Cold Atmospheric Plasma-Treated PBS Eliminates Immunosuppressive Pancreatic Stellate Cells and Induces Immunogenic Cell Death of Pancreatic Cancer Cells

 , , , , , , , , , and
, , , , , , , , , and
Abstract
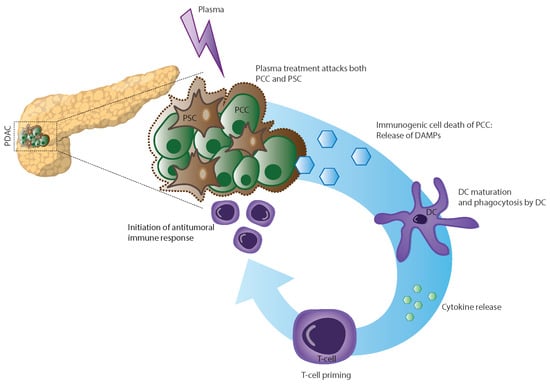
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
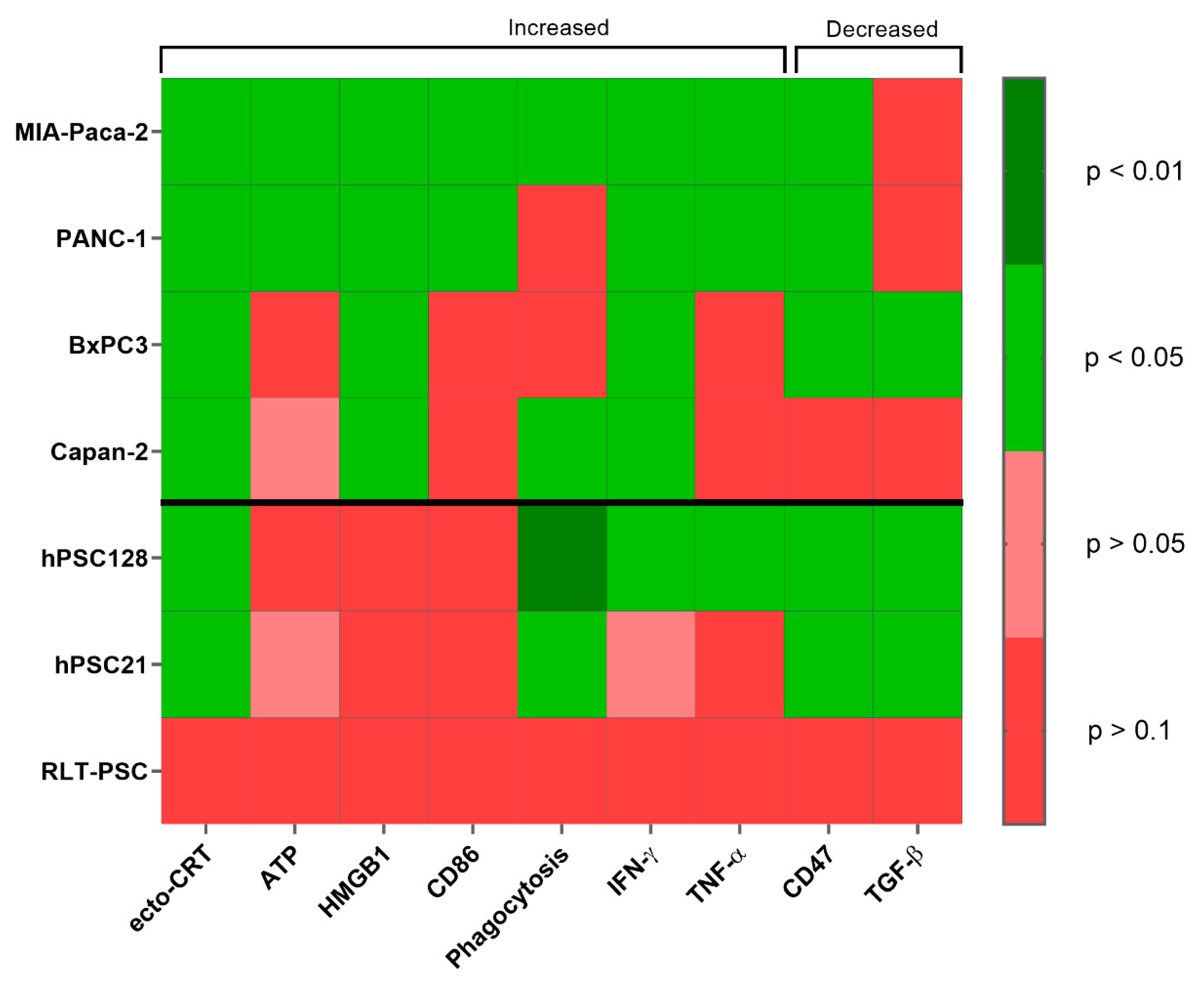
2. Results
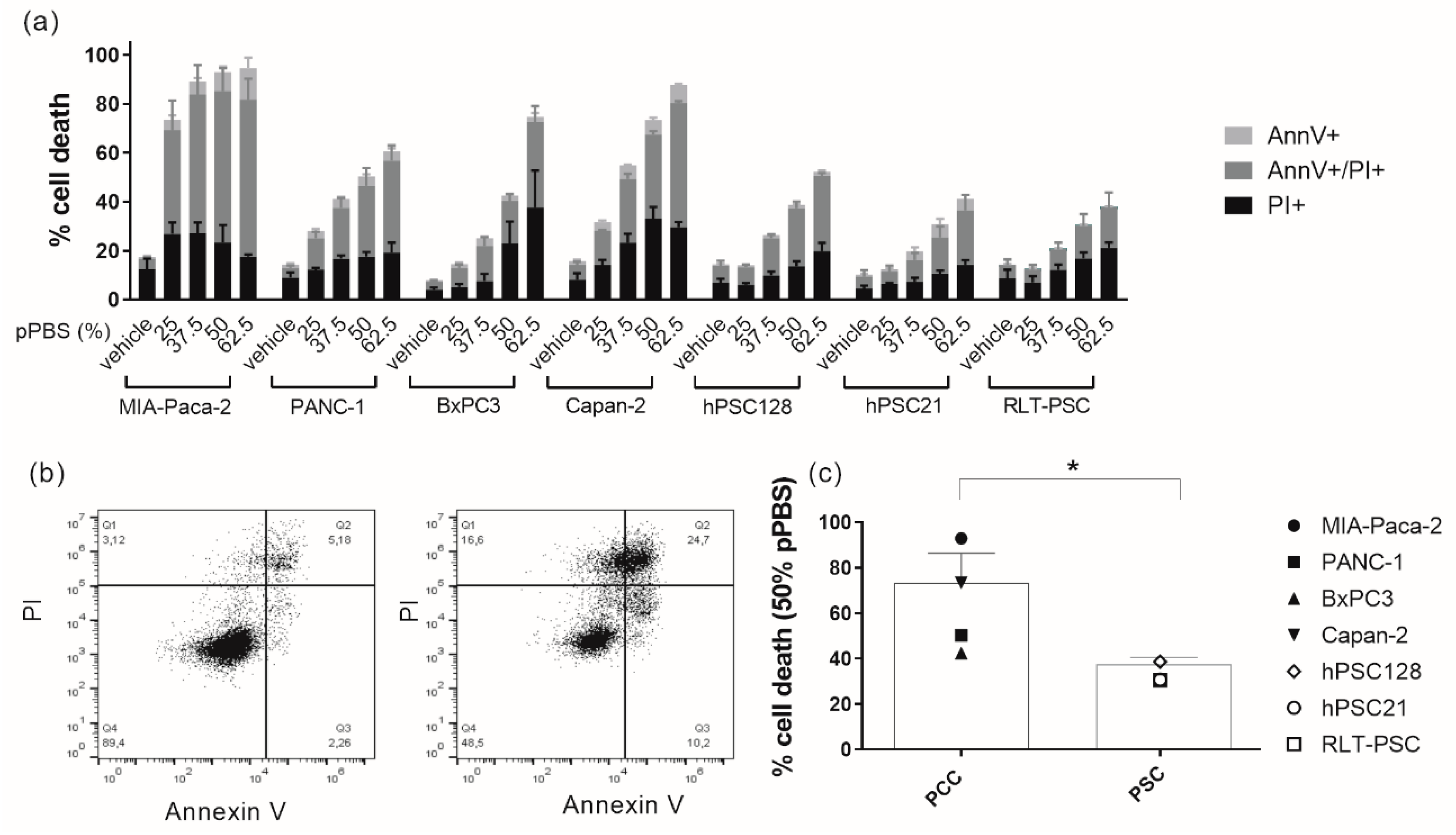
2.1. pPBS Induces Cell Death in Both PCCs and PSCs
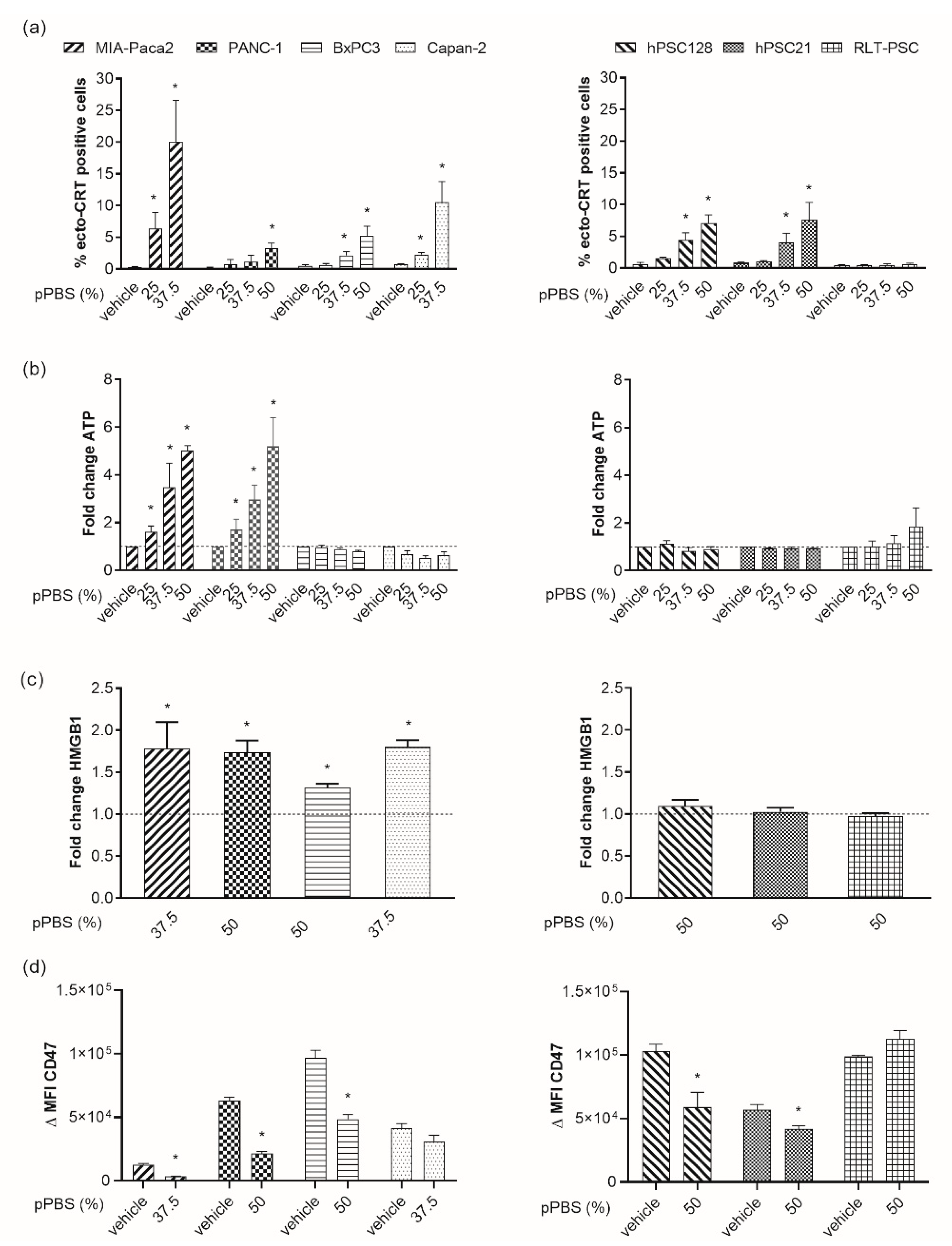
2.2. pPBS Induces ICD Markers on PCCs
2.3. pPBS-Treated Cells are Phagocytosed by DCs
2.4. pPBS Treatment of PCC Increases Maturation of DCs without Affecting Their Viability
2.5. Secretion of Cytokines after pPBS Treatment
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Treatment of PCC and PSC with Cold Atmospheric Plasma
4.3. Analysis of Cytotoxicity and ICD Markers
4.4. In Vitro Generation of Human Monocyte-Derived DCs
4.5. Coculture of DCs and Tumor Cells
4.6. Cytokine Secretion Profile
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ansari, D.; Gustafsson, A.; Andersson, R. Update on the Management of Pancreatic Cancer: Surgery is not Enough. World J. Gastroenterol. 2015, 21, 3157–3165. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; Coveler, A.L. Pancreatic Cancer: Optimizing Treatment Options, New, and Emerging Targeted Therapies. Drug Des. Dev. Ther. 2015, 9, 3529–3545. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Lohr, J.M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.L.; Heinemann, V. Addressing the Challenges of Pancreatic Cancer: Future Directions for Improving Outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2018. [Google Scholar]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti-PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Watt, J.; Kocher, H.M. The Desmoplastic Stroma of Pancreatic Cancer is a Barrier to Immune Cell Infiltration. Oncoimmunology 2013, 2, e26788. [Google Scholar] [CrossRef]
