Inhibition of Gap Junctions Sensitizes Primary Glioblastoma Cells for Temozolomide

 ,
,  , ,
, ,  ,
,  ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
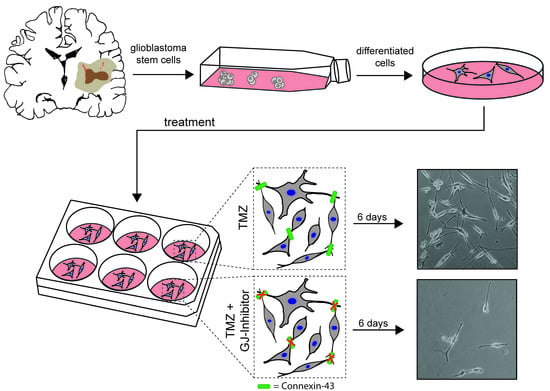
2.1. Characterization of Primary Human Glioblastoma Cell Populations
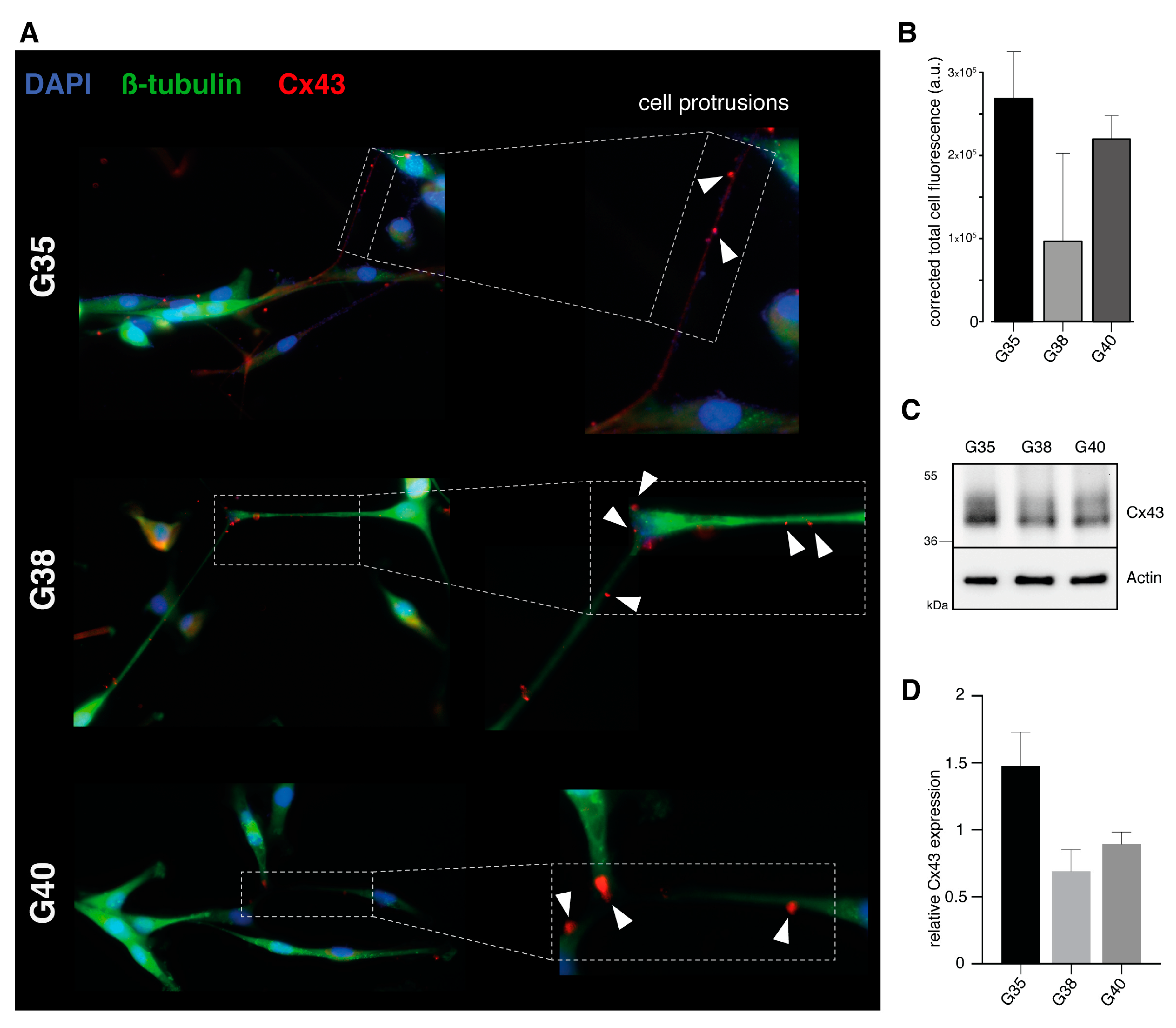
2.2. Verification of Connexin-43 Expression in Primary Glioblastoma Cell Populations
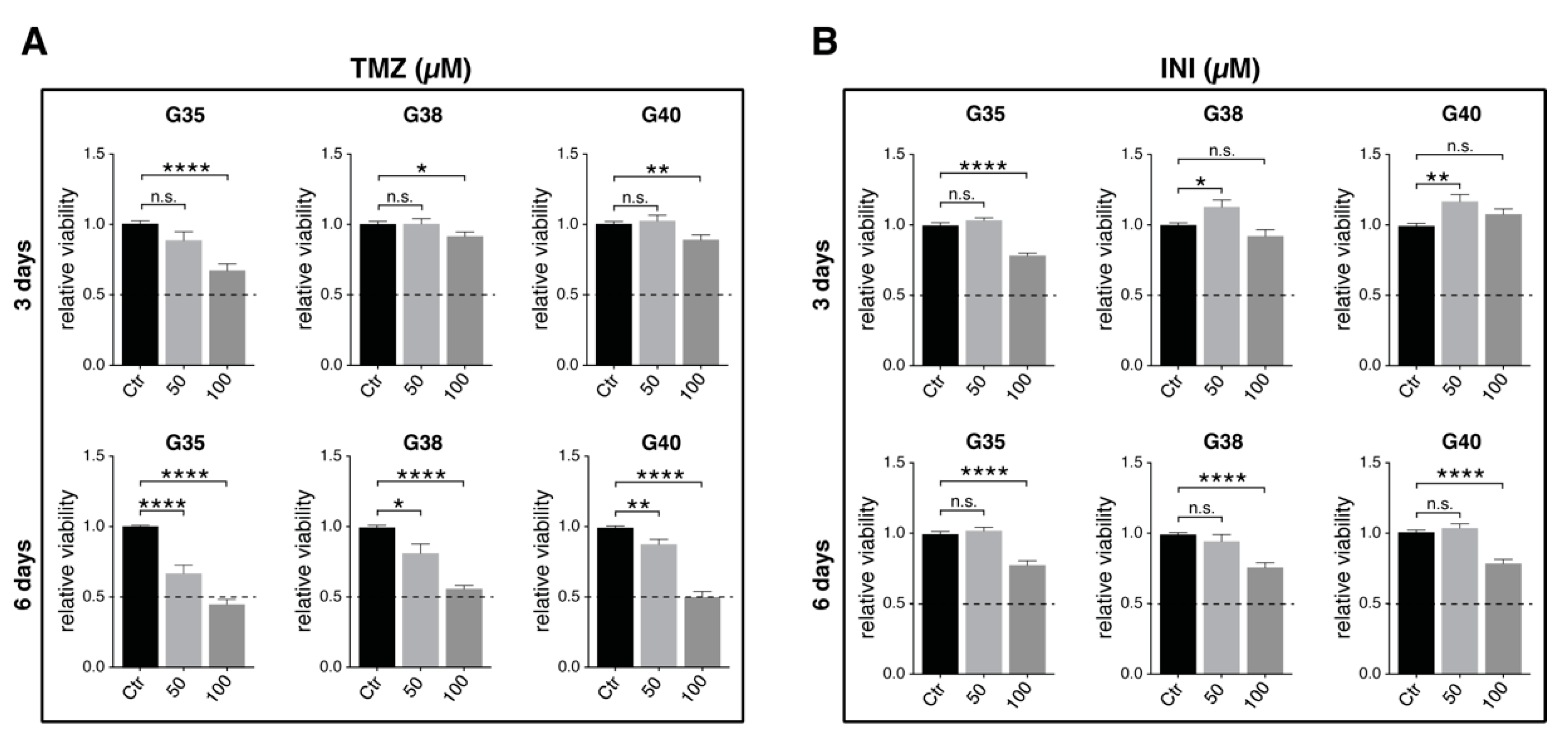
2.3. Determination of Effective Drug Concentrations
2.4. Additional Gap Junction-targeted Therapy Significantly Diminishes Glioblastoma Cell Confluence
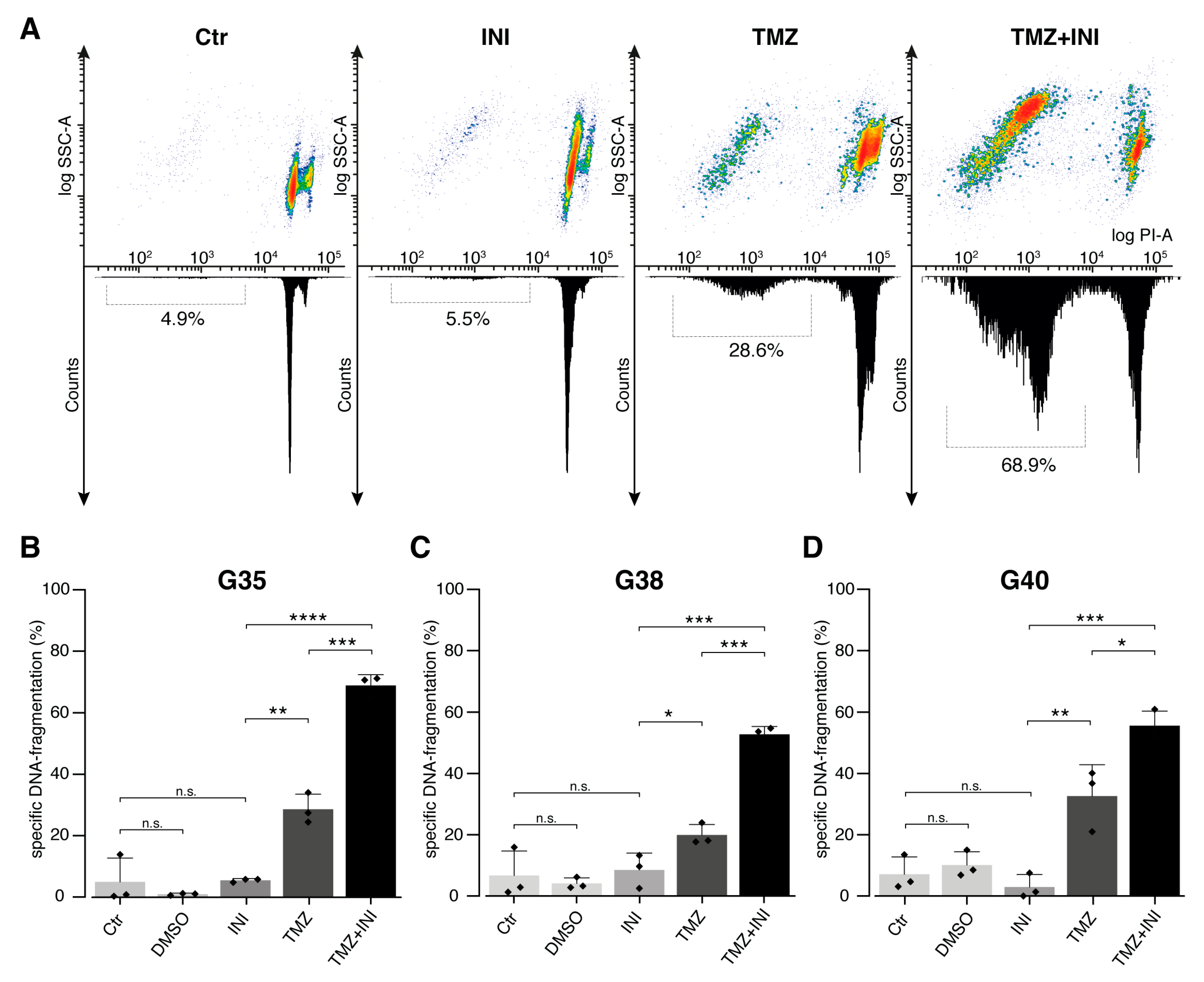
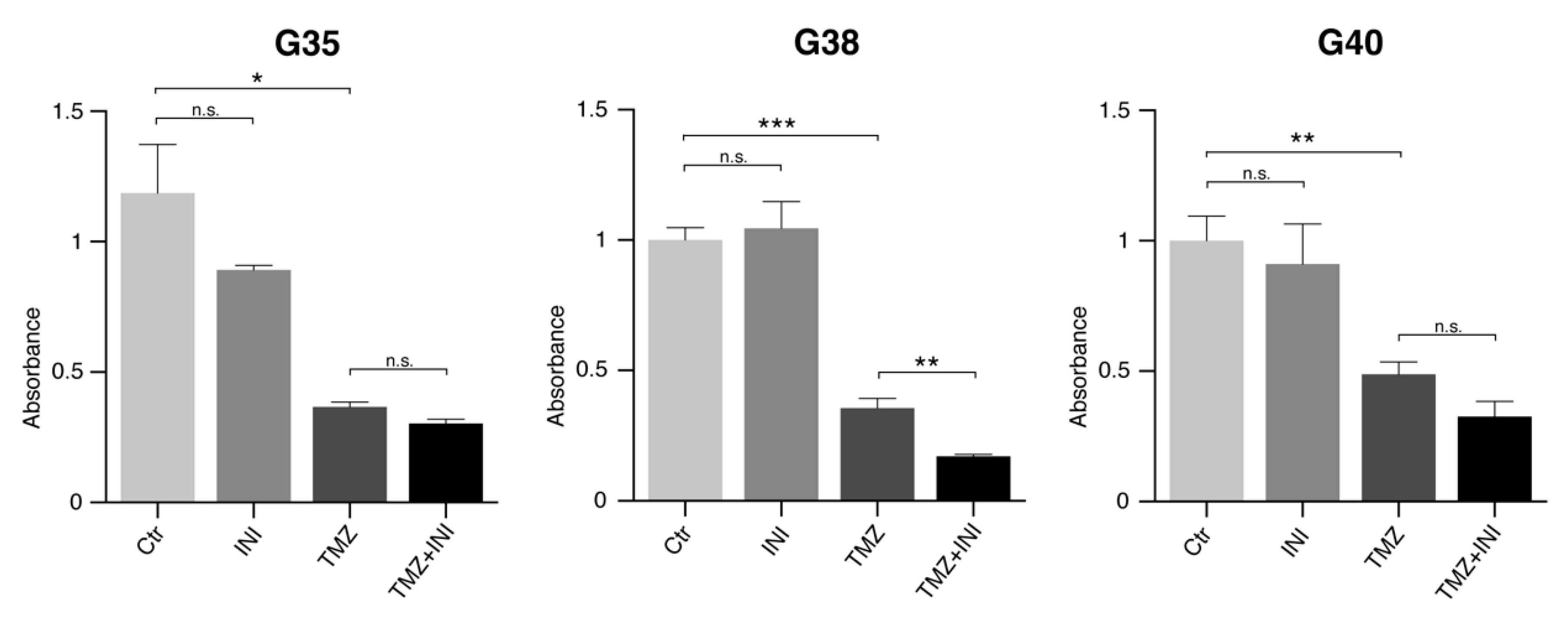
2.5. INI-0602 Sensitizes Glioblastoma Cells to Temozolomide-mediated Cell Death
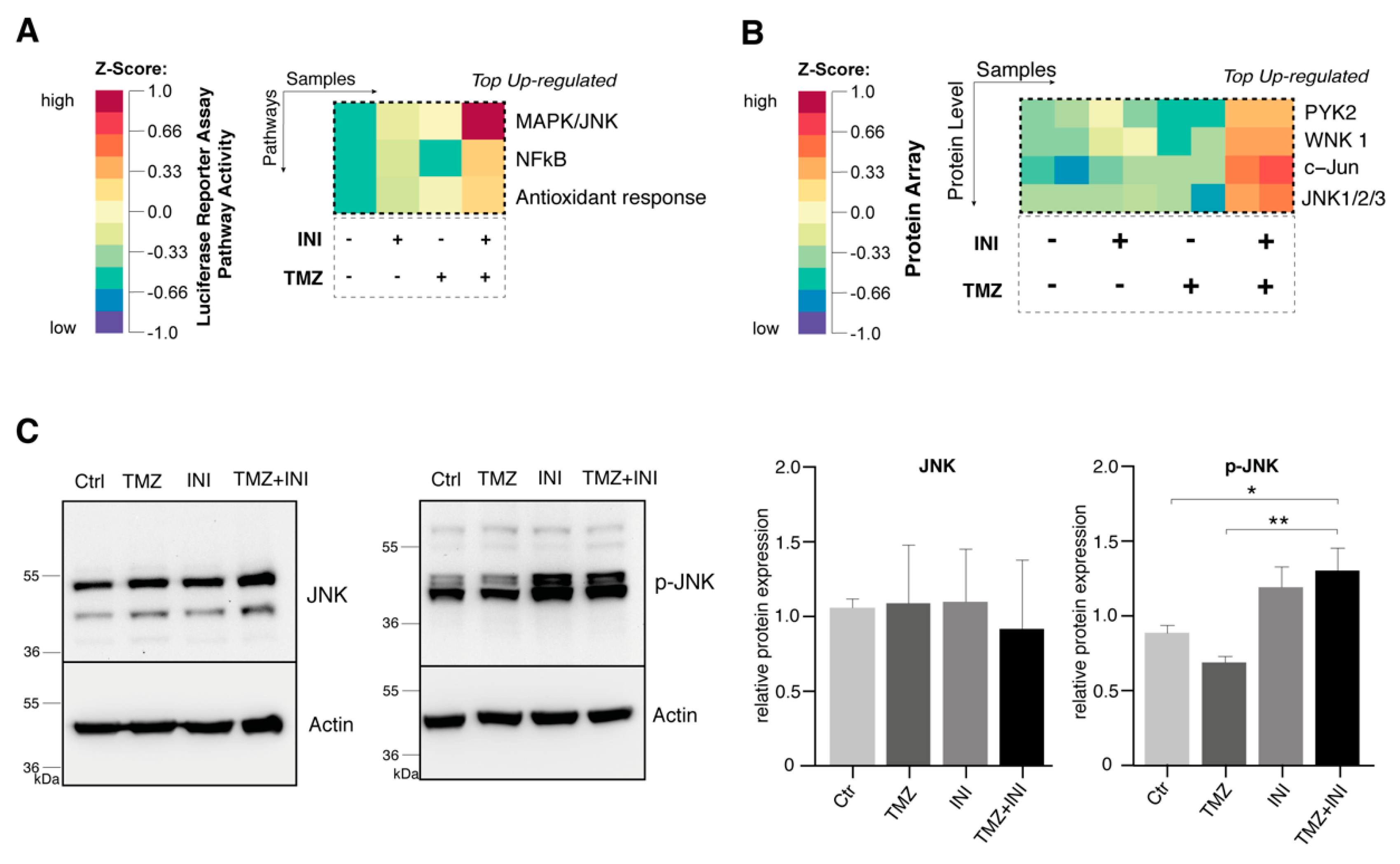
2.6. Additional Gap Junction Inhibition Leads to Elevated Activation of the JNK Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Lentiviral Transduction
4.2. Fluorescence Microscopy
4.3. IDH1 and IDH2 Mutation Analysis
4.4. MGMT Promoter Methylation Sequencing
4.5. Inhibitors and Drugs
4.6. Determination of Cellular Metabolic Activity
4.7. Cell Viability
4.8. Cell Confluence Measurement
4.9. Flow Cytometric Analysis of Cell Death
4.10. Cell Proliferation Measurement
4.11. Reporter Gene Assay
4.12. Proteome Array
4.13. Protein Immunoblotting
4.14. Statistical Analysis and Graphing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Preusser, M.; de Ribaupierre, S.; Wohrer, A.; Erridge, S.C.; Hegi, M.; Weller, M.; Stupp, R. Current concepts and management of glioblastoma. Ann. Neurol. 