
The Importance of M1-and M2-Polarized Macrophages in Glioma and as Potential Treatment Targets
 ,
,
Abstract
:1. Introduction

{kind=link}
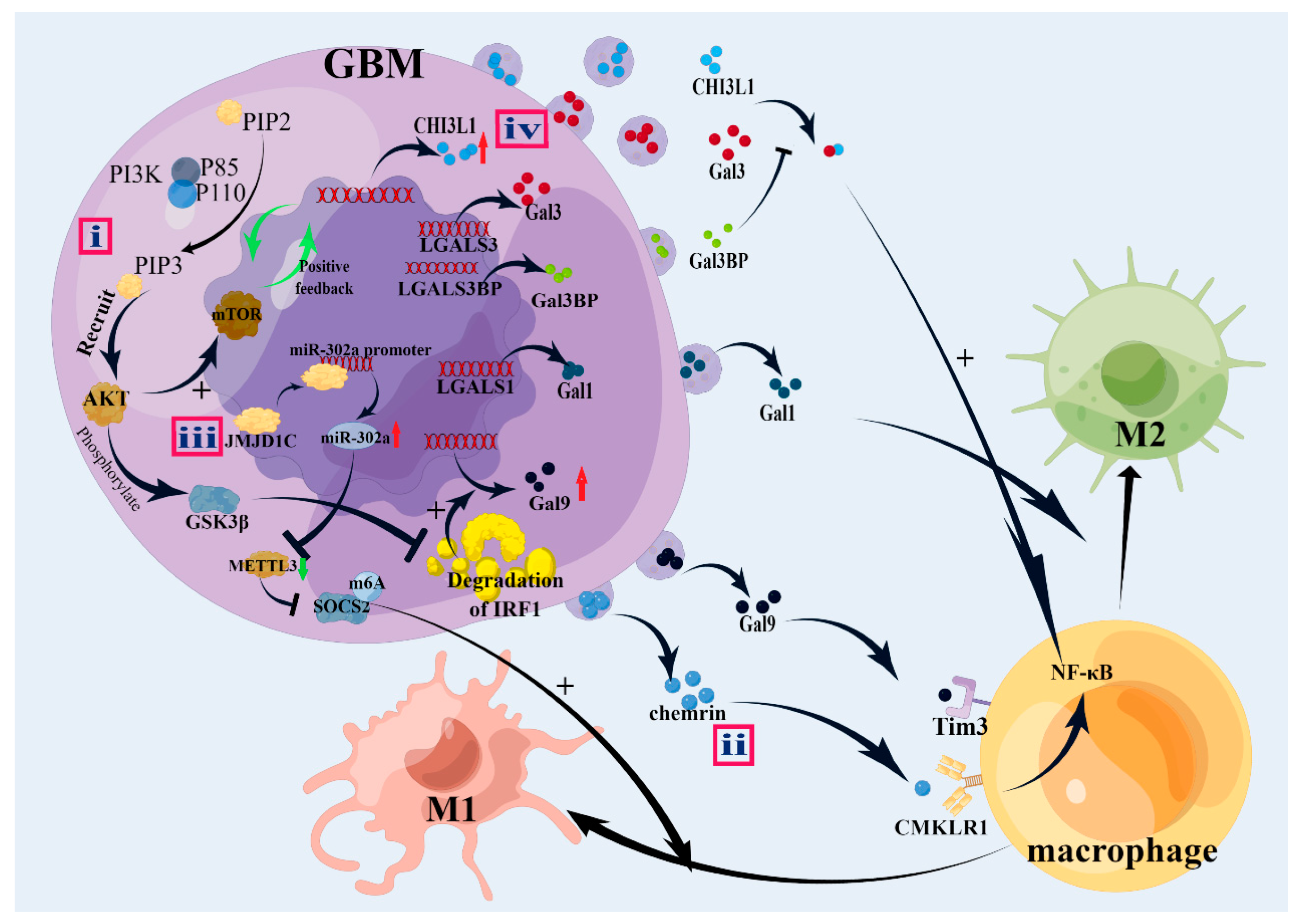{kind=link}
{kind=link}
{kind=link}
| M1 TAMs Markers | M2 TAMs Markers | ||||
|---|---|---|---|---|---|
| Study First Author | Year of Publication | Marker | Study First Author | Year of Publication | Marker |
| Kang et al. [11] | 2017 | iNOS | Codrici et al. [1] | 2022 | MRC1 |
| Peng et al. [24] | 2022 | