Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy


Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, NY 11724, USA
*
Authors to whom correspondence should be addressed.
Cancers 2018, 10(9), 298; https://doi.org/10.3390/cancers10090298
Submission received: 9 July 2018
/
Revised: 27 August 2018
/
Accepted: 28 August 2018
/
Published: 1 September 2018
Abstract
:A better understanding of mechanistic insights into genes and enzymes implicated in rare diseases provide a unique opportunity for orphan drug development. Advances made in identification of synthetic lethal relationships between rare disorder genes with oncogenes and tumor suppressor genes have brought in new anticancer therapeutic opportunities. Additionally, the rapid development of small molecule inhibitors against enzymes that participate in DNA damage response and repair has been a successful strategy for targeted cancer therapeutics. Here, we discuss the recent advances in our understanding of how many rare disease genes participate in promoting genome stability. We also summarize the latest developments in exploiting rare diseases to uncover new biological mechanisms and identify new synthetic lethal interactions for anticancer drug discovery that are in various stages of preclinical and clinical studies.
1. Introduction
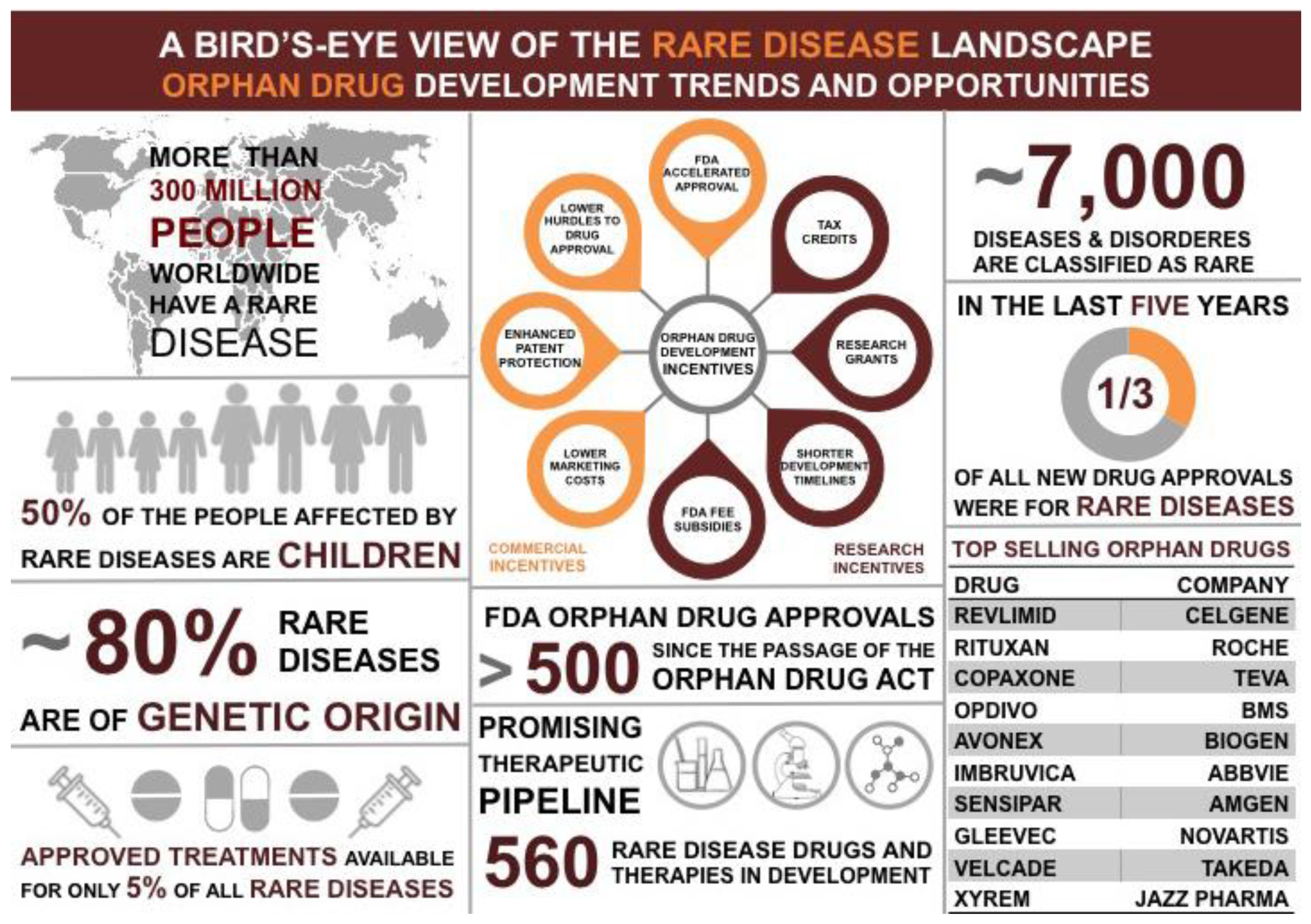
Rare diseases are defined as disorders that affect fewer than 200,000 people in the United States [1]. Although these disorders can often be chronic, debilitating, and life-limiting illnesses there are few effective therapies for these diseases (Figure 1) [1]. This is largely because pharmaceutical industries are unable to recover the cost associated with the R&D and marketing of such drugs [1]. The FDA Office of Orphan Products Development (OOPD) focuses on the development of drugs and products for rare diseases (Figure 1). Interestingly, many rare diseases are caused by the altered function of DNA damage response (DDR) genes causing genome instability—a hallmark of cancer. Since DDR genes are crucial for maintaining genome stability, mutations in these genes may have downstream cancer burden associated with them. Hence, understanding the biology of rare DNA repair diseases can help identify novel cancer therapies.
The DDR network includes several DNA repair pathways, which depending on the type of DNA lesion is employed by the cell to orchestrate repair [2,3]. There are several DNA lesions that can affect a cell: (A) single-stranded breaks, (B) base modifications (e.g., methylation, oxidation), (C) base mismatch (e.g., insertions, deletions, translocations), (D) incorporation of bulky adducts during replication (covalent attachment of aryl groups to the DNA), (E) incorporation of abasic sites (where a base is missing from the DNA), (F) interstrand and intrastrand covalent crosslinks, and (G) double-stranded breaks. Due to space limitations, in the following section we briefly discuss the repair of single-strand and double-strand DNA breaks.
1.1. Repair of Single-Strand DNA Breaks (SSBs)
SSBs, as the name suggests, afflicts one strand of the DNA double helix and is repaired by one of three pathways; base excision repair (BER), nucleotide excision repair (NER) and mismatch repair (MMR) [4]. BER, NER, and MMR are vital cellular pathways essential for the maintenance of genome integrity and cell survival (reviewed in [5,6,7,8]). Briefly, BER has evolved to repair endogenous DNA damage that can lead to modifications in the DNA bases such as alkylation, oxidation and deamination [5]. NER is distinguished from BER due to its versatility of sensing structural distortions in the double helix rather than specific base modifications [9,10]. “Bulky” lesions generated by UV radiation such as cyclobutane pyrimidine dimers (CPDs) and 6,4-photoproducts (6-4PPs) generate structural distortion in the DNA that obstruct progression of transcription and replication machineries that can lead to genome instability if left unrepaired [11]. MMR is involved in postreplication repair (PRR) and corrects DNA mismatches generated during DNA replication and recombination that escapes the proofreading function of DNA polymerases and can become permanent in dividing cells if left unrepaired. MMR thereby prevents both mutagenesis in the short term and oncogenesis in the long term [12,13].
