Antileukemic Efficacy in Vitro of Talazoparib and APE1 Inhibitor III Combined with Decitabine in Myeloid Malignancies



Abstract
:1. Introduction
2. Results
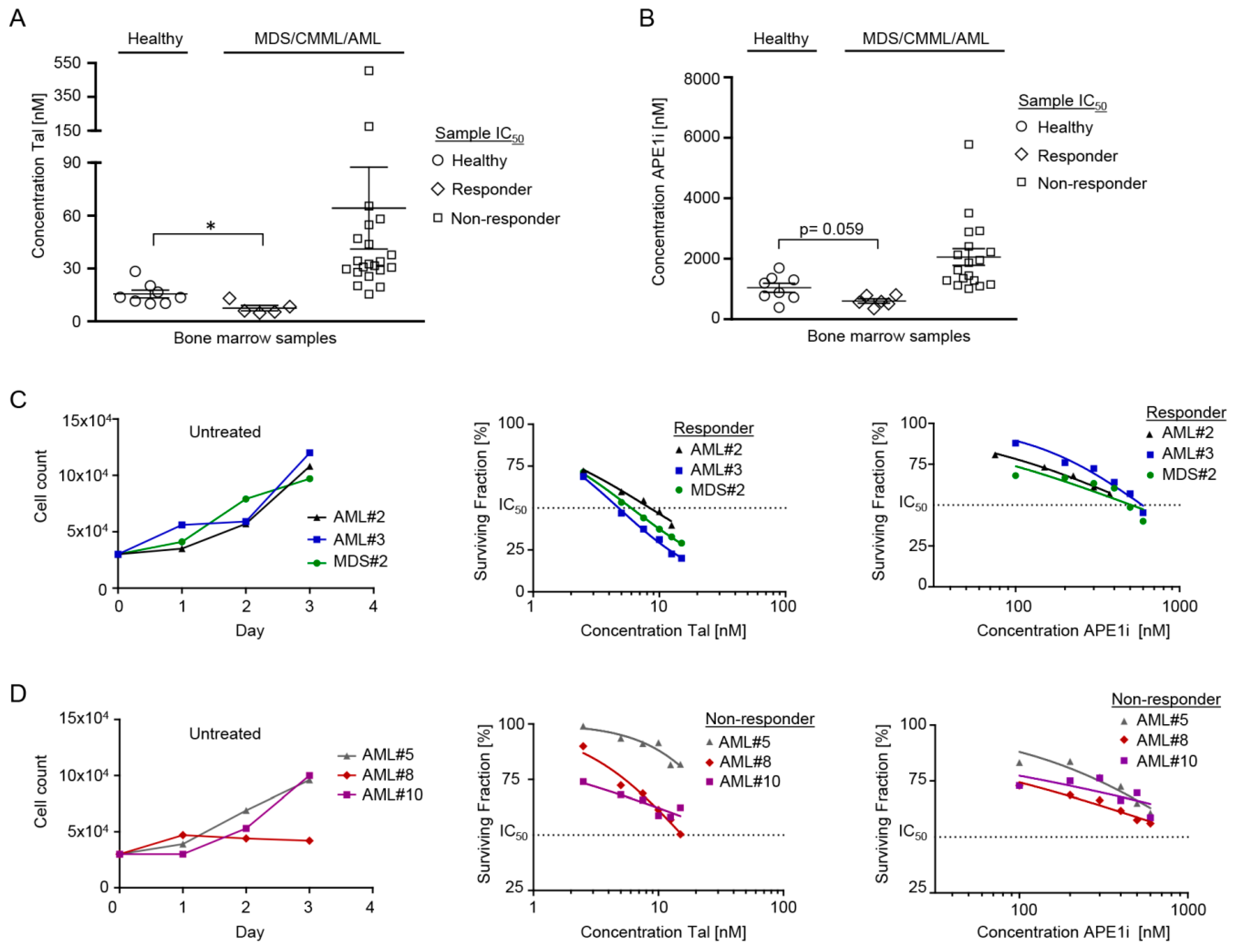
2.1. Cytotoxic Efficacy of Talazoparib and APE1 Inhibitor III in MDS/CMML and AML Cells
2.2. Cytotoxic Efficacy of Decitabine ± Talazoparib, Decitabine ± APE1 Inhibitor III, and Talazoparib ± APE1 Inhibitor III in MDS/CMML and AML Cells
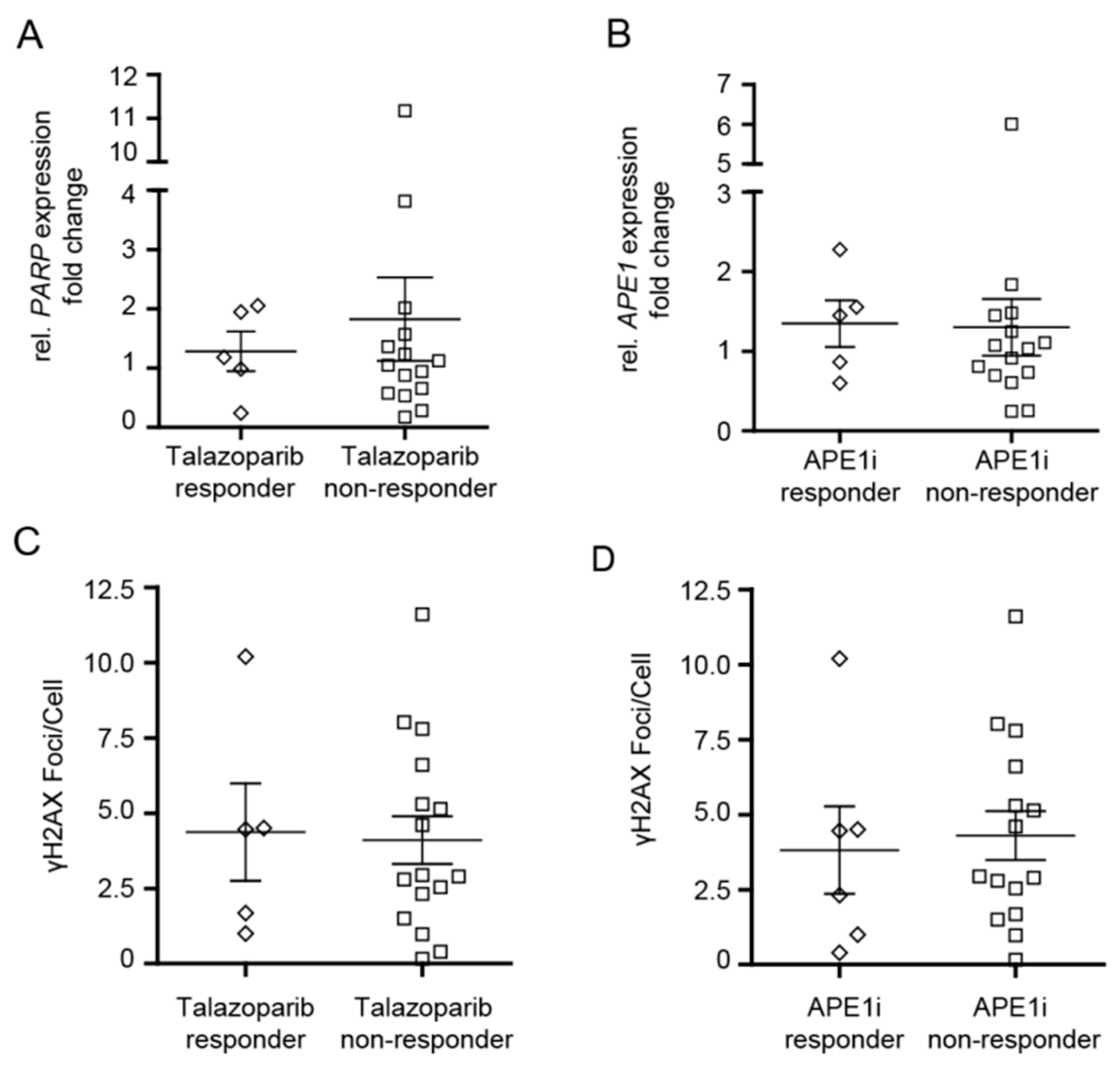
2.3. Analysis of PARP1 and APE1 mRNA Expression in MDS/CMML and AML Cells in Correlation to Cytotoxic Efficacy of Talazoparib and APE1 Inhibitor III
2.4. Analysis of γH2AX Foci in MDS/CMML and AML Cells in Correlation to Cytotoxic Efficacy of Talazoparib and APE1 Inhibitor III
2.5. Analysis of Chromosomal Aberrations and Gene Mutations in Correlation to Cytotoxic Efficacy of Talazoparib and APE1 Inhibitor III
3. Discussion
4. Materials and Methods
4.1. Bone Marrow Samples
4.2. Cytology, Flow Cytometry, Cytogenetics, and Gene Mutation Analysis
4.3. Cell Proliferation
4.4. Immunofluorescence Staining of γH2AX
4.5. PARP1 and APE1 mRNA Expression
4.6. Drug Stock Solutions
4.7. Cell Survival Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shah, A.; Andersson, T.M.; Rachet, B.; Bjorkholm, M.; Lambert, P.C. Survival and cure of acute myeloid leukaemia in England, 1971–2006: A population-based study. Br. J. Haematol. 