2011, 70, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Meinel, T.; Ewelt, C.; Martus, P.; Jakobs, O.; Felsberg, J.; Reifenberger, G. Prospective cohort study of radiotherapy with concomitant and adjuvant temozolomide chemotherapy for glioblastoma patients with no or minimal residual enhancing tumor load after surgery. J. Neuro-Oncol. 2012, 108, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 829–848. [Google Scholar] [CrossRef] [PubMed]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Giese, A.; Bjerkvig, R.; Berens, M.E.; Westphal, M. Cost of migration: Invasion of malignant gliomas and implications for treatment. J. Clin. Oncol. 2003, 21, 1624–1636. [Google Scholar] [CrossRef] [PubMed]
- Sinyuk, M.; Mulkearns-Hubert, E.E.; Reizes, O.; Lathia, J. Cancer Connectors: Connexins, Gap Junctions, and Communication. Front. Oncol. 2018, 8, 646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Batra, N.; Riquelme, M.A.; Jiang, J.X. Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 2012, 524, 2–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Katoh, F.; Kataoka, T.R.; Okada, M.; Tsubota, N.; Asada, H.; Yoshikawa, K.; Maeda, S.; Kitamura, Y.; Yamasaki, H.; et al. A role for heterologous gap junctions between melanoma and endothelial cells in metastasis. J. Clin. Investig. 2000, 105, 1189–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollmann, M.A.; Shao, Q.; Laird, D.W.; Sandig, M. Connexin 43 mediated gap junctional communication enhances breast tumor cell diapedesis in culture. Breast Cancer Res. 2005, 7, R522–R534. [Google Scholar] [CrossRef] [PubMed]