1.2. Repair of Double-Strand DNA Breaks (DSBR)
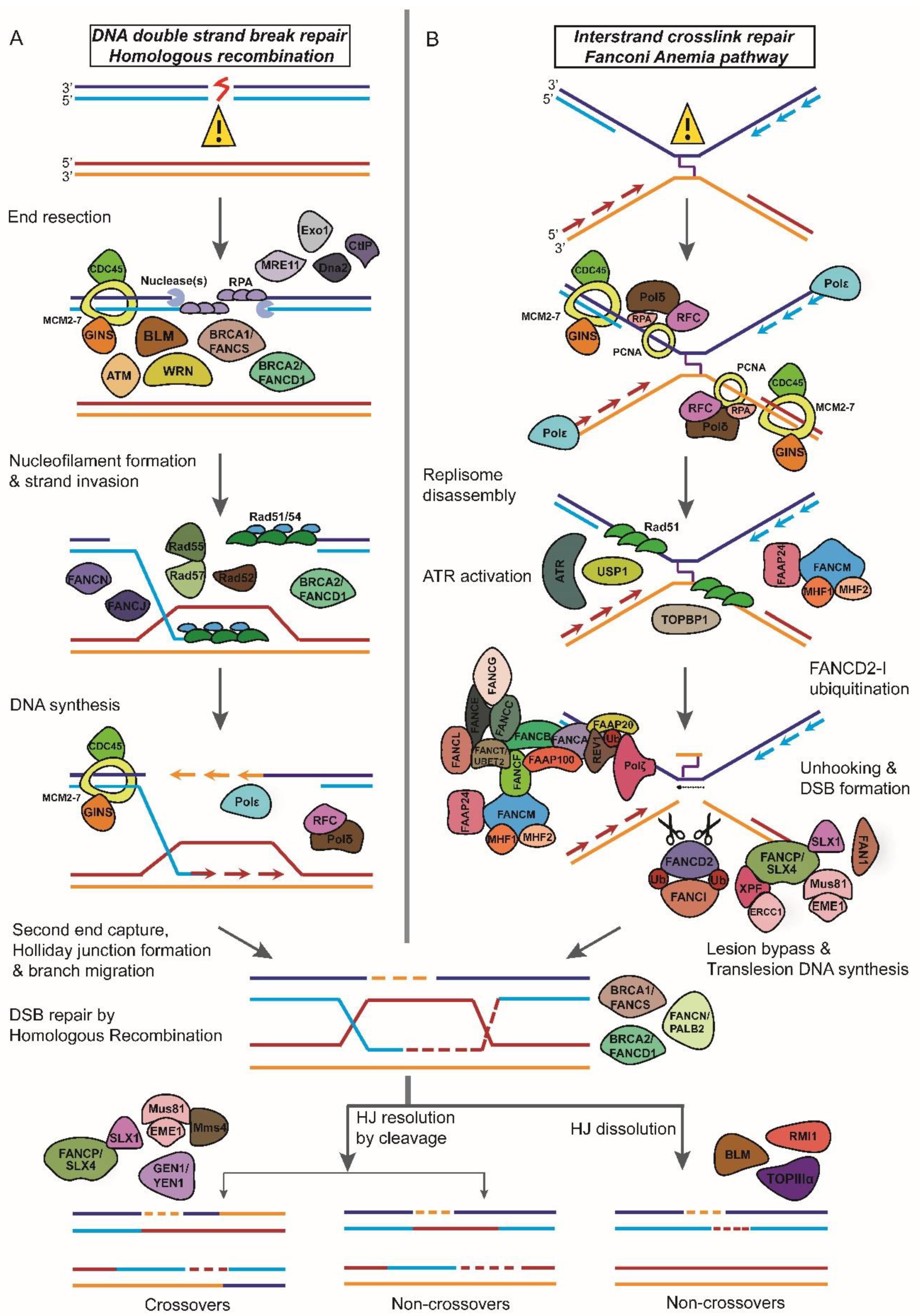
The two major DSBR pathways employed by the cell for the repair of broken DNA ends primarily differ in the use of a homologous DNA template for repair. Homologous recombination (HR) is an error-free, template-dependent pathway that uses DNA molecules of “identical or nearly identical nucleotide sequences” as template to copy the information across the double-strand break (DSB) (Figure 2A) [14]. Non-homologous end-joining (NHEJ), does not depend upon sequence homology to recover the lost genetic information at the site of the DNA break and is a mutagenic, error-prone mechanism of DNA repair [11]. A second NHEJ concomitant pathway often referred to as alternative-NHEJ (Alt-NHEJ)/Microhomology-mediated end-joining (MMEJ) differs from NHEJ most significantly in that it uses shorter homology overhangs (5–25 base pair (bp)) on the ends of the double-strand breaks during the alignment of the broken ends before religating them [15]. Though both these pathways are highly conserved throughout evolution, HR appears to be the predominant mechanism of DSBR during embryogenesis (and S/G2 phases of cell cycle), whereas NHEJ is a major pathway for repair in post-natal life and in the G0/G1 phase of the cell cycle [16].
In this review, we highlight how insights from understanding the biology of rare genetic diseases with defects in DNA damage response and repair can be translated to the clinic for cancer therapy. We discuss four diseases—Werner syndrome (WS), Bloom syndrome (BS), Ataxia telangiectasia (AT) and Fanconi Anemia (FA). We highlight the latest research in the development of small molecule inhibitors for these DNA damage repair enzymes that are being used in the clinic either as single agent or in combination with radio- and/or chemotherapy to exploit defects in DNA damage repair pathways in cancer cells.
2. Leveraging Insights from the Biology of Rare Genetic Diseases for Targeted Cancer Therapy
Geneticist Calvin Bridges first described synthetic lethality as a genetic interaction in which deficiency in a pair of genes results in cell death, while the disruption of either gene is viable [17]. The key impediment to exploiting synthetic lethality in cancer therapy is the identification of robust synthetic lethal genetic interactions [6]. What makes DNA repair genes attractive targets for cancer therapy using the synthetic lethality approach is; (1) many DNA repair genes have synthetic lethal relationships with oncogenes and tumor suppressor genes, (2) many repair genes and repair pathways are upregulated in tumors giving rise to a distinct genetic profile that can be exploited for targeted killing of cancer cells, (3) several cancer cells are dependent on DNA repair pathways for survival in response to genotoxin stress (e.g., radio/chemotherapy) [6,18,19,20,21,22,23,24]. In fact, in recent years, the success of poly(ADP ribose) polymerase inhibitors (PARPi): small molecules that inhibit the molecular and cellular function of PARP-1 and in doing so exploits the synthetic lethal relationship between PARP and BRCA1 or BRCA2 in BRCA1/2-null (ovarian or breast) tumors in response to platinum-based chemotherapy has made chemically induced synthetic lethality more mainstream in the development of new generation anti-cancer drugs [25,26].
PARP-1 is a nuclear enzyme that senses and binds to single-stranded DNA breaks and catalyzes the formation of PARP polymers by a process known as poly(ADP-ribosyl)ation (PARylation) [27]. PARP-1 uses the PAR chains to serve as a docking platform for the recruitment of DNA repair factors to the site of the break; in particular, PARP-1 recruits the BER core-complex proteins such as XRCC1, Polβ and DNA ligase III to orchestrate repair [27,28]. PARP-1 inhibition leads to an accumulation of SSBs, which if left unrepaired lead to collapse replication forks that are essentially DSBs and rely on HR for replication restart and repair [29]. BRCA1 and BRCA2 are tumor suppressor genes that are also key HR repair protein [29]. Due to their synthetic lethal relationship, in HR-defective BRCA1/2-mutant cells, that is, cancer cells, chemical inhibition of PARP-1 is selectively toxic in combination with conventional chemotherapy [29].
Applying the principle of chemically induced synthetic lethality to identify targets for cancer therapy has immense potential and relies on the identification of inhibitors that can selectively modulate DNA repair proteins that are either directly or indirectly involved in carcinogenesis. In the following section, we discuss four DNA repair genes/gene networks: Werner helicase, Bloom helicase, ATM kinase and Fanconi Anemia proteins and how pharmacologically inhibiting these gene products in combination with a cancer driver mutation is synthetically lethal and can be exploited for developing orphan drugs.
3. Werner Syndrome
WS is an adult-onset, autosomal-recessive, representative segmental progeroid syndrome [30]. Dr. Otto Werner at the Royal Albrecht University of Kiel first described WS in 1904 [31]. The clinical phenotypes of WS include an early onset of aging symptoms such as bilateral cataracts, abnormal glucose and lipid metabolism, hypogonadism, atherosclerosis, diabetes, skin ulcers, osteoporosis, and other age-related disorders [32,33]. Individuals with WS show no observable phenotype in their first decade. Generally, the first sign of WS is a lack of the pubertal growth spurt during teenage years [34]. WS is a rare genetic disorder with an estimated global incidence ranging between 1 in 1,000,000 and 1 in 10,000,000 births; however, demographically the incidence is higher in Japan at 1 in 100,000 births [35]. WS is caused by a mutation in the WRN gene that encodes a 180 kDa RecQ type DNA/RNA helicase that possesses both helicase and exonuclease activities (Figure 2A) [36]. WS is associated with abnormalities in many DNA metabolic pathways such as DNA repair, replication, and telomere maintenance [36].