2013, 162, 509–516. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Sole, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Tefferi, A. Chronic myelomonocytic leukemia: 2018 update on diagnosis, risk stratification and management. Am. J. Hematol. 2018, 93, 824–840. [Google Scholar] [CrossRef] [PubMed]
- Lubbert, M.; Suciu, S.; Baila, L.; Ruter, B.H.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; Labar, B.; Germing, U.; Salih, H.R.; et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: Final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J. Clin. Oncol. 2011, 29, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.P.; Chou, W.C.; Buckstein, R.; Cermak, J.; et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Issa, J.P.; Rosenfeld, C.S.; Bennett, J.M.; Albitar, M.; DiPersio, J.; Klimek, V.; Slack, J.; de Castro, C.; Ravandi, F.; et al. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer 2006, 106, 1794–1803. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Cashen, A.F.; Schiller, G.J.; O’Donnell, M.R.; DiPersio, J.F. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 556–561. [Google Scholar] [CrossRef]
- Jabbour, E.; Garcia-Manero, G.; Batty, N.; Shan, J.; O’Brien, S.; Cortes, J.; Ravandi, F.; Issa, J.P.; Kantarjian, H. Outcome of patients with myelodysplastic syndrome after failure of decitabine therapy. Cancer 2010, 116, 3830–3834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prebet, T.; Gore, S.D.; Esterni, B.; Gardin, C.; Itzykson, R.; Thepot, S.; Dreyfus, F.; Rauzy, O.B.; Recher, C.; Ades, L.; et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J. Clin. Oncol. 2011, 29, 3322–3327. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Gore, S.D.; Thepot, S.; Esterni, B.; Quesnel, B.; Beyne Rauzy, O.; Dreyfus, F.; Gardin, C.; Fenaux, P.; Vey, N. Outcome of acute myeloid leukaemia following myelodysplastic syndrome after azacitidine treatment failure. Br. J. Haematol. 2012, 157, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Harel, S.; Cherait, A.; Berthon, C.; Willekens, C.; Park, S.; Rigal, M.; Brechignac, S.; Thepot, S.; Quesnel, B.; Gardin, C.; et al. Outcome of patients with high risk Myelodysplastic Syndrome (MDS) and advanced Chronic Myelomonocytic Leukemia (CMML) treated with decitabine after azacitidine failure. Leuk. Res. 2015, 39, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. An Open-label, Multicenter Phase 1 Trial to Evaluate the Safety, Pharmacokinetics and Pharmacodynamics of Splicing Modulator H3B-8800 for Subjects with Myelodysplastic Syndromes, Acute Myeloid Leukemia, and Chronic Myelomonocytic Leukemia. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT02841540 (accessed on 16 August 2019).