- Heinemann, V.; Reni, M.; Ychou, M.; Richel, D.J.; Macarulla, T.; Ducreux, M. Tumour-Stroma Interactions in Pancreatic Ductal Adenocarcinoma: Rationale and Current Evidence for New Therapeutic Strategies. Cancer Treat. Rev. 2014, 40, 118–128. [Google Scholar] [CrossRef]
- Lunardi, S.; Muschel, R.J.; Brunner, T.B. The Stromal Compartments in Pancreatic Cancer: Are There Any Therapeutic Targets? Cancer Lett. 2014, 343, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mace, T.A.; Bloomston, M.; Lesinski, G.B. Pancreatic Cancer-Associated Stellate Cells: A Viable Target for Reducing Immunosuppression in the Tumor Microenvironment. Oncoimmunology 2013, 2, e24891. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.; Mahalingam, D. Immunotherapy in Pancreatic Adenocarcinoma-Overcoming Barriers to Response. J. Gastrointest. Oncol. 2018, 9, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like Receptor 4-Dependent Contribution of the Immune System to Anticancer Chemotherapy and Radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Garg, A.D.; Romano, E.; Rufo, N.; Agostinis, P. Immunogenic Versus Tolerogenic Phagocytosis during Anticancer Therapy: Mechanisms and Clinical Translation. Cell Death Differ. 2016, 23, 938–951. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Fucikova, J.; Moserova, I.; Truxova, I.; Hermanova, I.; Vancurova, I.; Partlova, S.; Fialova, A.; Sojka, L.; Cartron, P.F.; Houska, M.; et al. High Hydrostatic Pressure Induces Immunogenic Cell Death in Human Tumor Cells. Int. J. Cancer 2014, 135, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Hypericin-Based Photodynamic Therapy Induces Surface Exposure of Damage-Associated Molecular Patterns Like HSP70 and Calreticulin. Cancer Immunol. Immunother. 2012, 61, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, S.R.; Jammeh, M.L.; Wattenberg, M.M.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Radiation-Induced Immunogenic Modulation of Tumor Enhances Antigen Processing and Calreticulin Exposure, Resulting in Enhanced T-Cell Killing. Oncotarget 2014, 5, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A Novel Pathway Combining Calreticulin Exposure and ATP Secretion in Immunogenic Cancer Cell Death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [PubMed]