- Saito-Katsuragi, M.; Asada, H.; Niizeki, H.; Katoh, F.; Masuzawa, M.; Tsutsumi, M.; Kuniyasu, H.; Ito, A.; Nojima, H.; Miyagawa, S. Role for connexin 26 in metastasis of human malignant melanoma: Communication between melanoma and endothelial cells via connexin 26. Cancer 2007, 110, 1162–1172. [Google Scholar] [CrossRef]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hanggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro-Oncology 2017, 19, 1316–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, H.; Mizoguchi, H.; Doi, Y.; Jin, S.; Noda, M.; Liang, J.; Li, H.; Zhou, Y.; Mori, R.; Yasuoka, S.; et al. Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimer’s disease. PLoS ONE 2011, 6, e21108. [Google Scholar] [CrossRef]
- Nonnenmacher, L.; Westhoff, M.A.; Fulda, S.; Karpel-Massler, G.; Halatsch, M.E.; Engelke, J.; Simmet, T.; Corbacioglu, S.; Debatin, K.M. RIST: A potent new combination therapy for glioblastoma. Int. J. Cancer 2015, 136, E173–E187. [Google Scholar] [CrossRef]
- Strobele, S.; Schneider, M.; Schneele, L.; Siegelin, M.D.; Nonnenmacher, L.; Zhou, S.; Karpel-Massler, G.; Westhoff, M.A.; Halatsch, M.E.; Debatin, K.M. A Potential Role for the Inhibition of PI3K Signaling in Glioblastoma Therapy. PLoS ONE 2015, 10, e0131670. [Google Scholar] [CrossRef]
- Portnow, J.; Badie, B.; Chen, M.; Liu, A.; Blanchard, S.; Synold, T.W. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: Potential implications for the current approach to chemoradiation. Clin. Cancer Res. 2009, 15, 7092–7098. [Google Scholar] [CrossRef]
- Soroceanu, L.; Manning, T.J., Jr.; Sontheimer, H. Reduced expression of connexin-43 and functional gap junction coupling in human gliomas. Glia 2001, 33, 107–117. [Google Scholar] [CrossRef]
- Pu, P.; Xia, Z.; Yu, S.; Huang, Q. Altered expression of Cx43 in astrocytic tumors. Clin. Neurol. Neurosurg. 2004, 107, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Yulyana, Y.; Endaya, B.B.; Ng, W.H.; Guo, C.M.; Hui, K.M.; Lam, P.Y.; Ho, I.A. Carbenoxolone enhances TRAIL-induced apoptosis through the upregulation of death receptor 5 and inhibition of gap junction intercellular communication in human glioma. Stem Cells Dev. 2013, 22, 1870–1882. [Google Scholar] [CrossRef]
- Westhoff, M.A.; Zhou, S.; Bachem, M.G.; Debatin, K.M.; Fulda, S. Identification of a novel switch in the dominant forms of cell adhesion-mediated drug resistance in glioblastoma cells. Oncogene 2008, 27, 5169–5181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sin, W.C.; Crespin, S.; Mesnil, M. Opposing roles of connexin43 in glioma progression. Biochim. Biophys. Acta 2012, 1818, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.F.; Varghese, R.T.; Lamouille, S.; Guo, S.; Pridham, K.J.; Kanabur, P.; Osimani, A.M.; Sharma, S.; Jourdan, J.; Rodgers, C.M.; et al. Connexin 43 Inhibition Sensitizes Chemoresistant Glioblastoma Cells to Temozolomide. Cancer Res. 2016, 76, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Mettang, M.; Meyer-Pannwitt, V.; Karpel-Massler, G.; Zhou, S.; Carragher, N.O.; Fohr, K.J.; Baumann, B.; Nonnenmacher, L.; Enzenmuller, S.; Dahlhaus, M.; et al. Blocking distinct interactions between Glioblastoma cells and their tissue microenvironment: A novel multi-targeted therapeutic approach. Sci. Rep. 2018, 8, 5527. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, M.; Deleyrolle, L.P.; Mulkearns-Hubert, E.E.; Jarrar, A.; Li, M.; Sinyuk, M.; Otvos, B.; Brunet, S.; Flavahan, W.A.; Hubert, C.G.; et al. Differential connexin function enhances self-renewal in glioblastoma. Cell Rep. 2015, 11, 1031–1042. [Google Scholar] [CrossRef]
- Leshchenko, Y.; Likhodii, S.; Yue, W.; Burnham, W.M.; Velazquez, J.L.P. Carbenoxolone does not cross the blood brain barrier: An HPLC study. BMC Neurosci. 2006, 7, 3. [Google Scholar] [CrossRef]
- Umebayashi, D.; Natsume, A.; Takeuchi, H.; Hara, M.; Nishimura, Y.; Fukuyama, R.; Sumiyoshi, N.; Wakabayashi, T. Blockade of gap junction hemichannel protects secondary spinal cord injury from activated microglia-mediated glutamate exitoneurotoxicity. J. Neurotrauma 2014, 31, 1967–1974. [Google Scholar] [CrossRef]
- Sasaki, T.; Numano, R.; Yokota-Hashimoto, H.; Matsui, S.; Kimura, N.; Takeuchi, H.; Kitamura, T. A central-acting connexin inhibitor, INI-0602, prevents high-fat diet-induced feeding pattern disturbances and obesity in mice. Mol. Brain 2018, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- Loewenstein, W.R.; Kanno, Y. Intercellular communication and tissue growth. I. Cancerous growth. J. Cell Biol. 1967, 33, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Jamakosmanovic, A.; Loewenstein, W.R. Intercellular communication and tissue growth. 3. Thyroid cancer. J. Cell Biol. 1968, 38, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Matsui, Y. Cellular uncoupling in cancerous stomach epithelium. Nature 1968, 218, 775–776. [Google Scholar] [CrossRef] [PubMed]
