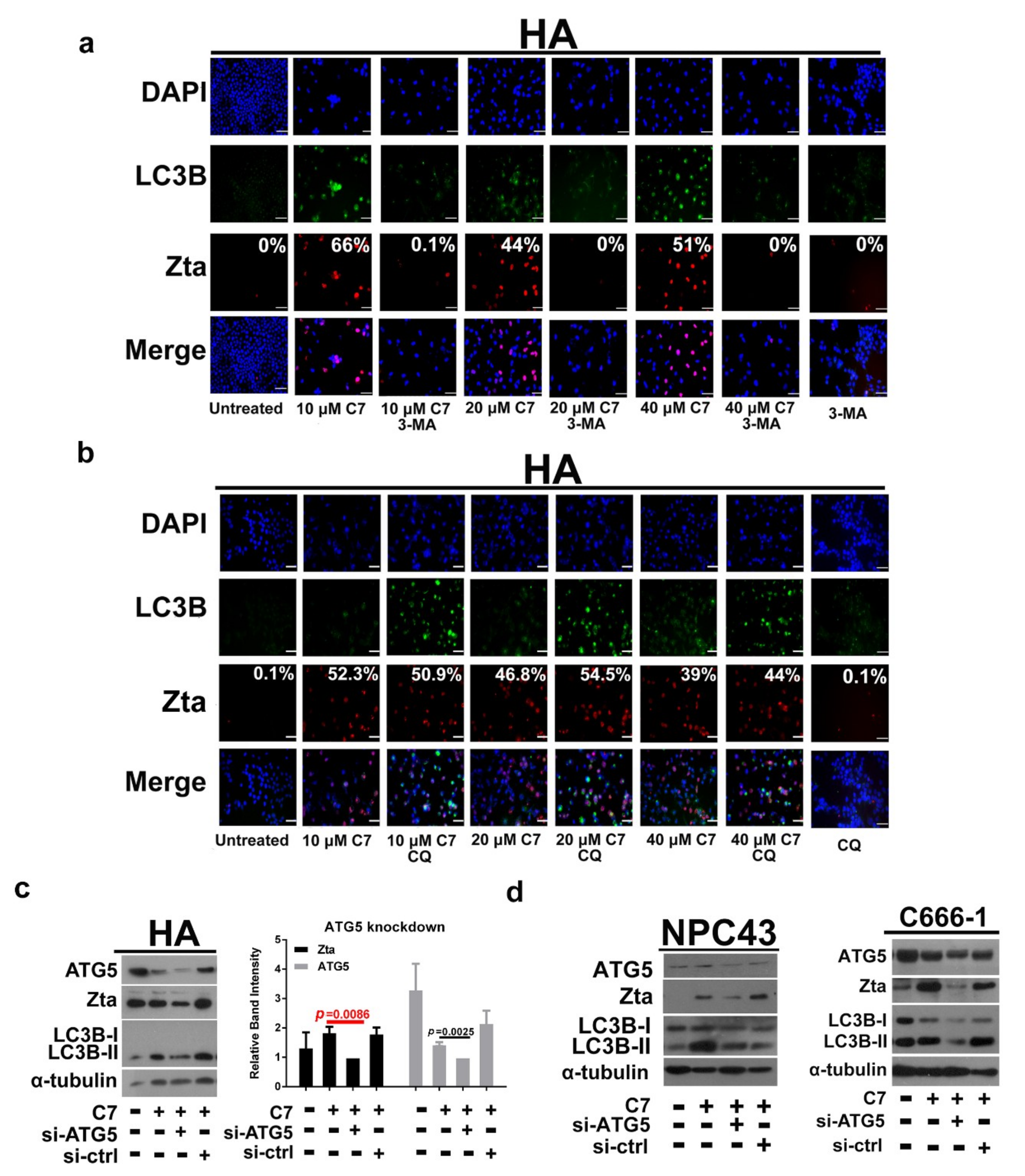
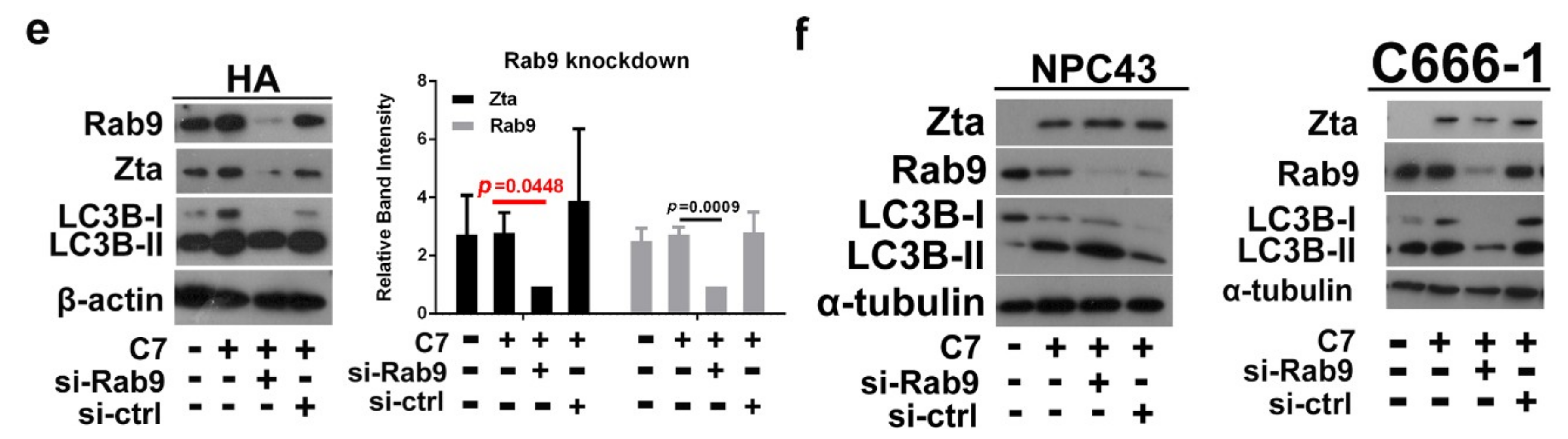
2.2. Autophagic Protein ATG5 Is Predominantly Required for EBV Lytic Reactivation upon C7 Treatment
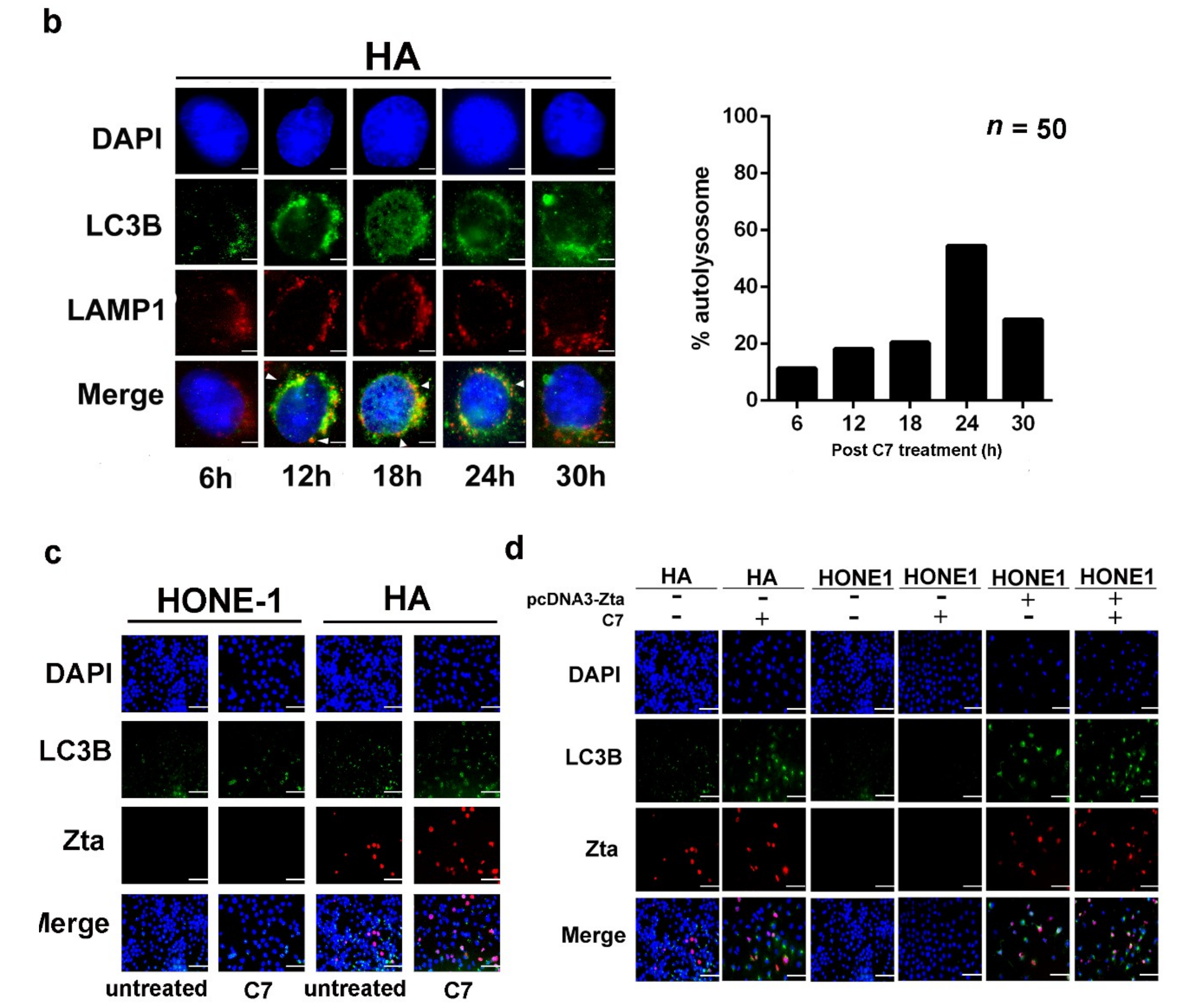
2.3. Positive Feedback Loop between Zta Protein and Autophagy Induction
2.4. Autophagy-Dependent and -Independent Reactivation of EBV Lytic Cycle by Lytic Cycle Inducers
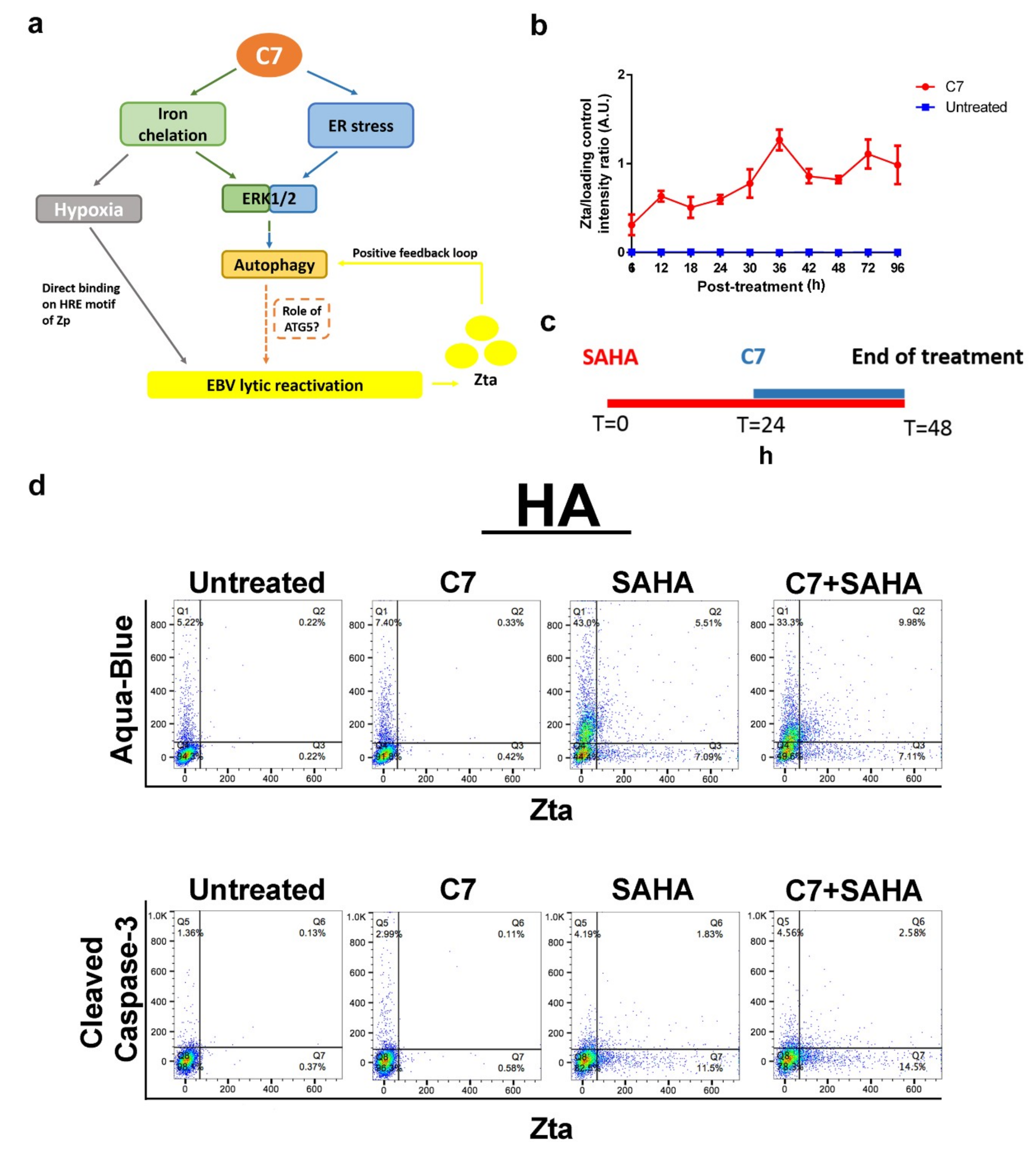
2.5. Combinatorial Treatment of C7 and SAHA Results in Synergistic Killing of NPC Cells
2.6. Combinatorial Treatment of C7 and SAHA at Their Corresponding Reactivation Kinetics Enhanced EBV Lytic Population
3. Discussion
4. Materials and Methods
4.1. Cells and Culturing Conditions
4.2. Cell Cycle Analysis
4.3. Drug Treatment
4.4. Immunofluorescent Staining
4.5. Cytosol Superoxide Measurement
4.6. MTT Assay
4.7. Transient Small Interference RNA and Short-Hairpin RNA Knockdown
4.8. Terminal Deoxynucleotidyl Transferase–Mediated dUTP Nick End Labeling (TUNEL) Assay
4.9. Western Blot Analysis
4.10. Quantification of Zta and Cleaved Caspase-3 by Flow Cytometry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Niedobitek, G.; Meru, N.; Delecluse, H.J. Epstein-Barr virus infection and human malignancies. Int. J. Exp. Pathol. 2001, 82, 149–170. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Chan, T.F.; Yang, W.; Shen, J.J.; Lam, K.P.; Kwok, H.; Sham, P.C.; Tsao, S.W.; Kwong, D.L.; Lung, M.L.; et al. High risk Epstein-Barr virus variants characterized by distinct polymorphisms in the EBER locus are strongly associated with nasopharyngeal carcinoma. Int. J. Cancer 2019, 144, 3031–3042. [Google Scholar] [CrossRef] [PubMed]