- Issa, J.J.; Roboz, G.; Rizzieri, D.; Jabbour, E.; Stock, W.; O’Connell, C.; Yee, K.; Tibes, R.; Griffiths, E.A.; Walsh, K.; et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: A multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015, 16, 1099–1110. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Roboz, G.J.; Kropf, P.L.; Yee, K.W.L.; O’Connell, C.L.; Tibes, R.; Walsh, K.J.; Podoltsev, N.A.; Griffiths, E.A.; Jabbour, E.; et al. Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: Phase 2 results from a multicentre, randomised, phase 1/2 trial. Lancet Oncol. 2017, 18, 1317–1326. [Google Scholar] [CrossRef]
- Renneville, A.; Abdelali, R.B.; Chevret, S.; Nibourel, O.; Cheok, M.; Pautas, C.; Dulery, R.; Boyer, T.; Cayuela, J.M.; Hayette, S.; et al. Clinical impact of gene mutations and lesions detected by SNP-array karyotyping in acute myeloid leukemia patients in the context of gemtuzumab ozogamicin treatment: Results of the ALFA-0701 trial. Oncotarget 2014, 5, 916–932. [Google Scholar] [CrossRef]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Altmeyer, M.; Messner, S.; Hassa, P.O.; Fey, M.; Hottiger, M.O. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009, 37, 3723–3738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrer, J.; Kranaster, R.; Altmeyer, M.; Marx, A.; Burkle, A. Quantitative analysis of the binding affinity of poly(ADP-ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res. 2007, 35, e143. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. 2013, 19, 5003–5015. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N. PARP inhibitors for anticancer therapy. Biochem. Soc. Trans. 2014, 42, 82–88. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents—A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef]
- Rai, G.; Vyjayanti, V.N.; Dorjsuren, D.; Simeonov, A.; Jadhav, A.; Wilson, D.M., 3rd; Maloney, D.J. Synthesis, biological evaluation, and structure-activity relationships of a novel class of apurinic/apyrimidinic endonuclease 1 inhibitors. J. Med. Chem. 2012, 55, 3101–3112. [Google Scholar] [CrossRef]
- Lindahl, T. DNA glycosylases, endonucleases for apurinic/apyrimidinic sites, and base excision-repair. Prog. Nucleic Acid Res. Mol. Biol. 1979, 22, 135–192. [Google Scholar]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell. Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Aggarwal, A.K.; Rechkoblit, O. Eukaryotic DNA polymerases. Curr. Opin. Struct. Biol. 2018, 53, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.; Poli, J.; Marians, K.J.; Pasero, P. Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; McNeill, D.R.; Abbotts, R.; Mohammed, M.Z.; Zdzienicka, M.Z.; Qutob, H.; Seedhouse, C.; Laughton, C.A.; Fischer, P.M.; Patel, P.M.; et al. Synthetic lethal targeting of DNA double-strand break repair deficient cells by human apurinic/apyrimidinic endonuclease inhibitors. Int. J. Cancer 2012, 131, 2433–2444. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Issa, J.P. DNA methylation as a therapeutic target in cancer. Clin. Cancer Res. 2007, 13, 1634–1637. [Google Scholar] [CrossRef]
- Patel, K.; Dickson, J.; Din, S.; Macleod, K.; Jodrell, D.; Ramsahoye, B. Targeting of 5-aza-2’-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res. 2010, 38, 4313–4324. [Google Scholar] [CrossRef]
- Esposito, M.T.; Zhao, L.; Fung, T.K.; Rane, J.K.; Wilson, A.; Martin, N.; Gil, J.; Leung, A.Y.; Ashworth, A.; So, C.W. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat. Med. 2015, 21, 1481–1490. [Google Scholar] [CrossRef]
- Gaymes, T.J.; Shall, S.; MacPherson, L.J.; Twine, N.A.; Lea, N.C.; Farzaneh, F.; Mufti, G.J. Inhibitors of poly ADP-ribose polymerase (PARP) induce apoptosis of myeloid leukemic cells: Potential for therapy of myeloid leukemia and myelodysplastic syndromes. Haematologica 2009, 94, 638–646. [Google Scholar] [CrossRef]
- Faraoni, I.; Compagnone, M.; Lavorgna, S.; Angelini, D.F.; Cencioni, M.T.; Piras, E.; Panetta, P.; Ottone, T.; Dolci, S.; Venditti, A.; et al. BRCA1, PARP1 and gammaH2AX in acute myeloid leukemia: Role as biomarkers of response to the PARP inhibitor olaparib. Biochim. Biophys. Acta 2015, 1852, 462–472. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Study of BMN 673, a PARP Inhibitor, in Patients with Advanced Hematological Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT01399840?term=BMN+673%2C+a+ PARP+inhibitor&rank=2 (accessed on 16 August 2019).
- ClinicalTrials.gov. Talazoparib for Cohesin-Mutated AML and MDS With Excess Blasts. Available online: https://clinicaltrials.gov/ct2/show/NCT03974217?term=talazoparib%2C+cohesin&rank=1 (accessed on 16 August 2019).
- ClinicalTrials.gov. Decitabine and Talazoparib in Untreated AML and R/R AML (1565GCC). Available online: https://clinicaltrials.gov/ct2/show/NCT02878785?term=Decitabine+and+Talazoparib+in+Untreated+AML+and+R%2FR+AML&rank=1 (accessed on 16 August 2019).
- Orta, M.L.; Hoglund, A.; Calderon-Montano, J.M.; Dominguez, I.; Burgos-Moron, E.; Visnes, T.; Pastor, N.; Strom, C.; Lopez-lazaro, M.; Helleday, T. The PARP inhibitor Olaparib disrupts base excision repair of 5-aza-2’-deoxycytidine lesions. Nucleic Acids Res. 2014, 42, 9108–9120. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Horton, J.K.; Chastain, P.D., 2nd; Gassman, N.R.; Freudenthal, B.D.; Hou, E.W.; Wilson, S.H. Suicidal cross-linking of PARP-1 to AP site intermediates in cells undergoing base excision repair. Nucleic Acids Res. 2014, 42, 6337–6351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Löffler, H.; Rastetter, J.; Haferlach, T. (Eds.) Light microscopic procedures. In Atlas of Clinical Hematology, 6th ed.; Springer: Heidelberg, Germany, 2005; p. 8. [Google Scholar]
- Gisselsson, D. Cytogenetic methods. In Cancer Cytogenetics, 3rd ed.; Heim, S., Mitelman, F., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2009; pp. 9–16. [Google Scholar]
- MLL. Request for Testing Form. Available online: https://www.mll.com/en.html (accessed on 16 August 2019).
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111. [Google Scholar] [CrossRef]
- Popp, H.D.; Brendel, S.; Hofmann, W.K.; Fabarius, A. Immunofluorescence Microscopy of γH2AX and 53BP1 for Analyzing the Formation and Repair of DNA Double-strand Breaks. J. Vis. Exp. 2017, 129. [Google Scholar] [CrossRef]
- Popp, H.D.; Naumann, N.; Brendel, S.; Henzler, T.; Weiss, C.; Hofmann, W.K.; Fabarius, A. Increase of DNA damage and alteration of the DNA damage response in myelodysplastic syndromes and acute myeloid leukemias. Leuk. Res. 2017, 57, 112–118. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| # | Age/Sex | Disease | Karyotype/FISH | Mutations | IC50 [nM] | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tal | APE1i | Dec | Dec + Tal | Dec + APE1i | Tal + APE1i | |||||
| HEALTHY#1 | 25/♂ | Healthy | 46,XY[20] | - | 20 | 1359 | 254 | 58 | 206 | 19 |
| HEALTHY#2 | 23/♀ | Healthy | 46,XX[20] | - | 28 | 392 | 23 | - | - | - |
| HEALTHY#3 | 77/♀ | Healthy | 46,XX[20] | - | 12 | 1195 | 156 | 52 | 128 | 10 |
| HEALTHY#4 | 26/♂ | Healthy | 46,XY[20] | - | 10 | 882 | 99 | 24 | 55 | 7 |
| HEALTHY#5 | 21/♂ | Healthy | 46,XY[20] | - | 14 | 724 | 113 | 14 | 102 | 8 |
| HEALTHY#6 | 22/♀ | Healthy | 46,XX[20] | - | 10 | 1697 | 111 | 26 | 111 | 10 |
| HEALTHY#7 | 21/♀ | Healthy | 46,XX[20] | - | 14 | 1305 | 147 | 35 | 123 | 12 |
| HEALTHY#8 | 25/♀ | Healthy | 46,XX[20] | - | 17 | 775 | 186 | 23 | 166 | 11 |
| MDS#1 | 59/♂ | MDS-EB-1 | 46,XY,-7,der(9)t(9;20)(p21;q13), der(20)t(9;20)(p21;q11),+der(20) t(9;20)(p21;q11)[17]/46,XY[3] | CBL, DNMT3A (VAR), EZH2 (VAR), FLT3-ITD | 20 | 1357 | 160 | 48 | 137 | 19 |
| MDS#2 | 73/♀ | MDS-MLD | 46,XX[20] | ASXL1, GATA2, RUNX1, U2AF1 | 6 | 509 | 9 | 3 | 8 | 5 |
| MDS#3 | 68/ ♂ | MDS-MLD | 45,X,-Y[8]/45,X,-Y,del(1)(p34p36)[12] | ASXL1, U2AF1 | 26 | 1281 | 107 | 24 | 60 | 21 |
| MDS#4 | 57♀ | MDS-EB-1 | 46,XX[20] | ASXL1 | 31 | 2921 | 185 | 89 | 181 | 36 |
| CMML#1 | 77/♂ | CMML-1 | 46,XY,t(2;2)(p23;q32)[10]/47,XY, t(2;2)(p23;q32),+8[2]/46,XY[9] | ASXL1, CEBPA, IDH1, RUNX1, SRSF2 | 28 | 1095 | 42 | 25 | 32 | 28 |
| CMML#2 | 73/♀ | CMML-2 | 46,XX[20] | ASXL1, CEBPA, EZH2 (VAR), TET2 | 13 | 1286 | 210 | 62 | 159 | 11 |
| CMML#3 | 84/♂ | CMML-0 | 46,XY[20] | DNMT3A, RUNX1, SRSF2, TET2 | 175 | 1008 | 121 | 42 | 67 | 47 |
| CMML#4 | 65/♂ | CMML-2 | 46,XY[20] | ASXL1, IDH2, NRAS, PTPN11, ZRSR2 | 47 | 1954 | 153 | 48 | 122 | 35 |
| AML#1 | 79/♂ | sAML | 46,XY[29] | ASXL1, FLT3-ITD, RUNX1, SF3B1 | 5 | 350 | 238 | 19 | 196 | 3 |
| AML#2 | 63/♂ | sAML | 46,XY[25] | JAK2, KMT2A-PTD (MLL-PTD), NRAS, SRSF2, TET2 (VAR) | 8 | 564 | 28 | - | - | - |
| AML#3 | 76/♂ | sAML | 46,XY[17] | BCOR, DNMT3A, KMT2A-PTD (MLL-PTD), NRAS, TET2, U2AF1 | 5 | 593 | 119 | 18 | 100 | 4 |
| AML#4 | 63/♀ | de novo AML | 46,XX,t(7;9)(q22;q34),add(17)(p12)[22]/46,XX[3] | ASXL1, DNMT3A, PTPN11, RUNX1 | 65 | 2407 | 172 | 93 | 137 | 84 |
| AML#5 | 78/♂ | sAML | 47,XY,+8[3]/46,XY[17] | ASXL1, IDH2, SRSF2 | 31 | 1148 | 135 | 73 | 96 | 17 |
| AML#6 | 83/♂ | de novo AML | 46,XY[20] | - | 30 | 793 | 268 | 223 | 293 | 28 |
| AML#7 | 72/♂ | sAML | 46,XY[20] | ASXL1, IDH2, SF3B1 | 44 | 1867 | 416 | 389 | 347 | 32 |
| AML#8 | 70/♀ | de novo AML | 46,XX[20] | FLT3-ITD, NPM1, TET2 | 16 | 1122 | 91 | 92 | 64 | 15 |
| AML#9 | 53/♀ | de novo AML | 46,XX[25] | DNMT3A, FLT3-TKD, IDH1, NPM1, NRAS | 33 | 5781 | 348 | 763 | 642 | 85 |
| AML#10 | 33/♀ | de novo AML | 46,XX[20] | DNMT3A (VAR), FLT3-ITD, KMT2A-PTD (MLL-PTD), KRAS | 38 | 3511 | 100 | 46 | 59 | 17 |
| AML#11 | 66/♂ | de novo AML | 47,XY,+8[13]/46,XY[7] | ASXL1, DNMT3A, IDH2, RUNX1, SRSF2 | 34 | 2893 | 51 | 34 | 35 | 25 |
| AML#12 | 68/♀ | de novo AML | 51,XX,+1,der(2)t(2;12),der(5) t(5;13),+8, +11,der(12),-der(13),+15,+19,+mar[25] | TP53 | 505 | 808 | 215 | - | 231 | - |
| AML#13 | 89/♂ | sAML | 47,XY,+8[23]/46,XY[2] | - | 34 | 2131 | 205 | 294 | 318 | 119 |
| AML#14 | 47/♂ | de novo AML | 42-46,XY,t(1;4)(p33;q35),del(3q),add(6q),-13, -15,-16,-17,-18,+19,+2-4mar [cp15] | DNMT3A, TP53 | 55 | - | - | - | - | - |
| AML#15 | 63/♂ | de novo AML | 46,XY[11] | IDH2, NPM1, SRSF2 | 32 | - | 756 | 306 | 3077 | 40 |
| AML#16 | 69/♂ | sAML | 47,XY,+21[6]/46,XY[14] | DNMT3A, KMT2A-PTD (MLL-PTD), RUNX1 | 19 | 1443 | 172 | 88 | 136 | 21 |
| AML#17 | 69/♂ | sAML | 46,XY[26] | FLT3-ITD, GATA2, WT1 | 58 | 2226 | 248 | 145 | 181 | 32 |
| AML#18 | 59/♀ | de novo AML | 45,XX[25] | BCOR, ETV6 (VAR), EZH2 (VAR), FLT3-ITD, NPM1, KRAS, TET2 (VAR) | 29 | 1626 | - | - | - | - |
| Talazoparib | APE1 inhibitor III | normal karyotype | -Y | -13 | -15 | -16 | -17 | -18 | Trisomy 1 | Monosomy 7 | Trisomy 8 | Trisomy 11 | Trisomy 15 | Trisomy 19 | Trisomy 21 | t(1;4) | t(2;2) | t(2;12) | t(5:13) | t(7;9) | t(9;20) | del(1)(p34p36) | del(3q) | del17p | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AML#6 | Responder | ||||||||||||||||||||||||
| AML#12 | |||||||||||||||||||||||||
| MDS#2 | Responder | ||||||||||||||||||||||||
| AML#1 | |||||||||||||||||||||||||
| AML#2 | |||||||||||||||||||||||||
| AML#3 | |||||||||||||||||||||||||
| CMML#2 | Non-responder | ||||||||||||||||||||||||
| MDS#1 | Non-responder | ||||||||||||||||||||||||
| MDS#3 | |||||||||||||||||||||||||
| MDS#4 | |||||||||||||||||||||||||
| CMML#1 | |||||||||||||||||||||||||
| CMML#3 | |||||||||||||||||||||||||
| CMML#4 | |||||||||||||||||||||||||
| AML#4 | |||||||||||||||||||||||||
| AML#5 | |||||||||||||||||||||||||
| AML#7 | |||||||||||||||||||||||||
| AML#8 | |||||||||||||||||||||||||
| AML#9 | |||||||||||||||||||||||||
| AML#10 | |||||||||||||||||||||||||
| AML#11 | |||||||||||||||||||||||||
| AML#13 | |||||||||||||||||||||||||
| AML#14 | |||||||||||||||||||||||||
| AML#15 | |||||||||||||||||||||||||
| AML#16 | |||||||||||||||||||||||||
| AML#17 | |||||||||||||||||||||||||
| AML#18 |
