
Polarizability of Kekulene, Septulene, and Nearest Non-Planar Polycyclic Aromatic Hydrocarbons


1
Laboratory of Mathematical Chemistry, Institute of Petrochemistry and Catalysis UFRC RAS, 450075 Ufa, Russia
2
Department of Oil and Gas Technology, Ufa State Petroleum Technological University, 450000 Ufa, Russia
*
Author to whom correspondence should be addressed.
C 2022, 8(4), 61; https://doi.org/10.3390/c8040061
Submission received: 17 October 2022
/
Revised: 3 November 2022
/
Accepted: 5 November 2022
/
Published: 7 November 2022
(This article belongs to the Section Carbon Skeleton)
Abstract
:The polarizability of polycyclic aromatic hydrocarbons (PAHs) is an important property that relates to their abundance in natural environments. To assess the differences in the mean polarizability of planar and non-planar polycyclic aromatic hydrocarbons (PAHs), we computationally studied the PAH series of circulenes (kekulene C48H24, [13]circulene C52H26, and septulene C56H28), a number of the nearest helicenes, and their “expanded” isomers. We mean under “nearest” a close number of aromatic rings: 11 (C46H26), 12 (C50H28), 13 (C54H30), and 14 (C58H32). For these PAHs, we performed the quantum chemical calculations of thermodynamic and polarizability parameters with the PBE/3ζ density functional theory method, which is widely used in the theoretical chemistry of fullerenes and PAHs. The calculated mean polarizabilities (in Å3) ranged from 80.1 for [11]helicene to 135.5 for septulene, and while the circulenes and expanded helicenes had similar values, the mean polarizability of the normal helicenes was markedly lower. In all four pairs of helical PAHs, the expanded helicene was energetically considerably more favorable than its standard helicene isomer. Herewith, the ratio of their polarizabilities was equal to 1.3.
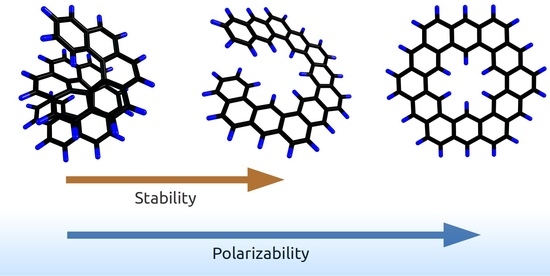
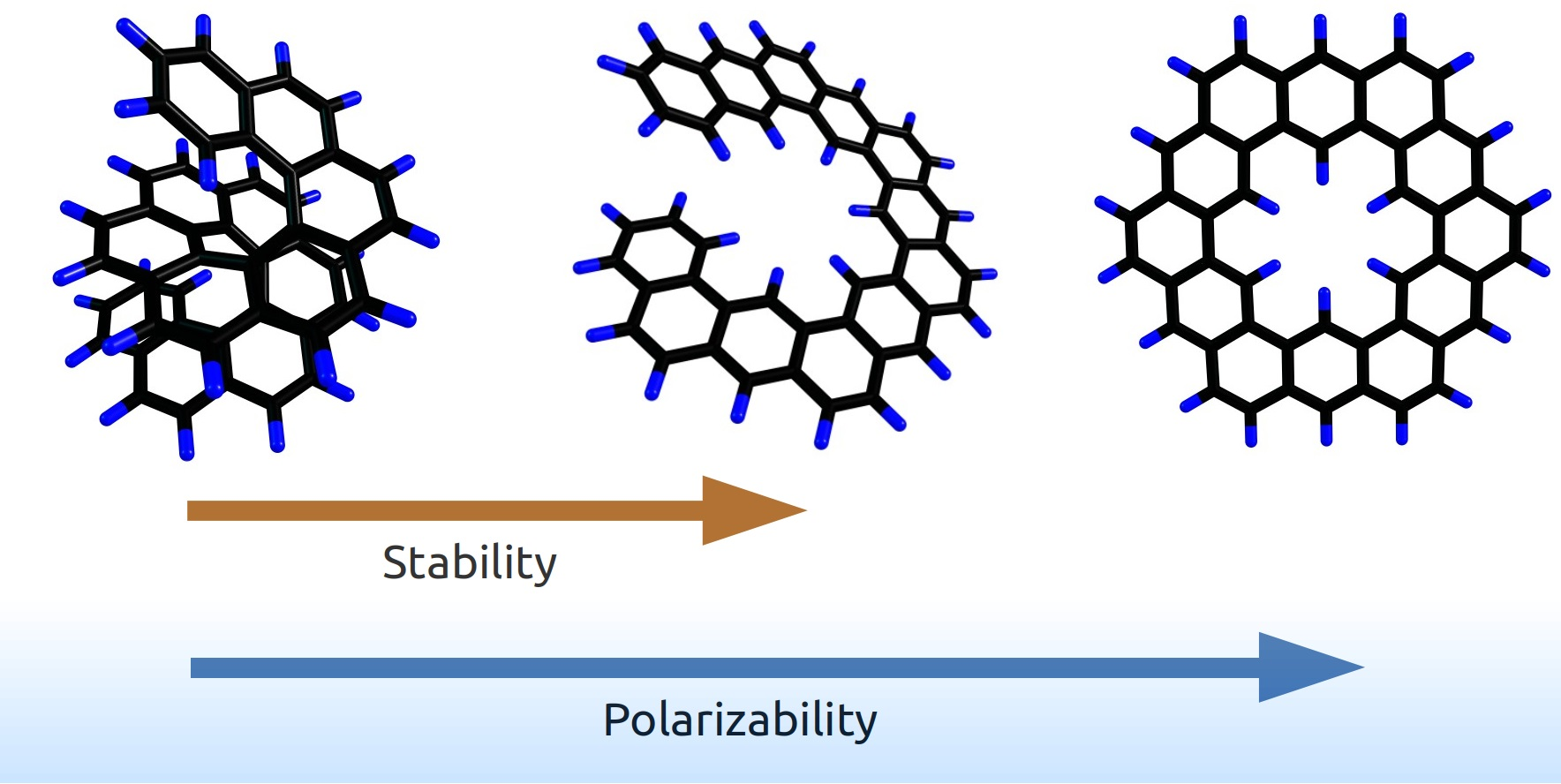
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs) make up a large group of organic compounds composed of two or more condensed aromatic rings. The PAHs’ properties, originating mainly from extensive delocalized π-conjugated systems, are one of the reasons for investigating their transformations [1,2,3,4]. The correlations between the structure