- Vermeylen, S.; De Waele, J.; Vanuytsel, S.; De Backer, J.; Van der Paal, J.; Ramakers, M.; Leyssens, K.; Marcq, E.; Van Audenaerde, J.; Smits, E.L.J.; et al. Cold Atmospheric Plasma Treatment of Melanoma and Glioblastoma Cancer Cells. Plasma Process Polym. 2016, 13, 1195–1205. [Google Scholar] [CrossRef]
- Yan, D.Y.; Sherman, J.H.; Keidar, M. Cold Atmospheric Plasma, A Novel Promising Anti-Cancer Treatment Modality. Oncotarget 2017, 8, 15977–15995. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, K.R.; Bekeschus, S.; Kaeding, A.; Hackbarth, C.; Kuehn, J.P.; Heidecke, C.D.; von Bernstorff, W.; von Woedtke, T.; Partecke, L.I. Non-Thermal Plasma-Treated Solution Demonstrates Antitumor Activity Against Pancreatic Cancer Cells in Vitro and in Vivo. Sci. Rep. 2017, 7, 8319. [Google Scholar] [CrossRef]
- Partecke, L.I.; Evert, K.; Haugk, J.; Doering, F.; Normann, L.; Diedrich, S.; Weiss, F.U.; Evert, M.; Huebner, N.O.; Guenther, C.; et al. Tissue Tolerable Plasma (TTP) Induces Apoptosis in Pancreatic Cancer Cells in Vitro and in Vivo. BMC Cancer 2012, 12, 473. [Google Scholar] [CrossRef]
- Kalghatgi, S.; Kelly, C.M.; Cerchar, E.; Torabi, B.; Alekseev, O.; Fridman, A.; Friedman, G.; Azizkhan-Clifford, J. Effects of Non-Thermal Plasma on Mammalian Cells. PLoS ONE 2011, 6, e16270. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Hilmi, M.; Bartholin, L.; Neuzillet, C. Immune Therapies in Pancreatic Ductal Adenocarcinoma: Where are We Now? World J. Gastroenterol. 2018, 24, 2137–2151. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, M.; Duewell, P.; Bauer, C.; Rothenfusser, S.; Lauber, K.; Endres, S.; Kobold, S. Strategies to Relieve Immunosuppression in Pancreatic Cancer. Immunotherapy 2015, 7, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Van Audenaerde, J. Interleukin-15 Stimulates Natural Killer Cell-Mediated Killing of Both Human Pancreatic Cancer and Stellate Cells. Oncotarget 2017, 8, 56968–56979. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.Y.; Cui, H.T.; Zhu, W.; Nourmohammadi, N.; Milberg, J.; Zhang, L.G.; Sherman, J.H.; Keidar, M. The Specific Vulnerabilities of Cancer Cells to the Cold Atmospheric Plasma-Stimulated Solutions. Sci. Rep. UK 2017, 7, 4479. [Google Scholar] [CrossRef]