3.1. Werner Helicase in Non-Homologous End-Joining and Base Excision Repair
WRN plays a regulatory role in pathway choice during DSBR by stimulating NHEJ and inhibiting MMEJ [37]. DNA-PKcs and its regulatory subunit, Ku70/80, initiates NHEJ repair in response to DNA damage that results in the formation of DSBs [38]. The Ku70/80 complex has been shown to physically interact with WRN and enhance its exonuclease activity without affecting its helicase activity [39]. The processing of the broken DNA ends by WRN generates substrates suitable for ligation by XRCC4 and ligaseIV [38].
WRN knockdown cells show sensitivity to DNA damaging agents (mitomycin C, camptothecin, and hydrogen peroxide) and reduced BER activity [40,41,42]. BER proceeds either via short-patch (SP-BER), in which one single nucleotide, or via long-patch (LP-BER), where greater than one nucleotide is incorporated during the repair process [43]. WRN has been shown to physically and functionally interact with proteins involved in both SP-BER and LP-BER. Notably, the strand displacement activity of DNA Polymerase β (Pol β), involved in both SP-BER and LP-BER is stimulated by WRN helicase activity [44,45,46]. WRN helicase activity cooperates with its (1) proofreading activity to assist Pol β during LP-BER and (2) exonuclease activity to process BER intermediates with 3′-mismatches [41,47]. WRN also interacts with other enzymes involved in SP-BER and LP-BER, including apurinic/apyrimidinic endonuclease 1 (APE1), flap endonuclease 1 (FEN-1) and poly(ADP-ribose) polymerase 1 (PARP-1), among others [48,49,50].
3.2. Small Molecule Inhibitors and Werner Helicase
Modulation of DNA repair proteins by small molecule inhibitors as targets for anticancer therapy has attracted great interest in recent years. The WRN helicase is one of these DNA repair factors. Two small molecule inhibitors from the National Cancer Institute (NCI) Diversity Set, NSC 19630 and NSC 617145 have been identified as pharmacological inhibitors of WRN helicase activity [51,52]. NSC 19630 [1-(propox-ymethyl)-maleimide] has been reported to inhibit growth of cell lines in various cancers with a notably strong representation in leukemia and renal cancer [52]. NSC 617145 [1,1′-(2,2-Dimethyl-1,3-propanediyl)bis[3,4-dichloro-1H-pyrrole-2,5-dione] is reported to be a candidate for chemically inducing synthetic lethality in Fanconi Anemia-deficient tumors harboring autosomal recessive mutations in either the FA group A (FANCA) or group D (FANCD2) genes [51]. FA-A and FA-D2 cells show hypersensitivity to low concentrations of MMC when co-treated with NSC 617145. This is because NSC 617145 acts synergistically with MMC and exacerbates MMC-induced DNA damage that accumulates in the absence of a functionally intact FA pathway. NSC 19630 and NSC 617145 are both attractive drugs for developing improved anticancer treatment regimens.
4. Bloom Syndrome
BS is an autosomal recessive chromosomal instability syndrome [53]. The clinical phenotypes of BS include prenatal and postnatal growth retardation, sunlight sensitivity, immune deficiency, fertility defects and a predisposition to the development of cancer, most commonly leukemia and lymphoma [54,55]. BS is a rare genetic disorder with fewer than 300 cases reported. However, epidemiologically, one-third of people with the disease are of Eastern European Jewish (Ashkenazi) decent and a disease prevalence of approximately 1:48,000 [53,56,57]. BS is due to mutations of the BLM gene (15q26.1), which encodes the BLM DNA helicase. BLM is a member of the conserved RecQ helicase family, an enzyme involved in maintenance of genomic integrity (Figure 2A) [58,59]. Due to its helicase activity, BLM, like WRN, functions to facilitate several metabolic processes that involve unwinding the DNA helix including DNA replication, RNA transcription, and DNA repair [60].
4.1. Bloom Helicase and Homologous Recombination
BLM protein displays a DNA-dependent ATPase activity and 3′–5′ DNA helicase activity that can unwind a variety of DNA substrates that arise during HR-mediated DNA repair. Accurate processing of these DNA intermediates is crucial for the maintenance of chromosomal stability and genomic integrity [61]. BLM, along with its conserved partner enzymes, including topoisomerase IIIα, RMI1 and 2 can also branch migrate recombination intermediates like DNA displacement loops (D-loop) and Holliday junctions (HJs) and channel these DNA molecules away from pathways leading to crossover products (Figure 2A) [62,63,64,65,66,67]. In the absence of BLM, HR is still functional albeit unregulated; this results in an increase in the number of sister chromatid exchanges (SCE) which arise by HR between sister chromatids during the S or G2 phases of the cell cycle that are characteristic of the syndrome [59,68,69].
4.2. Small Molecule Inhibitors and Bloom Helicase
The compound, designated ML216, identified from a high-throughput screen of a chemical compound library and medicinal chemistry optimization, is a potent small molecule inhibitor of BLM [54]. This inhibitor acts through competitive inhibition of the DNA binding activity of BLM [54]. In cell-based assays, using cultured human fibroblasts; ML216 induces SCEs, elevated sensitivity to aphidicolin (an inhibitor of replicative DNA polymerases) and reduced proliferation [54]. However, these phenotypes were not observed in otherwise isogenic, BLM-deficient cells [54]. These observations warrant further studies for the potential development of a new class of chemotherapy drugs to treat tumors that rely on BLM for proliferation.
Cu/Zn superoxide dismutase 1 (SOD1) is a non-essential gene that acts as a regulator of antioxidant defense [70]. Published reports suggest a synthetic lethal relationship between BLM and SOD1 in colorectal cancer (CRC) cell models [71]. Using siRNA-based silencing, chemical inhibition of SOD1 by ammonium tetrathiomolybdate (ATTM) and Lung Cancer Screen-1 (LCS-1), the authors report the induction of selective killing within BLM-deficient cells [71]. This data identifies SOD1 as a novel drug target in BLM CRC contexts.
5. Ataxia Telangiectasia
A-T is a rare, recessive genetic disorder that is primarily an immunodeficiency disease and affects a number of different organs in the body [72]. A-T is characterized by dilated blood vessels (telangiectasia) and a loss of voluntary movement coordination that includes a gait abnormality (ataxia) [72]. A-T is a rare genetic disorder with an estimated global incidence ranging between 1 out of 40,000 and 1 out of 100,000 births worldwide with no reported geographical or demographical epidemiology (data from NCI). A-T is due to mutations of the ATM gene, a member of the phosphatidyl inositol 3-kinase-like family of serine/threonine protein kinases (PIKKs) [73,74]. The ATM gene is located on chromosome 11q 22–23, includes 66 exons, and encodes a 370 kDa serine/threonine protein kinase [75]. ATM mediates checkpoint control by inducing cell cycle arrest at G1/S, S and G2/M phases in cells exposed to ionizing radiation (IR) and other agents that produce DSBs. In response to DNA damage, several proteins are phosphorylated at Ser/Thr-Glu motifs and additional sites in an ATM-dependent manner [76,77,78,79]. ATM also exerts cell cycle delay, in part, by phosphorylation and activation of downstream effector kinases, like Checkpoint kinase 2 (Chk2), which, in turn, phosphorylates p53 and several other targets [80,81].
5.1. ATM and Double-Strand Break Repair
In response to DSBs, Mre11-Rad50-Nbs1 (MRN) complex is one of the first factors recruited to the site (Figure 2A). Once recruited, MRN recruits ATM to the DNA lesion by binding to the C terminus of NBS1 and subsequently stimulates ATM kinase activity [82,83,84,85]. Following the activation of ATM by broken DNA ends, ATM rapidly phosphorylates S139 in the C-terminal tail of histone variant H2AX to form γH2AX [86]. The protein Mdc1 mediates the recruitment of ATM to the chromatin flanking the DSBs [87]. Through its C-terminal tandem BRCT domains, Mdc1 directly binds to γH2AX and through its FHA domain Mdc1 interacts with ATM [87,88,89,90]. In addition, Mdc1 recruits MRN to chromatin by an interaction with the FHA-BRCT region of NBS1, thereby stimulating the phosphorylation of ATM, which in turn promotes additional γH2AX formation along chromatin [91,92,93]. The phosphorylation of H2AX by ATM around DSBs triggers the chromatin-based signaling cascade involving phosphorylation, ubiquitylation, SUMOylation, and other post-translational modifications to promote recruitment of DNA repair proteins like BRCA1 and 53BP1 to the damage site [94,95,96,97,98,99,100,101,102,103,104,105,106,107].