- Stoletov, K.; Strnadel, J.; Zardouzian, E.; Momiyama, M.; Park, F.D.; Kelber, J.A.; Pizzo, D.P.; Hoffman, R.; VandenBerg, S.R.; Klemke, R.L. Role of connexins in metastatic breast cancer and melanoma brain colonization. J. Cell Sci. 2013, 126, 904–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.C.; Xiao, H.L.; Jiang, X.F.; Wang, Q.L.; Li, Y.; Yang, X.J.; Ping, Y.F.; Duan, J.J.; Jiang, J.Y.; Ye, X.Z.; et al. Connexin 43 reverses malignant phenotypes of glioma stem cells by modulating E-cadherin. Stem Cells 2012, 30, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Momiyama, M.; Omori, Y.; Ishizaki, Y.; Nishikawa, Y.; Tokairin, T.; Ogawa, J.; Enomoto, K. Connexin26-mediated gap junctional communication reverses the malignant phenotype of MCF-7 breast cancer cells. Cancer Sci. 2003, 94, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayan, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; Rodriguez-Cruz, V.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Temozolomide resistance in glioblastoma cells occurs partly through epidermal growth factor receptor-mediated induction of connexin 43. Cell Death Dis. 2014, 5, e1145. [Google Scholar] [CrossRef] [PubMed]
- Menachem, A.; Makovski, V.; Bodner, O.; Pasmanik-Chor, M.; Stein, R.; Shomron, N.; Kloog, Y. Intercellular transfer of small RNAs from astrocytes to lung tumor cells induces resistance to chemotherapy. Oncotarget 2016, 7, 12489–12504. [Google Scholar] [CrossRef] [Green Version]