- Kumar, N.; Attri, P.; Dewilde, S.; Bogaerts, A. Inactivation of Human Pancreatic Ductal Adenocarcinoma with Atmospheric Plasma Treated Media and Water: A Comparative Study. J. Phys. D 2018, 51, 255401. [Google Scholar] [CrossRef]
- Gorchs, L.; Hellevik, T.; Bruun, J.A.; Camilio, K.A.; Al-Saad, S.; Stuge, T.B.; Martinez-Zubiaurre, I. Cancer-Associated Fibroblasts from Lung Tumors Maintain Their Immunosuppressive Abilities after High-Dose Irradiation. Front. Oncol. 2015, 5, 87. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Tian, Y.; Zhang, J.; Zhang, H.; Gu, F.; Lu, Y.; Zou, S.; Chen, Y.; Sun, P.; Xu, M.; et al. Functions of Pancreatic Stellate Cell-Derived Soluble Factors in the Microenvironment of Pancreatic Ductal Carcinoma. Oncotarget 2017, 8, 102721–102738. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-Associated Fibroblasts Modulate Growth Factor Signaling and Extracellular Matrix Remodeling to Regulate Tumor Metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFbeta Pathway for Cancer Therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Lin, A.; Truong, B.; Pappas, A.; Kirifides, L.; Oubarri, A.; Chen, S.Y.; Lin, S.J.; Dobrynin, D.; Fridman, G.; Fridman, A.; et al. Uniform Nanosecond Pulsed Dielectric Barrier Discharge Plasma Enhances Anti-Tumor Effects by Induction of Immunogenic Cell Death in Tumors and Stimulation of Macrophages. Plasma Process Polym. 2015, 12, 1392–1399. [Google Scholar] [CrossRef]
- Lin, A.; Truong, B.; Patel, S.; Kaushik, N.; Choi, E.H.; Fridman, G.; Fridman, A.; Miller, V. Nanosecond-Pulsed DBD Plasma-Generated Reactive Oxygen Species Trigger Immunogenic Cell Death in A549 Lung Carcinoma Cells through Intracellular Oxidative Stress. Int. J. Mol. Sci. 2017, 18, 966. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.G.; Xiang, B.; Merlino, D.J.; Baybutt, T.R.; Sahu, J.; Fridman, A.; Snook, A.E.; Miller, V. Non-Thermal Plasma Induces Immunogenic Cell Death in Vivo in Murine CT26 Colorectal Tumors. Oncoimmunology 2018, 7, e1484978. [Google Scholar] [CrossRef]