and physicochemical properties were addressed in theoretical works [5,6,7,8,9,10]. The interest in molecular properties of PAHs is also due to their environmental chemistry: They are usually formed under the incomplete combustion of organic matter and, therefore, relate to ecological problems dealing with their potential cancerogenicity) [11]. Various PAHs are expected to be ubiquitous in the interstellar and circumstellar media. This is based on experimental works [12,13,14,15] and theoretical models [16].
Generally, most PAHs are planar, and the planar compounds are studied more extensively, compared with the non-planar ones. The latter include helicenes, which have a helical structure [3], and their isomers—the so-called “expanded helicenes”—whose structures also have a helix as a basis, just wider [4]. Currently, helicenes attract increasing attention because of the possibility of their formation in the interstellar medium, which has been predicted with theoretical models [10] and then confirmed with laboratory experiments [14]. We have previously shown that helicenes are less thermodynamically stable than their planar isomers but have lower mean polarizabilities. The latter feature enhances the survivability of helicenes under cosmic conditions that typically involve interactions of matters with irradiations [14,17]. Furthermore, we have formulated a qualitative correlation in the isomeric PAH series: non-planar isomers have higher total energy, lower mean polarizability, and should be more abundant in astronomical objects or their laboratory models (and vice versa in the case of flat PAHs) [17].
Polarizability is an important property of PAHs because it is connected to their structure, physicochemical properties, and chemical reactivity [9]. There are a number of publications dealing with the mean polarizabilities of PAHs computed with different theoretical methods: semiempirical [18], quantitative structure–property relationship (QSPR) [19], coupled cluster [20], and original and modified density functional theory [9,21]. Based on these works, the methods based on the Perdew–Burke–Ernzerhof density functional (PBE) [22] are applicable to study the links between structure, polarizability, and related properties of different PAHs.
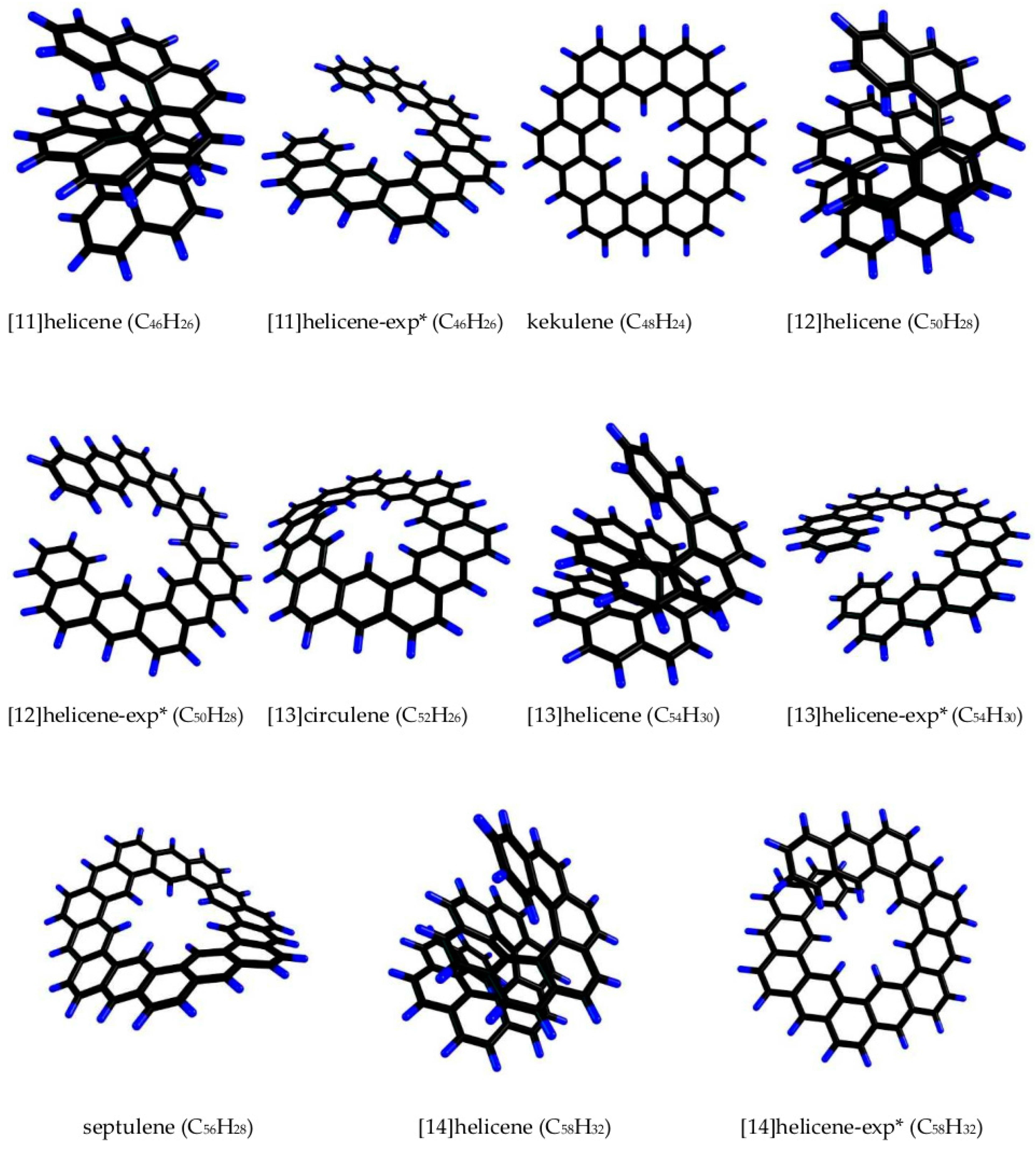
In the continuation of our systematic study on the PAH polarizability [17], we investigated the stability and polarizability of kekulene C48H24, septulene C56H28, and a number of their nearest non-planar PAHs: [13]circulene, [n]helicenes, and expanded [n]helicenes (in the following text, we will mark the latter with the suffix “-exp”), where n = 11, 12, 13, and 14. The optimized geometries of these molecules are shown in Figure 1 (the associated Cartesian coordinates of atomic positions can be found in the Supplementary Materials). A helix can be described by its diameter, number of turns, and pitch. [11]Helicene has only one complete turn of the helix, whereas the remaining helicenes have two. Expanded [n]helicenes ([n]helicenes-exp) are in a similar situation, but the first turn of their helix finishes at n = 12. The results are presented in this study.
2. Computation Details
Quantum chemical calculations were performed in the PRIRODA program (version 11) [23]. We used the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional with gradient correction [22] in combination with the split-valence, triple-zeta basis set 3ζ. As shown in previous works, the PBE/3ζ method reliably reproduces the experimental results on the polarizability, structural, and thermochemical properties of PAHs and fullerenes [10,21,24,25]. There are known limitations when using PBE and other pure DFT methods to describe extensive polyaromatic compounds [26]. These problems were studied in previous works on the examples of oligoacenes and phenacenes [9]. Analyzing our previous experience described in the abovementioned paper, we believe that the chosen method PBE/3ζ is applicable to the compounds of this work, as they are not in the list of “restricted” compounds.
The structures of the studied molecules were fully optimized without restrictions on symmetry. We calculated vibrational spectra for all optimized structures to ensure that they correspond to the minima on the potential energy surfaces (no imaginary frequencies are observed).
The reaction enthalpies were calculated as the difference between the total energies Etot of the products and the reactants, taking into account the zero-point vibrational energies (ZPVEs) and thermal corrections Hcorr:
The polarizability tensors were calculated according to the finite field approach whereby their elements αij were defined as the second derivatives of the total energy of the molecule over the homogeneous external electric field:
The mean polarizability α was calculated from the diagonal elements (we express α values in Å3 in the present work):
α = (αxx + αyy + αzz)/3.
The reliability of the method has been previously scrutinized. Here, we just note one example of the difference between the PAH polarizabilities. The calculated α values of anthracene (27.0 Å3) and phenanthrene molecules (25.7 Å3), obtained with the PBE/3ζ method, are in good agreement with the experimental references of 25.3 and 23.6 Å3, respectively (taken from [27]).
3. Results and Discussion
3.1. Objects under Study
Kekulene (first synthesized in 1978 [28]) is a flat molecule consisting of 12 aromatic rings (Figure 1). It could be considered as a loop of six phenanthrene fragments [29,30]. The highest possible point symmetry group for kekulene is D6h, which could be achieved in the case of full delocalization of the π-electrons. The real kekulene structure has a lower symmetry, viz. D3h, because its π-electrons are partially localized within rings, so kekulene consists of alternating aromatic and non-aromatic rings (this is in accordance with Clar’s rule). The alternation reduces idealized symmetry [29,30]. The chosen PBE/3ζ method reproduces this feature, and the optimization of the geometry results in a D3h symmetry structure.
As the nearest kekulene benzenoids, we considered the isomeric pairs of helicenes and expanded helicenes consisting of 11 (C46H26) and 12 (C50H28) aromatic rings. Both regular and expanded helicenes are optically active, though devoid of chiral centers. In our calculations, we used the (P)-enantiomers, which means that their helices are right-handed (plus), i.e., they are dextrorotatory. Note that both enantiomers have the same mean polarizabilities, and we plan to focus on the types of enantiomers in future studies.
Expanded helicenes with the odd number of aromatic rings (11 and 13 in our work) theoretically have two forms: (i) the three terminal rings at each end of the helix may form a kinked pattern as in phenanthrene or (ii) a straight one as in anthracene. The forms with the kinked ends of the helix are noticeably more favorable in terms of their total energy: by 11.5 and 11.9 kJ/mol for [11]helicene-exp and [13]helicene-exp, respectively.
Septulene (first synthesized in 2012) has two additional aromatic rings compared with kekulene (14 in total). Similar to kekulene, it can be considered as a loop of seven phenanthrene fragments where π-electrons remain localized over the rings. The molecule is non-planar and, as shown in synthetic research [31], has different conformations in the gas phase and crystalline form. The septulene crystal consists of molecules in the chair conformation, which is evidenced by X-ray analysis and force-field calculations. However, in the gas phase, the chair form is 22–25 kJ/mol less favorable, and one is left with a choice between different saddle conformations (because the flat D7h structure is similarly unfavorable, and its vibrational spectrum had imaginary frequencies in our calculations). We focused on the chair conformation of CS symmetry because both C2 and C1 symmetry structures have small imaginary frequencies, and the energy differences between the three structures are less than 2.6 kJ/mol.
Similar to kekulene, we considered the isomeric pairs of regular and expanded helicenes consisting of 13 (C54H30) and 14 (C58H32) aromatic rings as the benzenoids nearest to septulene. All chosen helicenes are (P)-enantiomers.
Between kekulene and septulene, there should be [13]circulene with similar properties. This compound has not been synthesized thus far. Its molecule is non-planar and has a symmetry plane that corresponds to the CS symmetry point group.
3.2. Thermodynamic Parameters of the Studied PAHs
When comparing the regular and expanded helicene isomers, we found the expanded helicene energetically more favorable in all studied pairs. The calculated differences between the isomers were 156.6, 169.9, 197.7, and 214.0 kJ/mol as the number of benzene rings increased from 11 to 14.
To assess the relative stabilities of our circulenes, we used the following hypothetical chemical reactions with ethylene that result in the corresponding helicenes:
and
where n = 12, 13, and 14. As found, all reactions (5) were exothermic, whereas reactions (4) were endothermic (Table 1). The enthalpies of both reactions of [13]circulene were noticeably lower than of the other circulenes, i.e., [13]circulene was less favorable than its closest (with the same number of rings) helicenes. This means that either [13]circulene is a less energetically favorable circulene, or both [13]helicene and [13]helicene-exp are especially favorable helicenes.
[n]circulene + C2H4 → [n]helicene
[n]circulene + C2H4 → [n]helicene-exp,
3.3. Mean Polarizabilities of the Studied PAHs
The calculated mean polarizabilities α for all studied PAHs were in the range from 80.1 Å3 for [11]helicene to 135.5 Å3 for septulene (Table 1). Since the mean polarizability of a molecule depends on its size, we calculated the changes in mean polarizability for the hypothetical reactions (4) and (5) using the following formula [32]:
If we group the studied PAH molecules by the number of aromatic rings and consider the size effects (Equation (6)), then circulene had the highest mean polarizability, and helicene had the lowest one inside each group.
As previously found, the mean polarizability relates to the yields of the products of chemical reactions (see, e.g., works [10,21,24,33,34]). In addition, there is the minimum polarizability principle that states that less polarizable isomers should be more thermodynamically stable [35,36]. In the classical theory of chemical structure, polarizability is associated with the volume of the electron cloud of a molecule and its deformability under external electric fields [37]. Hence, we can explain the low polarizability of helicenes by the smaller volume of their electron cloud, which is caused by the compactness of their structure relative to circulenes and expanded helicenes.
Considering the change in mean polarizability upon the chemical reactions of the molecules of the same type (Figure 2), we found that it was linearly correlated with the number of aromatic rings for all three types. Another finding was that the ratio of the polarizabilities of helicenes and helicenes-exp was constant and equaled 1.3 for all four pairs of helicene isomers.
3.4. General Remarks and Prospective
Very recently, we formulated a hypothesis that connects the structure, stability, and polarizability of PAHs depending on their planar or non-planar shape [17]. In general, non-planar PAHs have higher total energy and lower mean polarizability, compared with their planar isomers. Then, we started a systematic computational study to substantiate this statement with relevant examples. The case considered in the present study fits into the regularity. Indeed, if we consider isomers, the planarity increased in the following series: helicene < expanded helicene < planar PAHs. Accordingly, the stability and mean polarizability increased.
We assume that, in addition to astrochemical applications [10], our results could be useful for studying heavy oil fractions enriched with high-molecular aromatic hydrocarbons [38] and applications of PAHs in material science [39]. In a fundamental aspect, we will continue our research to quantify our qualitative correlations. We plan to find the regularities between structural descriptors and molecular properties [40]. There should be a more general regularity because our above-formulated statement is not exclusive to isomeric compounds (e.g., the ratio of “high planarity–high polarizability” is fulfilled in the case of the related PAHs connected with the transformations according to the Scholl reaction [41]).
4. Conclusions
Based on the performed density functional theory computations, we found that in a series of PAHs including kekulene, septulene, and the nearest benzenoids, the mean polarizability decreased with the molecule’s planarity, even though circulenes themselves were not always flat. This decrease was observed between both isomers and non-isomeric structurally related molecules. These findings serve as another example in favor of a previously found correlation that revealed higher mean polarizability for more planar PAHs than for non-planar isomers or structurally close molecules. The mean polarizability of helicenes was much lower than that of all other PAH types, which is explained by the significantly reduced volume of their electron clouds due to the compactness of the helical structure.
Another intriguing finding was that the expanded helicenes were found to be considerably more energetically favorable than the well-known and well-studied regular helicenes. This observation violates the minimum polarizability principle. We hope this theoretical study will enhance future interest in the expanded helicenes as a class of PAHs with non-trivial molecular properties.
Supplementary Materials
Cartesian coordinates of the optimized molecules, key energy, and polarizability parameters can be downloaded at: https://www.mdpi.com/article/10.3390/c8040061/s1.
Author Contributions
Conceptualization, D.S.S. and A.F.A.; methodology, D.S.S.; investigation, T.L.; validation, T.L.; writing—original draft preparation, T.L. and D.S.S.; writing—review and editing, T.L., A.F.A. and D.S.S.; visualization, T.L.; supervision, D.S.S.; funding acquisition, D.S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation, project “Information entropy of chemical reactions: A novel methodology for digital organic chemistry”, grant number 22-13-20095.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Clar, E. Polycyclic Hydrocarbons; Academic Press: New York, NY, USA, 1964. [Google Scholar] [CrossRef]
- Slayden, S.W.; Liebman, J.F. The energetics of aromatic hydrocarbons: An experimental thermochemical perspective. Chem. Rev. 2001, 101, 1541–1566. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Chen, C.-F. Helicenes: Synthesis and Applications. Chem. Rev. 2012, 112, 1463–1535. [Google Scholar] [CrossRef]
- Kiel, G.R.; Patel, S.C.; Smith, P.W.; Levine, D.S.; Tilley, T.D. Expanded Helicenes: A General Synthetic Strategy and Remarkable Supramolecular and Solid-State Behavior. J. Am. Chem. Soc. 2017, 139, 18456–18459. [Google Scholar] [CrossRef] [PubMed]
- Matta, C.; Hernández-Trujillo, J.; Tang, T.-H.; Bader, R.F.W. Hydrogen–hydrogen bonding: A stabilizing interaction in molecules and crystals. Chem. Eur. J. 2003, 9, 1940–1951. [Google Scholar] [CrossRef]
- Vashchenko, A.V.; Borodina, T.N. H–H interaction in phenanthrene: Attraction or repulsion? J. Struct. Chem. 2013, 54, 479–483. [Google Scholar] [CrossRef]
- Poater, J.; Visser, R.; Solà, M.; Bickelhaupt, F.M. Polycyclic benzenoids: Why kinked is more stable than straight. J. Org. Chem. 2007, 72, 1134–1142. [Google Scholar] [CrossRef]
- Portella, G.; Poater, J.; Bofill, J.M.; Alemany, P.; Solà, M. Local aromaticity of [n]acenes, [n]phenacenes, and [n]helicenes (n = 1–9). J. Org. Chem. 2005, 70, 2509–2521. [Google Scholar] [CrossRef]
- Sabirov, D. A correlation between the mean polarizability of the “kinked” polycyclic aromatic hydrocarbons and the number of H...H bond critical points predicted by Atoms-in-Molecules theory. Comput. Theor. Chem. 2014, 1030, 81–86. [Google Scholar] [CrossRef]
- Sabirov, D.; Garipova, R.R.; Cataldo, F. Polarizability of isomeric and related interstellar compounds in the aspect of their abundance. Mol. Astrophys. 2018, 12, 10–19. [Google Scholar] [CrossRef]
- Grandberg, I.I. Organic Chemistry; Mir: Moscow, Russia, 2002. [Google Scholar]
- Ehrenfreund, P.; Ruiterkamp, R.; Peeters, Z.; Foing, B.; Salama, F.; Martins, Z. The ORGANICS experiment on BIOPAN V: UV and space exposure of aromatic compounds. Planet. Space Sci. 2007, 55, 383–400. [Google Scholar] [CrossRef]
- Kwok, S.; Zhang, Y. Mixed aromatic–aliphatic organic nanoparticles as carriers of unidentified infrared emission features. Nature 2011, 479, 80–83. [Google Scholar] [CrossRef]
- Zhao, L.; Kaiser, R.I.; Xu, B.; Ablikim, U.; Lu, W.; Ahmed, M.; Evseev, M.M.; Bashkirov, E.K.; Azyazov, V.N.; Zagidullin, M.V.; et al. Gas phase synthesis of [4]-helicene. Nat. Commun. 2019, 10, 1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oña-Ruales, J.O.; Ruiz-Morales, Y.; Alvarez-Ramírez, F. The Helicenes: Potential carriers of diffuse interstellar bands. ACS Earth Space Chem. 2021, 5, 381–390. [Google Scholar] [CrossRef]
- Sadjadi, S.; Kwok, S.; Zhang, Y. Theoretical infrared spectra of MAON molecules. J. Phys. Conf. Ser. 2016, 728, 62003. [Google Scholar] [CrossRef] [Green Version]
- Sabirov, D.S.; Tukhbatullina, A.A.; Shepelevich, I.S. Polarizability in astrochemical studies of complex carbon-based compounds. ACS Earth Space Chem. 2022, 6, 1–17. [Google Scholar] [CrossRef]