- Stoker, S.D.; Novalic, Z.; Wildeman, M.A.; Huitema, A.D.; Verkuijlen, S.A.; Juwana, H.; Greijer, A.E.; Tan, I.B.; Middeldorp, J.M.; de Boer, J.P. Epstein-Barr virus-targeted therapy in nasopharyngeal carcinoma. J. Cancer Res. Clin. Oncol. 2015, 141, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Yiu, S.P.T.; Tam, K.P.; Chiang, A.K.S. Viral-Targeted Strategies Against EBV-Associated Lymphoproliferative Diseases. Front. Oncol. 2019, 9, 81. [Google Scholar] [CrossRef]
- Mak, N.K.; Lung, H.L.; Hui, K.F.; Chiang, A.K.S. Translational Studies: Drug Candidates and Preclinical Testing. In Nasopharyngeal Carcinoma: From Etiology to Clinical Practice; Lee, A., Lung, M., Ng, W., Eds.; Academic Press: Cambridge, UK, 2019; pp. 109–126. [Google Scholar]
- Yiu, S.P.T.; Hui, K.F.; Choi, C.K.; Kao, R.Y.T.; Ma, C.W.; Yang, D.; Chiang, A.K.S. Intracellular Iron Chelation by a Novel Compound, C7, Reactivates Epstein(-)Barr Virus (EBV) Lytic Cycle via the ERK-Autophagy Axis in EBV-Positive Epithelial Cancers. Cancers 2018, 10, 505. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Wang, L.; Ye, X.; Zhao, T. The physiological roles of autophagy in the mammalian life cycle. Biol. Rev. Camb. Philos. Soc. 2019, 94, 503–516. [Google Scholar] [CrossRef]
- McKnight, N.C.; Zhenyu, Y. Beclin 1, an Essential Component and Master Regulator of PI3K-III in Health and Disease. Curr. Pathobiol. Rep. 2013, 1, 231–238. [Google Scholar] [CrossRef]
- Gao, W.; Chen, Z.; Wang, W.; Stang, M.T. E1-like activating enzyme Atg7 is preferentially sequestered into p62 aggregates via its interaction with LC3-I. PLoS ONE 2013, 8, e73229. [Google Scholar] [CrossRef]
- Hong, S.B.; Kim, B.W.; Kim, J.H.; Song, H.K. Structure of the autophagic E2 enzyme Atg10. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Bose, P.; Patel, K.; Roy, S.G.; Gain, C.; Gowda, H.; Robertson, E.S.; Saha, A. Transcriptional and epigenetic modulation of autophagy promotes EBV oncoprotein EBNA3C induced B-cell survival. Cell Death Dis. 2018, 9, 605. [Google Scholar] [CrossRef]
- Lee, D.Y.; Sugden, B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 2008, 27, 2833–2842. [Google Scholar] [CrossRef]
- Pujals, A.; Favre, L.; Pioche-Durieu, C.; Robert, A.; Meurice, G.; Le Gentil, M.; Chelouah, S.; Martin-Garcia, N.; Le Cam, E.; Guettier, C.; et al. Constitutive autophagy contributes to resistance to TP53-mediated apoptosis in Epstein-Barr virus-positive latency III B-cell lymphoproliferations. Autophagy 2015, 11, 2275–2287. [Google Scholar] [CrossRef]
- Fotheringham, J.A.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 2 induces autophagy to promote abnormal acinus formation. J. Virol. 2015, 89, 6940–6944. [Google Scholar] [CrossRef]
- Hung, C.H.; Chen, L.W.; Wang, W.H.; Chang, P.J.; Chiu, Y.F.; Hung, C.C.; Lin, Y.J.; Liou, J.Y.; Tsai, W.J.; Hung, C.L.; et al. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J. Virol. 2014, 88, 12133–12145. [Google Scholar] [CrossRef]