- Zong, L.; Zhu, Y.; Liang, R.; Zhao, H.B. Gap junction mediated miRNA intercellular transfer and gene regulation: A novel mechanism for intercellular genetic communication. Sci. Rep. 2016, 6, 19884. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, M.A.; Zhou, S.; Nonnenmacher, L.; Karpel-Massler, G.; Jennewein, C.; Schneider, M.; Halatsch, M.E.; Carragher, N.O.; Baumann, B.; Krause, A.; et al. Inhibition of NF-kappaB signaling ablates the invasive phenotype of glioblastoma. Mol. Cancer Res. 2013, 11, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Ebelt, N.D.; Cantrell, M.A.; Van Den Berg, C.L. c-Jun N-Terminal Kinases Mediate a Wide Range of Targets in the Metastatic Cascade. Genes Cancer 2013, 4, 378–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cellurale, C.; Weston, C.R.; Reilly, J.; Garlick, D.S.; Jerry, D.J.; Sluss, H.K.; Davis, R.J. Role of JNK in a Trp53-dependent mouse model of breast cancer. PLoS ONE 2010, 5, e12469. [Google Scholar] [CrossRef] [PubMed]
- Cellurale, C.; Girnius, N.; Jiang, F.; Cavanagh-Kyros, J.; Lu, S.; Garlick, D.S.; Mercurio, A.M.; Davis, R.J. Role of JNK in mammary gland development and breast cancer. Cancer Res. 2012, 72, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Maeda, S.; Chang, L.; Karin, M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA 2006, 103, 10544–10551. [Google Scholar] [CrossRef]
- Das, M.; Garlick, D.S.; Greiner, D.L.; Davis, R.J. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011, 25, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lin, A. Role of JNK activation in apoptosis: A double-edged sword. Cell Res. 2005, 15, 36–42. [Google Scholar] [CrossRef]
- Schneider, M.; Strobele, S.; Nonnenmacher, L.; Siegelin, M.D.; Tepper, M.; Stroh, S.; Hasslacher, S.; Enzenmuller, S.; Strauss, G.; Baumann, B.; et al. A paired comparison between glioblastoma “stem cells” and differentiated cells. Int. J. Cancer 2016, 138, 1709–1718. [Google Scholar] [CrossRef]