- Freund, E.; Liedtke, K.R.; van der Linde, J.; Metelmann, H.R.; Heidecke, C.D.; Partecke, L.I.; Bekeschus, S. Physical Plasma-Treated Saline Promotes an Immunogenic Phenotype in CT26 Colon Cancer Cells in Vitro and in Vivo. Sci. Rep. 2019, 9, 634. [Google Scholar] [CrossRef]
- Azzariti, A.; Iacobazzi, R.M.; Di Fonte, R.; Porcelli, L.; Gristina, R.; Favia, P.; Fracassi, F.; Trizio, I.; Silvestris, N.; Guida, G.; et al. Plasma-Activated Medium Triggers Cell Death and the Presentation of Immune Activating Danger Signals in Melanoma and Pancreatic Cancer Cells. Sci. Rep. 2019, 9, 4099. [Google Scholar] [CrossRef]
- Di Blasio, S.; Wortel, I.M.; van Bladel, D.A.; de Vries, L.E.; Duiveman-de Boer, T.; Worah, K.; de Haas, N.; Buschow, S.I.; de Vries, I.J.; Figdor, C.G.; et al. Human CD1c(+) DCs are Critical Cellular Mediators of Immune Responses Induced by Immunogenic Cell Death. Oncoimmunology 2016, 5, e1192739. [Google Scholar] [CrossRef]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic Cell Vaccines Based on Immunogenic Cell Death Elicit Danger Signals and T Cell-Driven Rejection of High-Grade Glioma. Sci. Transl. Med. 2016, 8, 328ra27. [Google Scholar] [CrossRef]
- Pawaria, S.; Binder, R.J. CD91-Dependent Programming of T-Helper Cell Responses Following Heat Shock Protein Immunization. Nat. Commun. 2011, 2, 521. [Google Scholar] [CrossRef]
- Garg, A.D.; Dudek, A.M.; Ferreira, G.B.; Verfaillie, T.; Vandenabeele, P.; Krysko, D.V.; Mathieu, C.; Agostinis, P. ROS-Induced Autophagy in Cancer Cells Assists in Evasion from Determinants of Immunogenic Cell Death. Autophagy 2013, 9, 1292–1307. [Google Scholar] [CrossRef]