- Granato, M.; Santarelli, R.; Farina, A.; Gonnella, R.; Lotti, L.V.; Faggioni, A.; Cirone, M. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J. Virol. 2014, 88, 12715–12726. [Google Scholar] [CrossRef]
- Nowag, H.; Guhl, B.; Thriene, K.; Romao, S.; Ziegler, U.; Dengjel, J.; Munz, C. Macroautophagy Proteins Assist Epstein Barr Virus Production and Get Incorporated Into the Virus Particles. EBioMedicine 2014, 1, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Gonnella, R.; Granato, M.; Farina, A.; Santarelli, R.; Faggioni, A.; Cirone, M. PKC theta and p38 MAPK activate the EBV lytic cycle through autophagy induction. Biochim. Biophys. Acta 2015, 1853, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Frudd, K.; Burgoyne, T.; Burgoyne, J.R. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat. Commun. 2018, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S. Biological Roles of Alternative Autophagy. Mol. Cells 2018, 41, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Cheung, A.K.; Choi, C.K.; Yeung, P.L.; Middeldorp, J.M.; Lung, M.L.; Tsao, S.W.; Chiang, A.K. Inhibition of class I histone deacetylases by romidepsin potently induces Epstein-Barr virus lytic cycle and mediates enhanced cell death with ganciclovir. Int. J. Cancer 2016, 138, 125–136. [Google Scholar] [CrossRef]