- De Witt Hamer, P.C.; Van Tilborg, A.A.; Eijk, P.P.; Sminia, P.; Troost, D.; Van Noorden, C.J.; Ylstra, B.; Leenstra, S. The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene 2008, 27, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Bock, C.; El-Maarri, O.; Hubner, A.; Ehrentraut, D.; Schramm, J.; Felsberg, J.; Kahl, P.; Buttner, R.; Pietsch, T.; et al. Optimization of quantitative MGMT promoter methylation analysis using pyrosequencing and combined bisulfite restriction analysis. J. Mol. Diagn. 2007, 9, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potthoff, A.-L.; Heiland, D.H.; Evert, B.O.; Almeida, F.R.; Behringer, S.P.; Dolf, A.; Güresir, Á.; Güresir, E.; Joseph, K.; Pietsch, T.; et al. Inhibition of Gap Junctions Sensitizes Primary Glioblastoma Cells for Temozolomide. Cancers 2019, 11, 858. https://doi.org/10.3390/cancers11060858
Potthoff A-L, Heiland DH, Evert BO, Almeida FR, Behringer SP, Dolf A, Güresir Á, Güresir E, Joseph K, Pietsch T, et al. Inhibition of Gap Junctions Sensitizes Primary Glioblastoma Cells for Temozolomide. Cancers. 2019; 11(6):858. https://doi.org/10.3390/cancers11060858
Chicago/Turabian StylePotthoff, Anna-Laura, Dieter Henrik Heiland, Bernd O. Evert, Filipe Rodrigues Almeida, Simon P. Behringer, Andreas Dolf, Ági Güresir, Erdem Güresir, Kevin Joseph, Torsten Pietsch, and et al. 2019. "Inhibition of Gap Junctions Sensitizes Primary Glioblastoma Cells for Temozolomide" Cancers 11, no. 6: 858. https://doi.org/10.3390/cancers11060858