- Lin, A.; Gorbanev, Y.; De Backer, J.; Van Loenhout, J.; Van Boxem, W.; Lemière, F.; Cos, P.; Dewilde, S.; Smits, E.; Bogaerts, A. Non-Thermal Plasma as a Unique Delivery System of Short-Lived Reactive Oxygen and Nitrogen Species for Immunogenic Cell Death in Melanoma Cells. Adv. Sci. 2019, 1802062. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, M.; Then, F.; Galambos, P.; Scholz, C.; Siegmund, B.; Endres, S.; Eigler, A. Extracellular ATP and TNF-alpha Synergize in the Activation and Maturation of Human Dendritic Cells. J. Immunol. 2000, 165, 4704–4709. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Ueno, H.; Fay, J.; Banchereau, J. Dendritic Cells and Immunity against Cancer. J. Intern. Med. 2011, 269, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Deicher, A.; Andersson, R.; Tingstedt, B.; Lindell, G.; Bauden, M.; Ansari, D. Targeting Dendritic Cells in Pancreatic Ductal Adenocarcinoma. Cancer Cell Int. 2018, 18, 85. [Google Scholar] [CrossRef] [PubMed]
- Bekeschus, S.; Kolata, J.; Winterbourn, C.; Kramer, A.; Turner, R.; Weltmann, K.D.; Broker, B.; Masur, K. Hydrogen Peroxide: A Central Player in Physical Plasma-Induced Oxidative Stress in Human Blood Cells. Free Radic. Res. 2014, 48, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Seres, T.; Knickelbein, R.G.; Warshaw, J.B.; Johnston, R.B., Jr. The Phagocytosis-Associated Respiratory Burst in Human Monocytes is Associated with Increased Uptake of Glutathione. J. Immunol. 2000, 165, 3333–3340. [Google Scholar] [CrossRef] [PubMed]
- Jesnowski, R.; Furst, D.; Ringel, J.; Chen, Y.; Schrodel, A.; Kleeff, J.; Kolb, A.; Schareck, W.D.; Lohr, M. Immortalization of Pancreatic Stellate Cells As an in Vitro Model of Pancreatic Fibrosis: Deactivation is Induced by Matrigel and N-Acetylcysteine. Lab. Investig. 2005, 85, 1276–1291. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Yoshida, N.; Takikawa, T.; Shimosegawa, T. IL-6/STAT3 Plays a Regulatory Role in the Interaction Between Pancreatic Stellate Cells and Cancer Cells. Dig. Dis. Sci. 2016, 61, 1561–1571. [Google Scholar] [CrossRef]
- Bekeschus, S.; Schmidt, A.; Weltmann, K.D.; von Woedtke, T. The Plasma jet kINPen—A Powerful Tool for Wound Healing. Clin. Plasma Med. 2016, 4, 19–28. [Google Scholar] [CrossRef]