- Lee, H.H.; Chang, S.S.; Lin, S.J.; Chua, H.H.; Tsai, T.J.; Tsai, K.; Lo, Y.C.; Chen, H.C.; Tsai, C.H. Essential role of PKCdelta in histone deacetylase inhibitor-induced Epstein-Barr virus reactivation in nasopharyngeal carcinoma cells. J. Gen. Virol. 2008, 89, 878–883. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Hui, K.F.; Chiang, A.K. Suberoylanilide hydroxamic acid induces viral lytic cycle in Epstein-Barr virus-positive epithelial malignancies and mediates enhanced cell death. Int. J. Cancer 2010, 126, 2479–2489. [Google Scholar] [CrossRef]
- Hui, K.F.; Ho, D.N.; Tsang, C.M.; Middeldorp, J.M.; Tsao, G.S.; Chiang, A.K. Activation of lytic cycle of Epstein-Barr virus by suberoylanilide hydroxamic acid leads to apoptosis and tumor growth suppression of nasopharyngeal carcinoma. Int. J. Cancer 2012, 131, 1930–1940. [Google Scholar] [CrossRef]
- Sun, K.; Deng, W.; Zhang, S.; Cai, N.; Jiao, S.; Song, J.; Wei, L. Paradoxical roles of autophagy in different stages of tumorigenesis: Protector for normal or cancer cells. Cell Biosci. 2013, 3, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorburn, A.; Thamm, D.H.; Gustafson, D.L. Autophagy and cancer therapy. Mol. Pharmacol. 2014, 85, 830–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, H.; Nguyen, T.T.; Chow, K.H.; Tan, P.H.; Soo, K.C.; Tran, E. Over-expression of the mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK in hepatocellular carcinoma: Its role in tumor progression and apoptosis. BMC Gastroenterol. 2003, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Han, W.; Sui, X.; Fang, Y.; Pan, H. Autophagy: A novel therapeutic target for hepatocarcinoma (Review). Oncol. Lett. 2014, 7, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Manov, I.; Pollak, Y.; Broneshter, R.; Iancu, T.C. Inhibition