| Talazoparib | APE1 inhibitor III | ASXL1 | ASXL2 | BCOR | CBL | CEBPA | DNMT3A | ETV6 | EZH2 | FLT3-ITD | FLT3-TKD | GATA1 | GATA2 | IDH1 | IDH2 | JAK2 | KIT | KMT2A-PTD (MLL-PTD) | NPM1 | NRAS | KRAS | PTPN11 | RUNX1 | SF3B1 | SRSF2 | TET2 | TP53 | U2AF1 | WT1 | ZRSR2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AML#6 | Responder | not available | |||||||||||||||||||||||||||||
| AML#12 | |||||||||||||||||||||||||||||||
| MDS#2 | Responder | ||||||||||||||||||||||||||||||
| AML#1 | |||||||||||||||||||||||||||||||
| AML#2 | VAR | ||||||||||||||||||||||||||||||
| AML#3 | |||||||||||||||||||||||||||||||
| CMML#2 | Non-responder | VAR | |||||||||||||||||||||||||||||
| MDS#1 | Non-responder | VAR | VAR | ||||||||||||||||||||||||||||
| MDS#3 | |||||||||||||||||||||||||||||||
| MDS#4 | |||||||||||||||||||||||||||||||
| CMML#1 | |||||||||||||||||||||||||||||||
| CMML#3 | |||||||||||||||||||||||||||||||
| CMML#4 | |||||||||||||||||||||||||||||||
| AML#4 | |||||||||||||||||||||||||||||||
| AML#5 | |||||||||||||||||||||||||||||||
| AML#7 | |||||||||||||||||||||||||||||||
| AML#8 | |||||||||||||||||||||||||||||||
| AML#9 | |||||||||||||||||||||||||||||||
| AML#10 | VAR | ||||||||||||||||||||||||||||||
| AML#11 | |||||||||||||||||||||||||||||||
| AML#13 | not available | ||||||||||||||||||||||||||||||
| AML#14 | |||||||||||||||||||||||||||||||
| AML#15 | |||||||||||||||||||||||||||||||
| AML#16 | |||||||||||||||||||||||||||||||
| AML#17 | |||||||||||||||||||||||||||||||
| AML#18 | VAR | VAR | VAR | ||||||||||||||||||||||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohl, V.; Flach, J.; Naumann, N.; Brendel, S.; Kleiner, H.; Weiss, C.; Seifarth, W.; Nowak, D.; Hofmann, W.-K.; Fabarius, A.; et al. Antileukemic Efficacy in Vitro of Talazoparib and APE1 Inhibitor III Combined with Decitabine in Myeloid Malignancies. Cancers 2019, 11, 1493. https://doi.org/10.3390/cancers11101493
Kohl V, Flach J, Naumann N, Brendel S, Kleiner H, Weiss C, Seifarth W, Nowak D, Hofmann W-K, Fabarius A, et al. Antileukemic Efficacy in Vitro of Talazoparib and APE1 Inhibitor III Combined with Decitabine in Myeloid Malignancies. Cancers. 2019; 11(10):1493. https://doi.org/10.3390/cancers11101493
Chicago/Turabian StyleKohl, Vanessa, Johanna Flach, Nicole Naumann, Susanne Brendel, Helga Kleiner, Christel Weiss, Wolfgang Seifarth, Daniel Nowak, Wolf-Karsten Hofmann, Alice Fabarius, and et al. 2019. "Antileukemic Efficacy in Vitro of Talazoparib and APE1 Inhibitor III Combined with Decitabine in Myeloid Malignancies" Cancers 11, no. 10: 1493. https://doi.org/10.3390/cancers11101493