ATM has roles both in NHEJ and HR. NHEJ requires several proteins such as ATM, the MRN complex, DNA-PKcs, and Artemis [108]. ATM is required for the phosphorylation and activation of DNA-PKcs [109]. ATM and DNA-PKcs are also essential for Artemis activation and recruitment to DNA ends [110]. ATM promotes HR by phosphorylating and activating CtIP, a downstream effector molecule important for DSB end resection (Figure 2A). Presence of CtIP at the DNA lesion facilitates the recruitment of replication protein A (RPA) to initiate HR [111,112,113]. Moreover, ATM-dependent phosphorylation of CtIP promotes the removal of Ku from single-ended DSBs and sets in motion Rad51-mediated strand invasion step of HR [114].
5.2. Small Molecule Inhibitors and ATM-CHK2 Helicases
Due to their central role in promoting DNA repair and facilitating the resistance of cancer cells to genotoxin treatment, the ATM-CHK2 kinases are excellent candidates for modulation by small molecules inhibitors for targeted cancer therapy. Table 1 summarizes a comprehensive list of small molecule inhibitors of ATM and CHK2 that are in preclinical or clinical development for cancer therapy being tested either as single agents or in combination with radio-and/or chemotherapy.
6. Fanconi Anemia
First described by the Swiss pediatrician Guido Fanconi, FA is a rare cancer-susceptibility syndrome caused by biallelic mutations in one of the 21 known complementation groups and is inherited as recessive autosomal or X-linked genetic disease [127,128,129,130,131]. The clinical phenotypes of FA include developmental disorders and physical abnormalities, progressive aplastic anemia, chromosomal fragility, congenital abnormalities and susceptibility to cancer: particularly myeloid leukemia and solid tumors including squamous cell carcinomas of the head and neck, liver tumors, and gynecologic malignancies [129,132,133,134,135]. The incidence rate of FA is estimated to be about 1 in 136,000 live births. This rare condition has a higher incidence among people of Ashkenazi Jewish descent, the Roma population of Spain, and black South Africans due to founder mutations [133]. FA can result from mutations in at least 21 genes. These genes encode for proteins, which along with several associated genes, form a nuclear multiprotein network that orchestrates DNA repair in a common pathway called the FA pathway (Figure 2B) [136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151].
6.1. Fanconi Anemia Proteins and DNA Interstrand Crosslink Repair
DNA interstrand crosslinks (ICLs) are toxic lesions that block replication and transcription by inhibiting the translocation of enzymes on the replication fork by covalently linking strands of the double helix leading to genome instability [136]. There are two major pathways for the repair of ICLs: (1) replication-independent ICL repair and (2) replication-dependent ICL repair [152]. The replication-dependent repair of ICLs is dependent on FA-mediated HR repair (Figure 2B) [152].
The FA proteins have been divided into three groups and they act in a sequential manner to promote ICL repair [136]. Group I (also known as the core complex) protein FANCM, a helicase and DNA translocase together with accessory factors Fanconi anemia-associated protein 24 (FAAP24), FAAP 100 and the histone fold proteins MHF1 (FAAP16 or CENPS) and MHF2 (FAAP10 or CENPX) recognizes the ICL and recruits other Group I proteins also known as the core complex consisting of FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FANCT, and FAAP20 to the ICL site (Figure 2B) [153]. Following recruitment to the ICL, two core complex components FANCL (the E3 ligase) and FANCT (ubiquitin E2 ligase) ubiquitinates downstream Group II proteins FANCD2–I (also known as the ID complex) (Figure 2B) [154,155]. The recruitment of the ID complex to the ICL is mediated by UHRF1 (ubiquitin-like with PHD and RING finger domains 1) protein, involved in ICL sensing (Figure 2B) [156]. Ubiquination of the FANCD2–I complex is essential for the recruitment of nucleases to the ICL for promoting nucleolytic incision flanking the crosslink, a phenomenon referred to as ‘unhooking’ of the ICL (Figure 2B) [157]. FANCD2-Ub first recruits the nuclease scaffolding protein SLX4 (FANCP) to the ICL, which then promotes the recruitment of other structure-specific endonucleases like XPF-ERCC1, MUS81-EME1, FAN1, and SLX1 to the site of the ICL (Figure 2B) [158,159,160,161,162]. Following the unhooking step, translesion DNA polymerases such as REV7 (FANCV), and polymerase η inserts a base opposite the unhooked lesion and polymerase ζ extends DNA synthesis from the misincorporated base to fill the ssDNA gaps resulting from ICL unhooking in order to complete replication (Figure 2B) [163,164]. The ssDNA gaps are converted to dsDNA breaks by MUS81–EME1, which is subsequently repaired by HR (Figure 2B) [165].
6.2. Small Molecule Inhibitors and Fanconi Anemia Proteins
Chemotherapeutic drugs like cisplatin and its derivatives, carboplatin and oxaliplatin act by inhibiting DNA replication by forming ICLs that impede the progression of the replication fork [166]. The crucial role played by FA proteins in the cellular resistance to ICLs makes them excellent targets for inhibition by small molecule inhibitors that may sensitize cancer cells to radio- and/or chemotherapy [166]. Table 2 summarizes a list of small molecule inhibitors of FANCD2 ubiquitination and foci formation; a marker of FA pathway abrogation that are in preclinical or clinical development for cancer therapy.
7. Conclusions
Identifying target genes/enzymes, and understanding the mechanism(s) underlying the progression of rare cancers provides us with the opportunity to: (A) exploit the disease-causing gene as a therapeutic target, and (B) expand the repertoire of targets for orphan drug discovery and development. In recent years, the success of small-molecule inhibitors for cancer therapy over conventional chemotherapy has directed scientific interest in identifying targetable enzymes that can be successfully modulated to improve the efficacy of current drugs. As presented in this review, enzymes implicated in rare genetic disease have been excellent targets for such modulation by small-molecule inhibitors and exploiting vulnerabilities in cancer. It is important to note that this approach also has certain limitations. First, there is a very high failure rate while designing new drugs particularly in late stages of clinical trials and second, many small-molecule inhibitors have high rates of cell toxicity making them unusable in therapy. However, with the advancements in next-generation genome sequencing technologies, identification of reliable biomarkers and large-scale gene profiling methods, the possibility of developing new personalized drugs for cancer patients is on the horizon.
Author Contributions
All authors were involved in the conception, design, and drafting of the manuscript. All authors read and approved the final manuscript.
Funding
This research received no external funding.
Acknowledgments
We apologize to those colleagues whose work has not been cited due to space limitation. We thank Saikat Saha for helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Alt-NHEJ | Alternative-NHEJ |
| ATTM | Ammonium tetrathiomolybdate |
| AT | Ataxia telangiectasia |
| BER | Base excision repair |
| BS | Bloom syndrome |
| CRC | Colorectal cancer |
| CPD | Cyclobutane pyrimidine dimers |
| D-loops | Displacement loops |
| DDR | DNA damage response |
| DSBs | Double-strand breaks |
| DSBR | Double-strand break repair |
| FA | Fanconi Anemia |
| OOPD | FDA Office of Orphan Products Development |
| HJ | Holliday junction |
| HR | Homologous recombination |
| ICLs | Interstrand crosslinks |
| LCS-1 | Lung Cancer Screen-1 |
| MMEJ | Microhomology-mediated end-joining |
| MMR | Mismatch repair |
| MRN | Mre11-Rad50-Nbs1 |
| NHEJ | Non-homologous end-joining |
| NER | Nucleotide excision repair |
| PARP | Poly(ADP ribose) polymerase |
| PRR | Post-replication repair |
| RPA | Replication protein A |
| SSBs | Single-strand breaks |
| SCE | Sister chromatid exchanges |
| SOD1 | Superoxide dismutase 1 |
| WS | Werner syndrome |
References
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Fishel, R. Mismatch repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef] [PubMed]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Scharer, O.D. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006, 106, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Castel, S.E.; Ren, J.; Bhattacharjee, S.; Chang, A.Y.; Sanchez, M.; Valbuena, A.; Antequera, F.; Martienssen, R.A. Dicer Promotes Transcription Termination at Sites of Replication Stress to Maintain Genome Stability. Cell 2014, 159, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Marsischky, G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999, 9, 89–96. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Meers, C.; Keskin, H.; Storici, F. DNA repair by RNA: Templated, or not templated, that is the question. DNA Repair (Amst.) 2016, 44, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decottignies, A. Alternative end-joining mechanisms: A historical perspective. Front. Genet. 2013, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell. Biol. 2003, 4, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.B. The origin of variation. Amer. Nat. 1922, 56, 51–63. [Google Scholar] [CrossRef]
- Biss, M.; Xiao, W. Selective tumor killing based on specific DNA-damage response deficiencies. Cancer Biol. Ther. 2012, 13, 239–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Furgason, J.M.; Bahassi El, M. Targeting DNA repair mechanisms in cancer. Pharmacol. Ther. 2013, 137, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Dietlein, F.; Thelen, L.; Reinhardt, H.C. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014, 30, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. Choices have consequences: The nexus between DNA repair pathways and genomic instability in cancer. Clin. Transl. Med. 2016, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.T.; Yu, X.C. Functions of PARylation in DNA Damage Repair Pathways. Genom. Proteom. Bioinform. 2016, 14, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W.; Aoufouchi, S.; Johnson, P.; Shall, S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly(ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick-sensor’ in vitro. Nucleic Acids Res. 1996, 24, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, M. Werner’s syndrome: From clinics to genetics. Clin. Exp. Rheumatol. 2000, 18, 760–766. [Google Scholar] [PubMed]
- O, W. On cataract in conjunction with scleroderma. Otto Werner, doctoral dissertation, 1904, Royal Ophthalmology Clinic, Royal Christian Albrecht University of Kiel. Adv. Exp. Med. Biol. 1985, 190, 1–14. [Google Scholar]
- Huang, S.R.; Li, B.M.; Gray, M.D.; Oshima, J.; Mian, S.I.; Campisi, J. The premature ageing syndrome protein, WRN, is a 3′→5′ exonuclease. Nat. Genet. 1998, 20, 114–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, C.J.; Martin, G.M.; Schultz, A.L.; Motulsky, A.G. Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore) 1966, 45, 177–221. [Google Scholar] [CrossRef] [PubMed]
- Muftuoglu, M.; Oshima, J.; von Kobbe, C.; Cheng, W.H.; Leistritz, D.F.; Bohr, V.A. The clinical characteristics of Werner syndrome: Molecular and biochemical diagnosis. Hum. Genet. 2008, 124, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Masala, M.V.; Scapaticci, S.; Olivieri, C.; Pirodda, C.; Montesu, M.A.; Cuccuru, M.A.; Pruneddu, S.; Danesino, C.; Cerimele, D. Epidemiology and clinical aspects of Werner’s syndrome in North Sardinia: Description of a cluster. Eur J. Dermatol. 2007, 17, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Oshima, J. Werner Syndrome. J. Biomed. Biotechnol. 2002, 2, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamanna, R.A.; Lu, H.M.; de Freitas, J.K.; Tian, J.; Croteau, D.L.; Bohr, V.A. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat. Commun. 2016, 7, 13785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Zhang, T.; Ren, Y.; Wang, Z.; Ling, C.C.; He, F.; Li, G.C.; Wang, C.; Wen, B. Inhibiting DNA-PKcs in a non-homologous end-joining pathway in response to DNA double-strand breaks. Oncotarget 2017, 8, 22662–22673. [Google Scholar] [CrossRef] [PubMed]
- Li, B.M.; Comai, L. Functional interaction between Ku and the Werner syndrome protein in DNA end processing. J. Biol. Chem. 2000, 275, 39800. [Google Scholar] [CrossRef] [PubMed]
- Von Kobbe, C.; May, A.; Grandori, C.; Bohr, V.A. Werner syndrome cells escape hydrogen peroxide-induced cell proliferation arrest. FASEB J. 2004, 18, 1970–1972. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Wilson, D.M., 3rd; Prasad, R.; Opresko, P.L.; Beck, G.; May, A.; Wilson, S.H.; Bohr, V.A. The Werner syndrome protein operates in base excision repair and cooperates with DNA polymerase beta. Nucleic Acids Res. 2006, 34, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.L.; Ghosh, A.K.; Bohr, V.A. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst.) 2010, 9, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattler, U.; Frit, P.; Salles, B.; Calsou, P. Long-patch DNA repair synthesis during base excision repair in mammalian cells. EMBO Rep. 2003, 4, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dianov, G.L.; Prasad, R.; Wilson, S.H.; Bohr, V.A. Role of DNA polymerase beta in the excision step of long patch mammalian base excision repair. J. Biol. Chem. 1999, 274, 13741–13743. [Google Scholar] [CrossRef] [PubMed]
- Sobol, R.W.; Horton, J.K.; Kuhn, R.; Gu, H.; Singhal, R.K.; Prasad, R.; Rajewsky, K.; Wilson, S.H. Requirement of mammalian DNA polymerase-beta in base-excision repair (vol 379, pg 183, 1996). Nature 1996, 383, 457. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Opresko, P.L.; von Kobbe, C.; Kedar, P.S.; Prasad, R.; Wilson, S.H.; Bohr, V.A. The Werner syndrome protein stimulates DNA polymerase beta strand displacement synthesis via its helicase activity. J. Biol. Chem. 2003, 278, 22686–22695. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Fan, J.; Momand, J.; Perrino, F.W.; Bohr, V.A.; Wilson, D.M. WRN exonuclease activity is blocked by DNA termini harboring 3′ obstructive groups. Mech. Ageing Dev. 2007, 128, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosh, R.M.; Driscoll, H.C.; Dianov, G.L.; Sommers, J.A. Biochemical characterization of the WRN-FEN-1 functional interaction. Biochemistry 2002, 41, 12204–12216. [Google Scholar] [CrossRef] [PubMed]
- von Kobbe, C.; Harrigan, J.A.; May, A.; Opresko, P.L.; Dawut, L.; Cheng, W.H.; Bohr, V.A. Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol. Cell. Biol. 2003, 23, 8601–8613. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.; Harrigan, J.A.; Indig, F.E.; Wilson, D.M.; Bohr, V.A. Regulation of WRN helicase activity in human base excision repair. J. Biol. Chem. 2004, 279, 53465–53474. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Banerjee, T.; Sommers, J.A.; Brosh, R.M., Jr. Targeting an Achilles’ heel of cancer with a WRN helicase inhibitor. Cell Cycle 2013, 12, 3329–3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M., Jr. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- German, J.; Sanz, M.M.; Ciocci, S.; Ye, T.Z.; Ellis, N.A. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum. Mutat. 2007, 28, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.H.; Dexheimer, T.S.; Rosenthal, A.S.; Chu, W.K.; Singh, D.K.; Mosedale, G.; Bachrati, C.Z.; Schultz, L.; Sakurai, M.; Savitsky, P.; et al. A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem. Biol. 2013, 20, 55–62. [Google Scholar] [CrossRef] [PubMed]
- German, J. Bloom’s syndrome. XX. The first 100 cancers. Cancer Genet. Cytogenet. 1997, 93, 100–106. [Google Scholar] [CrossRef]
- Roa, B.B.; Savino, C.V.; Richards, C.S. Ashkenazi Jewish population frequency of the Bloom syndrome gene 2281 delta 6ins7 mutation. Genet. Test. 1999, 3, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Eng, C.; Desnick, R.J.; German, J.; Ellis, N.A. Carrier frequency of the Bloom syndrome blmAsh mutation in the Ashkenazi Jewish population. Mol. Genet. Metab. 1998, 64, 286–290. [Google Scholar] [CrossRef] [PubMed]
- German, J.; Roe, A.M.; Leppert, M.F.; Ellis, N.A. Bloom syndrome: An analysis of consanguineous families assigns the locus mutated to chromosome band 15q26.1. Proc. Natl. Acad. Sci. USA 1994, 91, 6669–6673. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.K.; Hickson, I.D. RecQ helicases: Multifunctional genome caretakers. Nat. Rev. Cancer 2009, 9, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol. 2017, 8, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Mohaghegh, P.; Karow, J.K.; Brosh, R.M., Jr.; Bohr, V.A.; Hickson, I.D. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001, 29, 2843–2849. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M., Jr.; Li, J.L.; Kenny, M.K.; Karow, J.K.; Cooper, M.P.; Kureekattil, R.P.; Hickson, I.D.; Bohr, V.A. Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J. Biol. Chem. 2000, 275, 23500–23508. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Bellaoui, M.; Zhang, C.; Desai, R.; Morozov, P.; Delgado-Cruzata, L.; Rothstein, R.; Freyer, G.A.; Boone, C.; Brown, G.W. RMI1/NCE4, a suppressor of genome instability, encodes a member of the RecQ helicase/Topo III complex. EMBO J. 2005, 24, 2024–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meetei, A.R.; Sechi, S.; Wallisch, M.; Yang, D.; Young, M.K.; Joenje, H.; Hoatlin, M.E.; Wang, W. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol. Cell. Biol. 2003, 23, 3417–3426. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Davies, S.L.; Levitt, N.C.; Hickson, I.D. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem. 2001, 276, 19375–19381. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chan, K.L.; Ralf, C.; Bernstein, D.A.; Garcia, P.L.; Bohr, V.A.; Vindigni, A.; Janscak, P.; Keck, J.L.; Hickson, I.D. The HRDC domain of BLM is required for the dissolution of double Holliday junctions. EMBO J. 2005, 24, 2679–2687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Smith, K.; Waldman, B.C.; Waldman, A.S. Depletion of the bloom syndrome helicase stimulates homology-dependent repair at double-strand breaks in human chromosomes. DNA Repair (Amst.) 2011, 10, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Salah, G.; Hadj Salem, I.; Masmoudi, A.; Kallabi, F.; Turki, H.; Fakhfakh, F.; Ayadi, H.; Kamoun, H. A novel frameshift mutation in BLM gene associated with high sister chromatid exchanges (SCE) in heterozygous family members. Mol. Biol. Rep. 2014, 41, 7373–7380. [Google Scholar] [CrossRef] [PubMed]
- Chaganti, R.S.; Schonberg, S.; German, J. A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc. Natl. Acad. Sci. USA 1974, 71, 4508–4512. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A.R.; Culotta, V.C. SOD1 integrates signals from oxygen and glucose to repress respiration. Cell 2013, 152, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Sajesh, B.V.; McManus, K.J. Targeting SOD1 induces synthetic lethal killing in BLM- and CHEK2-deficient colorectal cancer cells. Oncotarget 2015, 6, 27907–27922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.M.R.; Lam, Z.; Last, J.I.; Byrd, P.J. Ataxia telangiectasia: More variation at clinical and cellular levels. Clin. Genet. 2015, 87, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Lempiainen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovejoy, C.A.; Cortez, D. Common mechanisms of PIKK regulation. DNA Repair (Amst.) 2009, 8, 1004–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednarski, J.J.; Sleckman, B.P. Integrated signaling in developing lymphocytes The role of DNA damage responses. Cell Cycle 2012, 11, 4129–4134. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.B.; Albuquerque, C.P.; Chen, S.H.; Zhou, H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 10364–10369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell. 2012, 46, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal. 2010, 3, rs3. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007, 11, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Andegeko, Y.; Moyal, L.; Mittelman, L.; Tsarfaty, I.; Shiloh, Y.; Rotman, G. Nuclear retention of ATM at sites of DNA double strand breaks. J. Biol. Chem. 2001, 276, 38224–38230. [Google Scholar] [PubMed]
- Bekker-Jensen, S.; Lukas, C.; Kitagawa, R.; Melander, F.; Kastan, M.B.; Bartek, J.; Lukas, J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 2006, 173, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; Le Beau, M.; Yates, J.R.; Hays, L.; Morgan, W.F.; Petrini, J.H.J. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell 1998, 93, 477–486. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Wang, B.; Bignell, C.R.; Taylor, A.M.; Elledge, S.J. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003, 421, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Edwards, R.A.; Thede, G.L.; Glover, J.N. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J. Biol. Chem. 2005, 280, 32053–32056. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Minter-Dykhouse, K.; Franco, S.; Gostissa, M.; Rivera, M.A.; Celeste, A.; Manis, J.P.; van Deursen, J.; Nussenzweig, A.; Paull, T.T.; et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell. 2006, 21, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Jackson, S.P. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008, 9, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melander, F.; Bekker-Jensen, S.; Falck, J.; Bartek, J.; Mailand, N.; Lukas, J. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J. Cell Biol. 2008, 181, 213–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spycher, C.; Miller, E.S.; Townsend, K.; Pavic, L.; Morrice, N.A.; Janscak, P.; Stewart, G.S.; Stucki, M. Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J. Cell Biol. 2008, 181, 227–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, R.; Xie, A. Double strand break repair functions of histone H2AX. Mutat. Res. 2013, 750, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huen, M.S.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Huang, J.; Chen, J.J. CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nat. Struct. Mol. Biol. 2007, 14, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chen, J.; Yu, X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 2007, 316, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Elledge, S.J. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 20759–20763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Sobhian, B.; Shao, G.; Lilli, D.R.; Culhane, A.C.; Moreau, L.A.; Xia, B.; Livingston, D.M.; Greenberg, R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007, 316, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Polo, S.; Miller, K.M.; Jackson, S.P. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 2009, 462, 935–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, G.S. Solving the RIDDLE of 53BP1 recruitment to sites of damage. Cell Cycle 2009, 8, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, S.; Rendtlew Danielsen, J.; Fugger, K.; Gromova, I.; Nerstedt, A.; Lukas, C.; Bartek, J.; Lukas, J.; Mailand, N. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell. Biol. 2010, 12, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Meerang, M.; Ritz, D.; Paliwal, S.; Garajova, Z.; Bosshard, M.; Mailand, N.; Janscak, P.; Hubscher, U.; Meyer, H.; Ramadan, K. The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat. Cell Biol. 2011, 13, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.H.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Riballo, E.; Kuhne, M.; Rief, N.; Doherty, A.; Smith, G.C.; Recio, M.J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A.; et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell. 2004, 16, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.P.; Uematsu, N.; Kobayashi, J.; Lerenthal, Y.; Krempler, A.; Yajima, H.; Lobrich, M.; Shiloh, Y.; Chen, D.J. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J. Biol. Chem. 2007, 282, 6582–6587. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Yu, Y.; Riballo, E.; Douglas, P.; Walker, S.A.; Ye, R.; Harer, C.; Marchetti, C.; Morrice, N.; Jeggo, P.A.; et al. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J. 2006, 25, 3880–3889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.L.; Shi, L.Z.; Wong, C.C.L.; Han, X.M.; Hwang, P.Y.H.; Truong, L.N.; Zhu, Q.Y.; Shao, Z.P.; Chen, D.J.; Berns, M.W.; et al. The Interaction of CtIP and Nbs1 Connects CDK and ATM to Regulate HR-Mediated Double-Strand Break Repair. PLoS Genet. 2013, 9, e1003277. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Lobrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, U509-U506. [Google Scholar] [CrossRef] [PubMed]
- Chanut, P.; Britton, S.; Coates, J.; Jackson, S.P.; Calsou, P. Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat. Commun. 2016, 7, 12889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zuo, B.; Wang, H.; Ren, L.; Yang, P.; Zeng, M.; Duan, D.; Liu, C.; Li, M. CGK733 enhances multinucleated cell formation and cytotoxicity induced by taxol in Chk1-deficient HBV-positive hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2012, 422, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Rainey, M.D.; Charlton, M.E.; Stanton, R.V.; Kastan, M.B. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008, 68, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Alao, J.P.; Sunnerhagen, P. The ATM and ATR inhibitors CGK733 and caffeine suppress cyclin D1 levels and inhibit cell proliferation. Radiat. Oncol. 2009, 4, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batey, M.A.; Zhao, Y.; Kyle, S.; Richardson, C.; Slade, A.; Martin, N.M.; Lau, A.; Newell, D.R.; Curtin, N.J. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol. Cancer Ther. 2013, 12, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Cmielova, J.; Havelek, R.; Kohlerova, R.; Soukup, T.; Bruckova, L.; Suchanek, J.; Vavrova, J.; Mokry, J.; Rezacova, M. The effect of ATM kinase inhibition on the initial response of human dental pulp and periodontal ligament mesenchymal stem cells to ionizing radiation. Int. J. Radiat. Biol. 2013, 89, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Jo, Y.H.; Cho, C.H.; Choe, W.; Kang, I.; Baik, H.H.; Yoon, K.S. ATM-deficient human fibroblast cells are resistant to low levels of DNA double-strand break induced apoptosis and subsequently undergo drug-induced premature senescence. Biochem. Biophys. Res. Commun. 2013, 430, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Mao, C.; Wu, J.; Li, S.; Ma, R.; Cao, H.; Ji, M.; Jing, C.; Tang, J. Improved ataxia telangiectasia mutated kinase inhibitor KU60019 provides a promising treatment strategy for non-invasive breast cancer. Oncol. Lett. 2014, 8, 2043–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobson, A.G.; Lountos, G.T.; Lorenzi, P.L.; Llamas, J.; Connelly, J.; Cerna, D.; Tropea, J.E.; Onda, A.; Zoppoli, G.; Kondapaka, S.; et al. Cellular inhibition of checkpoint kinase 2 (Chk2) and potentiation of camptothecins and radiation by the novel Chk2 inhibitor PV1019 [7-nitro-1H-indole-2-carboxylic acid {4-[1-(guanidinohydrazone)-ethyl]-phenyl}-amide]. J. Pharmacol. Exp. Ther. 2009, 331, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.E.; Walton, M.I.; Eve, P.D.; Boxall, K.J.; Antoni, L.; Caldwell, J.J.; Aherne, W.; Pearl, L.H.; Oliver, A.W.; Collins, I.; et al. CCT241533 is a potent and selective inhibitor of CHK2 that potentiates the cytotoxicity of PARP inhibitors. Cancer Res. 2011, 71, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Riesterer, O.; Matsumoto, F.; Wang, L.; Pickett, J.; Molkentine, D.; Giri, U.; Milas, L.; Raju, U. A novel Chk inhibitor, XL-844, increases human cancer cell radiosensitivity through promotion of mitotic catastrophe. Investig. New Drugs 2011, 29, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.J.; Yakes, F.M.; Chen, J.; Tadano, M.; Bornheim, L.; Clary, D.O.; Tai, A.; Wagner, J.M.; Miller, N.; Kim, Y.D.; et al. Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle 2007, 6, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Fanconi, G. Familial constitutional panmyelocytopathy, Fanconi’s anemia (F.A.). I. Clinical aspects. Semin. Hematol. 1967, 4, 233–240. [Google Scholar] [PubMed]
- Lobitz, S.; Velleuer, E. Guido Fanconi (1892–1979): A jack of all trades. Nat. Rev. Cancer 2006, 6, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Mathew, C.G. Fanconi anaemia genes and susceptibility to cancer. Oncogene 2006, 25, 5875–5884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meetei, A.R.; Levitus, M.; Xue, Y.T.; Medhurst, A.L.; Zwaan, M.; Ling, C.; Rooimans, M.A.; Bier, P.; Hoatlin, M.; Pals, G.; et al. X-linked inheritance of Fanconi anemia complementation group B. Nat. Genet. 2004, 36, 1219–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantinou, A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma 2012, 121, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P. Fanconi anemia and the development of leukemia. Best Pract. Res. Clin. Haematol. 2014, 27, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, P.S.; Tamary, H.; Alter, B.P. How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel. Am. J. Med. Genet. 2011, 155A, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Daly, M.; Arends, M.J.; Patel, K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012, 489, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.S.; Pondarre, C.; et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012, 11, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway. Cell Commun. Signal. 2017, 15. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, S.; Wu, Z. Fanconi anemia pathway defects in inherited and sporadic cancers. Transl. Pediatr. 2014, 3, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell. Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.S.; Castella, M.; Abeyta, A.; Gafken, P.R.; Tucker, N.; Taniguchi, T. Ubiquitination-Linked Phosphorylation of the FANCI S/TQ Cluster Contributes to Activation of the Fanconi Anemia I/D2 Complex. Cell Rep. 2017, 19, 2432–2440. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Pradhan, A.; Singh, T.R.; Du, C.H.; Li, J.; Wahengbam, K.; Grassman, E.; Auerbach, A.D.; Pang, Q.S.; Meetei, A.R. FAAP20: A novel ubiquitin-binding FA nuclear core-complex protein required for functional integrity of the FA-BRCA DNA repair pathway. Blood 2012, 119, 3285–3294. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Osman, F.; Feeney, L.; Lorenz, A.; Bryer, C.; Whitby, M.C. MHF1-2/CENP-S-X performs distinct roles in centromere metabolism and genetic recombination (Retracted article. See vol. 8, art no. 180010, 2018). Open Biol. 2013, 3, 130102. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Ling, C.; Coulthard, R.; Yan, Z.J.; Xue, Y.T.; Meetei, A.R.; Laghmani, E.H.; Joenje, H.; McDonald, N.; de Winter, J.P.; et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol. Cell 2007, 25, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.B.; Nebert, D.W.; Bruford, E.A.; Thompson, D.C.; Joenje, H.; Vasiliou, V. Update of the human and mouse Fanconi anemia genes. Hum. Genomics 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yang, K.L.; Dejsuphong, D.; D’Andrea, A.D. Regulation of Rev1 by the Fanconi anemia core complex. Nat. Struct. Mol. Biol. 2012, 19, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Ishiai, M.; Ali, A.M.; Medhurst, A.L.; Neveling, K.; Kalb, R.; Yan, Z.J.; Xue, Y.T.; Oostra, A.B.; Auerbach, A.D.; et al. FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO J. 2007, 26, 2104–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandi, S.; Whitby, M.C. The ATPase activity of Fml1 is essential for its roles in homologous recombination and DNA repair. Nucleic Acids Res. 2012, 40, 9584–9595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Nandi, S.; Osman, F.; Ahn, J.S.; Jakovleska, J.; Lorenz, A.; Whitby, M.C. The FANCM Ortholog Fml1 Promotes Recombination at Stalled Replication Forks and Limits Crossing Over during DNA Double-Strand Break Repair. Mol. Cell 2008, 32, 118–128. [Google Scholar] [CrossRef] [PubMed]
- de Oca, R.M.; Andreassen, P.R.; Margossian, S.P.; Gregory, R.C.; Taniguchi, T.; Wang, X.Z.; Houghtaling, S.; Grompe, M.; D’Andrea, A.D. Regulated interaction of the Fanconi anemia protein, FANCD2, with chromatin. Blood 2005, 105, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Dorsman, J.C.; Levitus, M.; Rockx, D.; Rooimans, M.A.; Oostra, A.B.; Haitjema, A.; Bakker, S.T.; Steltenpool, J.; Schuler, D.; Mohan, S.; et al. Identification of the Fanconi anemia complementation group I gene, FANCI. Cell. Oncol. 2007, 29, 211–218. [Google Scholar] [PubMed]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Mamrak, N.E.; Shimamura, A.; Howlett, N.G. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. 2017, 31, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.J.; Huang, T.T. The Fanconi anemia pathway in replication stress and DNA crosslink repair. Cell. Mol. Life Sci. 2012, 69, 3963–3974. [Google Scholar] [CrossRef] [PubMed]
- Palovcak, A.; Liu, W.; Yuan, F.; Zhang, Y. Maintenance of genome stability by Fanconi anemia proteins. Cell Biosci. 2017, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Hira, A.; Yoshida, K.; Sato, K.; Okuno, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Miyano, S.; Shimamoto, A.; Tahara, H.; et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am. J. Hum. Genet. 2015, 96, 1001–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, J.A.; Frost, M.G.; Carroll, E.; Rowe, M.L.; Howard, M.J.; Sidhu, A.; Chaugule, V.K.; Alpi, A.F.; Walden, H. The Fanconi Anemia DNA Repair Pathway Is Regulated by an Interaction between Ubiquitin and the E2-like Fold Domain of FANCL. J. Biol. Chem. 2015, 290, 20995–21006. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Zhan, B.; Yoshikawa, Y.; Haas, W.; Gygi, S.P.; Cohn, M.A. UHRF1 is a sensor for DNA interstrand crosslinks and recruits FANCD2 to initiate the Fanconi anemia pathway. Cell Rep. 2015, 10, 1947–1956. [Google Scholar] [CrossRef] [PubMed]
- Knipscheer, P.; Raschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.V.; Scharer, O.D.; Elledge, S.J.; Walter, J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009, 326, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.N.; Kobayashi, S.; Tsuda, M.; Kurumizaka, H.; Takata, M.; Kono, K.; Jiricny, J.; Takeda, S.; Hirota, K. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 6492–6496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Spitz, G.S.; Veturi, U.; Lach, F.P.; Auerbach, A.D.; Smogorzewska, A. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood 2013, 121, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Klein Douwel, D.; Boonen, R.A.; Long, D.T.; Szypowska, A.A.; Raschle, M.; Walter, J.C.; Knipscheer, P. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell. 2014, 54, 460–471. [Google Scholar] [CrossRef] [PubMed]
- McMahon, L.W.; Sangerman, J.; Goodman, S.R.; Kumaresan, K.; Lambert, M.W. Human α Spectrin II and the FANCA, FANCC, and FANCG Proteins Bind to DNA Containing Psoralen Interstrand Cross-Links†. Biochemistry 2001, 40, 7025–7034. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.W. Nuclear alpha spectrin: Critical roles in DNA interstrand cross-link repair and genomic stability. Exp. Biol. Med. (Maywood) 2016, 241, 1621–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef] [PubMed]
- Murakumo, Y. The property of DNA polymerase ζ: REV7 is a putative protein involved in translesion DNA synthesis and cell cycle control. Mutat. Res. 2002, 510, 37–44. [Google Scholar] [CrossRef]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Clauson, C.; Scharer, O.D.; Niedernhofer, L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012732. [Google Scholar] [CrossRef] [PubMed]
- Chirnomas, D.; Taniguchi, T.; de la Vega, M.; Vaidya, A.P.; Vasserman, M.; Hartman, A.R.; Kennedy, R.; Foster, R.; Mahoney, J.; Seiden, M.V.; et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol. Cancer Ther. 2006, 5, 952–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkitt, K.; Ljungman, M. Phenylbutyrate interferes with the Fanconi anemia and BRCA pathway and sensitizes head and neck cancer cells to cisplatin. Mol. Cancer 2008, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Dexheimer, T.S.; Ai, Y.; Liang, Q.; Villamil, M.A.; Inglese, J.; Maloney, D.J.; Jadhav, A.; Simeonov, A.; Zhuang, Z. Selective and cell-active inhibitors of the USP1/UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem. Biol. 2011, 18, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Dexheimer, T.S.; Zhang, P.; Rosenthal, A.S.; Villamil, M.A.; You, C.; Zhang, Q.; Chen, J.; Ott, C.A.; Sun, H.; et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat. Chem. Biol. 2014, 10, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Hsieh, G.; Buhrlage, S.J.; Huang, M.; Park, E.; Cuny, G.D.; Galinsky, I.; Stone, R.M.; Gray, N.S.; D’Andrea, A.D.; et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol. Cancer Ther. 2013, 12, 2651–2662. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004, 18, 1958–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Opportunities and Challenges in developing orphan drugs to prevent, diagnose and treat rare diseases.