of doxorubicin-induced autophagy in hepatocellular carcinoma Hep3B cells by sorafenib—The role of extracellular signal-regulated kinase counteraction. FEBS J. 2011, 278, 3494–3507. [Google Scholar] [CrossRef]
- Lee, S.B.; Tong, S.Y.; Kim, J.J.; Um, S.J.; Park, J.S. Caspase-independent autophagic cytotoxicity in etoposide-treated CaSki cervical carcinoma cells. DNA Cell Biol. 2007, 26, 713–720. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yiu, S.P.T.; Hui, K.F.; Münz, C.; Lo, K.-W.; Tsao, S.W.; Kao, R.Y.T.; Yang, D.; Chiang, A.K.S. Autophagy-Dependent Reactivation of Epstein-Barr Virus Lytic Cycle and Combinatorial Effects of Autophagy-Dependent and Independent Lytic Inducers in Nasopharyngeal Carcinoma. Cancers 2019, 11, 1871. https://doi.org/10.3390/cancers11121871
Yiu SPT, Hui KF, Münz C, Lo K-W, Tsao SW, Kao RYT, Yang D, Chiang AKS. Autophagy-Dependent Reactivation of Epstein-Barr Virus Lytic Cycle and Combinatorial Effects of Autophagy-Dependent and Independent Lytic Inducers in Nasopharyngeal Carcinoma. Cancers. 2019; 11(12):1871. https://doi.org/10.3390/cancers11121871
Chicago/Turabian StyleYiu, Stephanie Pei Tung, Kwai Fung Hui, Christian Münz, Kwok-Wai Lo, Sai Wah Tsao, Richard Yi Tsun Kao, Dan Yang, and Alan Kwok Shing Chiang. 2019. "Autophagy-Dependent Reactivation of Epstein-Barr Virus Lytic Cycle and Combinatorial Effects of Autophagy-Dependent and Independent Lytic Inducers in Nasopharyngeal Carcinoma" Cancers 11, no. 12: 1871. https://doi.org/10.3390/cancers11121871