- Alparone, A.; Librando, V.; Minniti, Z. Validation of semiempirical PM6 method for the prediction of molecular properties of polycyclic aromatic hydrocarbons and fullerenes. Chem. Phys. Lett. 2008, 460, 151–154. [Google Scholar] [CrossRef]
- Martin, D.; Sild, S.; Maran, U.; Karelson, M. QSPR modeling of the polarizability of polyaromatic hydrocarbons and fullerenes. J. Phys. Chem. C 2008, 112, 4785–4790. [Google Scholar] [CrossRef]
- Smith, S.M.; Markevitch, A.N.; Romanov, D.A.; Li, X.; Levis, R.J.; Schlegel, H.B. Static and dynamic polarizabilities of conjugated molecules and their cations. J. Phys. Chem. A 2004, 108, 11063–11072. [Google Scholar] [CrossRef] [Green Version]
- Sabirov, D.S.; Terentyev, A.O.; Bulgakov, R.G. Polarizability of fullerene [2+2]-dimers: A DFT study. Phys. Chem. Chem. Phys. 2014, 16, 14594–14600. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Laikov, D.N.; Ustynyuk, Y.A. PRIRODA-04: A quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing. Russ. Chem. Bull. 2005, 54, 820–826. [Google Scholar] [CrossRef]
- Sabirov, D.S. Polarizability of C60 fullerene dimer and oligomers: The unexpected enhancement and its use for rational design of fullerene-based nanostructures with adjustable properties. RSC Adv. 2013, 3, 19430–19439. [Google Scholar] [CrossRef]
- Sabirov, D.S.; Terentyev, A.O.; Cataldo, F. Bisadducts of the C60 and C70 fullerenes with anthracene: Isomerism and DFT estimation of stability and polarizability. Comput. Theor. Chem. 2016, 1081, 44–48. [Google Scholar] [CrossRef]
- Nénon, S.; Champagne, B.R.; Spassova, M. Assessing long-range corrected functionals with physically-adjusted range-separated parameters for calculating the polarizability and the second hyperpolarizability of polydiacetylene and polybutatriene chains. Phys. Chem. Chem. Phys. 2014, 16, 7083–7088. [Google Scholar] [CrossRef] [PubMed]
- Le Fèvre, R.J.W.; Sundaram, K.M.S. Molecular polarisability. The molar Kerr constants, polarisations, etc., of ten polynuclear hydrocarbons as solutes in benzene. J. Chem. Soc. 1963, 4442–4446. [Google Scholar] [CrossRef]
- Diederich, F.; Staab, H.A. Benzenoid versus annulenoid aromaticity: Synthesis and properties of kekulene. Angew. Chem. Int. Ed. Engl. 1978, 17, 372–374. [Google Scholar] [CrossRef]
- Poater, J.; Paauwe, J.; Pan, S.; Merino, G.; Fonseca-Guerra, C.; Bickelhaupt, F.M. Kekulene: Structure, stability and nature of H…H interactions in large PAHs. Mol. Astrophys. 2017, 8, 19–26. [Google Scholar] [CrossRef]
- Haags, A.; Reichmann, A.; Fan, Q.; Egger, L.; Kirschner, H.; Naumann, T.; Werner, S.; Vollgraff, T.; Sundermeyer, J.; Eschmann, L.; et al. Kekulene: On-surface synthesis, orbital structure, and aromatic stabilization. ACS Nano 2020, 14, 15766–15775. [Google Scholar] [CrossRef]
- Kumar, B.; Viboh, R.L.; Bonifacio, M.C.; Thompson, W.B.; Buttrick, J.C.; Westlake, B.C.; Kim, M.-S.; Zoellner, R.W.; Varganov, S.A.; Mörschel, P.; et al. Septulene: The Heptagonal Homologue of Kekulene. Angew. Chem. Int. Ed. 2012, 51, 12795–12800. [Google Scholar] [CrossRef]
- Ghanty, T.K.; Ghosh, S.K. A density functional approach to hardness, polarizability, and valency of molecules in chemical reactions. J. Phys. Chem. 1996, 100, 12295–12298. [Google Scholar] [CrossRef]
- Sabirov, D.; Garipova, R.R.; Kinzyabaeva, Z.S. Fullerene–1,4-dioxane adducts: A DFT study of the structural features and molecular properties. Fuller. Nanotub. Carbon Nanostruct. 2020, 28, 154–159. [Google Scholar] [CrossRef]
- Sharipov, A.S.; Loukhovistky, B.I. Small atomic clusters: Quantum chemical research of isomeric composition and physical properties. Struct. Chem. 2019, 30, 2057–2084. [Google Scholar] [CrossRef]
- Blair, S.A.; Thakkar, A.J. How often is the minimum polarizability principle violated? Chem. Phys. Lett. 2013, 556, 346–349. [Google Scholar] [CrossRef]
- Hohm, U. Is there a minimum polarizability principle in chemical reactions? J. Phys. Chem. A 2000, 104, 8418–8423. [Google Scholar] [CrossRef]
- Vereshchagin, A.N. Polarizability of Molecules; Nauka: Moscow, Russia, 1980. [Google Scholar]
- Dolomatov, M.Y.; Burangulov, D.Z.; Dolomatova, M.M.; Osipenko, D.F.; Zaporin, V.P.; Tukhbatullina, A.A.; Akhmetov, A.F.; Sabirov, D.S. Low-sulphur vacuum gasoil of Western Siberia oil: The impact of its structural and chemical features on the properties of the produced needle coke. C 2022, 8, 19. [Google Scholar] [CrossRef]
- Sugawara, Y.; Kaji, Y.; Ogawa, K.; Eguchi, R.; Oikawa, S.; Gohda, H.; Fujiwara, A.; Kubozono, Y. Characteristics of field-effect transistors using the one-dimensional extended hydrocarbon [7]phenacene. Appl. Phys. Lett. 2011, 98, 013303. [Google Scholar] [CrossRef]
- Sabirov, D.S.; Ori, O.; Tukhbatullina, A.A.; Shepelevich, I.S. Structural descriptors of benzenoid hydrocarbons: A mismatch between the estimates and parity effects in helicenes. C 2022, 8, 42. [Google Scholar] [CrossRef]
- Lukmanov, T.I.; Shepelevich, I.S.; Sabirov, D.S. Polarizability of polycyclic aromatic hydrocarbon compounds from the intermediate stages of the oxidative condensation of hexaphenylbenzene into hexa-peri-benzocoronene. Vestn. Bashkir. Univ. 2022, 27, 98–101. [Google Scholar] [CrossRef]
Figure 1.
All polycyclic aromatic hydrocarbons under study. The energetically unfavorable conformations are not shown. Note: * Suffix “-exp” indicates expanded helicene structures.
Figure 1.
All polycyclic aromatic hydrocarbons under study. The energetically unfavorable conformations are not shown. Note: * Suffix “-exp” indicates expanded helicene structures.

Figure 2.
The relationship between the mean polarizability and the number of aromatic rings for selected PAHs.
Figure 2.
The relationship between the mean polarizability and the number of aromatic rings for selected PAHs.


{kind=link}
{kind=link}
{kind=link}
Table 1.
The calculated thermochemical parameters and mean polarizabilities of the studied PAHs.
| Molecule | α (Å3) | ΔαR (Å3) a | (kJ/mol) b |
|---|---|---|---|
| [11]helicene | 80.1 | n/a | n/a |
| [11]helicene-exp | 102.9 | n/a | n/a |
| kekulene | 111.1 | n/a | n/a |
| [12]helicene | 86.3 | −28.6 | 126.9 |
| [12]helicene-exp | 112.6 | −2.3 | −43.0 |
| [13]circulene | 123.0 | n/a | n/a |
| [13]helicene | 92.3 | −34.5 | 90.5 |
| [13]helicene-exp | 119.1 | −7.7 | −107.2 |
| septulene | 135.5 | n/a | n/a |
| [14]helicene | 98.5 | −40.8 | 141.4 |
| [14]helicene-exp | 127.8 | −11.4 | −72.6 |
a The change in the mean polarizability for a reaction is calculated in the same way as the change in total energy; see Equation (6). b Heat effects of reactions (4) and (5).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lukmanov, T.; Akhmetov, A.F.; Sabirov, D.S. Polarizability of Kekulene, Septulene, and Nearest Non-Planar Polycyclic Aromatic Hydrocarbons. C 2022, 8, 61. https://doi.org/10.3390/c8040061
AMA Style
Lukmanov T, Akhmetov AF, Sabirov DS. Polarizability of Kekulene, Septulene, and Nearest Non-Planar Polycyclic Aromatic Hydrocarbons. C. 2022; 8(4):61. https://doi.org/10.3390/c8040061
Chicago/Turabian StyleLukmanov, Timur, Arslan F. Akhmetov, and Denis Sh. Sabirov. 2022. "Polarizability of Kekulene, Septulene, and Nearest Non-Planar Polycyclic Aromatic Hydrocarbons" C 8, no. 4: 61. https://doi.org/10.3390/c8040061
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.