Figure 1.
Opportunities and Challenges in developing orphan drugs to prevent, diagnose and treat rare diseases.

Figure 2.
A schematic for DNA damage response and repair Werner, Bloom, ATM, and Fanconi Anemia proteins are members of the DNA damage response network. (A) Werner and Bloom helicases and ATM serine/threonine kinase are involved in DNA end processing after the double-strand break is formed. (B) Fanconi Anemia proteins are involved in DNA interstrand crosslink sensing and repair by homologous recombination.
Figure 2.
A schematic for DNA damage response and repair Werner, Bloom, ATM, and Fanconi Anemia proteins are members of the DNA damage response network. (A) Werner and Bloom helicases and ATM serine/threonine kinase are involved in DNA end processing after the double-strand break is formed. (B) Fanconi Anemia proteins are involved in DNA interstrand crosslink sensing and repair by homologous recombination.


{kind=link}
{kind=link}
Table 1.
Small molecule inhibitors of ATM-CHK2 Kinases in preclinical or clinical development for cancer therapy.
Table 1.
Small molecule inhibitors of ATM-CHK2 Kinases in preclinical or clinical development for cancer therapy.
| Target | Molecule | Stage of Testing | Tumor Type | Reference/ ClinicalTrial.gov Identifier |
|---|---|---|---|---|
| CHK1/CHK2 | AZD7762 | Phase 1 Administered as single intravenous unit and in combination with gemcitabine | Solid Tumors | NCT00413686 |
| CHK1/CHK2 | CBP501 | Phase 1 Administered in combination with cisplatin/nivolumab | Advanced Solid Tumors | NCT03113188 |
| CHK1/CHK2 | LY2606368 | Phase 1 Administered in combination with cisplatin/cetuximab/pemetrexed/fluorouracil | Neoplasm Metastasis Colorectal Neoplasm Breast Cancer | NCT02124148 |
| CHK1/CHK2 | LY2606368 | Phase 2 Single agent | Ovarian Cancer Breast Cancer Prostate Cancer | NCT02203513 |
| ATM/ATR | CGK733 | Preclinical testing using Chk1-deficient HBV-positive hepatocellular carcinoma cells | Hepatocellular carcinoma | [115] |
| ATM | CP466722 | Preclinical testing using multiple cell lines in combination with infrared radiation (IR) | Cervical cancer Fibroblasts | [116] |
| ATM/ATR | Caffeine | Preclinical testing as single agent using human cancer cells and non-transformed mouse fibroblast cell lines | Breast Cancer Prostate cancer | [117] |
| ATM | KU59403 | Preclinical testing using p53 functional and dysfunctional models of human cancer in combination with camptothecin, doxorubicin or etoposide | Osteosarcoma | [118] |
| ATM | KU55933 | Preclinical testing using human mesenchymal stem cells in combination with IR | Mesenchymal stem cells | [119] |
| ATM | KU55933 | Preclinical testing using ATM-defective and normal human fibroblast cells in combination with doxorubicin | Fibroblast | [120] |
| ATM | KU-60019 | Preclinical testing using human glioma cells in combination with IR | Glioma | [121] |
| ATM | KU-60019 | Preclinical testing using non-invasive breast cancer cells in combination with doxorubicin | Breast Cancer | [122] |
| CHK2 | PV1019 | Preclinical testing using human tumor cell lines in combination with topotecan, camptothecin or radiation | Ovarian Carcinoma | [123] |
| CHK2 | CCT241533 | Preclinical testing using human tumor cell lines in combination with PARP inhibitors | Colon Cancer Breast Cancer Glioblastoma Osteosarcoma Lung Cancer Cervical Cancer | [124] |
| CHK1/CHK2 | XL-844 | Preclinical testing using HT-29 cell line in combination with IR | Colon Cancer | [125] |
| CHK1/CHK2 | XL-844 | Preclinical testing using multiple cell lines in combination with gemcitabine | Pancreatic Cancer Cervical Cancer Ovarian Cancer | [126] |
Table 2.
Small molecule inhibitors of FANCD2 mono-ubiquitination in preclinical or clinical development for cancer therapy.
Table 2.
Small molecule inhibitors of FANCD2 mono-ubiquitination in preclinical or clinical development for cancer therapy.
| Target | Molecule | Stage of Testing | Tumor Type | Reference/ ClinicalTrial.gov Identifier |
|---|---|---|---|---|
| 26S proteasome | Bortezomib | Phase 3 Administered in combination with Daratumumab/bortezomib/dexamethasone | Relapsed or Refractory Multiple Myeloma | NCT03234972 |
| 26S proteasome | Bortezomib | Phase 3 Administered in combination with daratumumab/cyclophosphamide bortezomib/dexamethasone | Amyloidosis | NCT03201965 |
| 26S proteasome | Bortezomib | Phase 2 As single agent | Multiple Myeloma | NCT00153920 |
| 26S proteasome | Bortezomib | Phase 2 As single agent | Multiple Myeloma Stage I, II, and III | NCT00075881 |
| 26S proteasome | Bortezomib | Phase 2 As single agent | Primary Peritoneal Cavity Cancer and Recurrent Ovarian Epithelial Cancer | NCT00023712 |
| 26S proteasome | Bortezomib | Phase 2 Administered in combination with bortezomib/vorinostat | Acute Lymphoblastic Leukemia | NCT02553460 |
| FANCF | Curcumin | Preclinical testing using cell lines in combination with cisplatin | Ovarian and Breast Cancer | [167] |
| FANCS | Phenylbutyrate | Preclinical testing using cell lines in combination with cisplatin | Head and Neck Cancer | [168] |
| USP1 | GW7647 | Preclinical testing using cell lines in combination with cisplatin | Non-small-cell Lung Cancer | [169] |
| USP1 | Pimozide | Preclinical testing using cell lines in combination with cisplatin | Non-small-cell Lung Cancer | [169] |
| USP1-UAF1 | ML323 | Preclinical testing using cell lines in combination with cisplatin | Non-small-cell Lung Cancer and Osteosarcoma | [170] |
| USP1 | C527 | Preclinical testing using cell lines in combination with MMC and camptothecin | Leukemia | [171] |
| ATR | Wortmannin | Preclinical testing using HeLa cell lines in combination with IR or mitomycin C (MMC) | Cervical Cancer | [172] |
| PKA PKC PKG | H-9 | Preclinical testing using HeLa cell lines in combination with IR | Cervical Cancer | [167] |
| CDK GSK3 | Alsterpaullone | Preclinical testing using HeLa cell lines in combination with IR | Cervical Cancer | [167] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bhattacharjee, S.; Nandi, S. Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy. Cancers 2018, 10, 298. https://doi.org/10.3390/cancers10090298
AMA Style
Bhattacharjee S, Nandi S. Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy. Cancers. 2018; 10(9):298. https://doi.org/10.3390/cancers10090298
Chicago/Turabian StyleBhattacharjee, Sonali, and Saikat Nandi. 2018. "Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy" Cancers 10, no. 9: 298. https://doi.org/10.3390/cancers10090298
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.