- Smits, E.L.; Ponsaerts, P.; Van de Velde, A.L.; Van Driessche, A.; Cools, N.; Lenjou, M.; Nijs, G.; Van Bockstaele, D.R.; Berneman, Z.N.; Van Tendeloo, V.F. Proinflammatory Response of Human Leukemic Cells to dsRNA Transfection Linked to Activation of Dendritic Cells. Leukemia 2007, 21, 1691–1699. [Google Scholar] [CrossRef]
- Lion, E.; Anguille, S.; Berneman, Z.N.; Smits, E.L.; Van Tendeloo, V.F. Poly(I:C) Enhances the Susceptibility of Leukemic Cells to NK Cell Cytotoxicity and Phagocytosis by DC. PLoS ONE 2011, 6, e20952. [Google Scholar] [CrossRef] [PubMed]
- De Waele, J.; Marcq, E.; Van Audenaerde, J.R.; Van Loenhout, J.; Deben, C.; Zwaenepoel, K.; Van de Kelft, E.; Van der Planken, D.; Menovsky, T.; Van den Bergh, J.M.; et al. Poly(I:C) Primes Primary Human Glioblastoma Cells for an Immune Response Invigorated by PD-L1 Blockade. Oncoimmunology 2018, 7, e1407899. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Loenhout, J.; Flieswasser, T.; Freire Boullosa, L.; De Waele, J.; Van Audenaerde, J.; Marcq, E.; Jacobs, J.; Lin, A.; Lion, E.; Dewitte, H.; et al. Cold Atmospheric Plasma-Treated PBS Eliminates Immunosuppressive Pancreatic Stellate Cells and Induces Immunogenic Cell Death of Pancreatic Cancer Cells. Cancers 2019, 11, 1597. https://doi.org/10.3390/cancers11101597
Van Loenhout J, Flieswasser T, Freire Boullosa L, De Waele J, Van Audenaerde J, Marcq E, Jacobs J, Lin A, Lion E, Dewitte H, et al. Cold Atmospheric Plasma-Treated PBS Eliminates Immunosuppressive Pancreatic Stellate Cells and Induces Immunogenic Cell Death of Pancreatic Cancer Cells. Cancers. 2019; 11(10):1597. https://doi.org/10.3390/cancers11101597
Chicago/Turabian StyleVan Loenhout, Jinthe, Tal Flieswasser, Laurie Freire Boullosa, Jorrit De Waele, Jonas Van Audenaerde, Elly Marcq, Julie Jacobs, Abraham Lin, Eva Lion, Heleen Dewitte, and et al. 2019. "Cold Atmospheric Plasma-Treated PBS Eliminates Immunosuppressive Pancreatic Stellate Cells and Induces Immunogenic Cell Death of Pancreatic Cancer Cells" Cancers 11, no. 10: 1597. https://doi.org/10.3390/cancers11101597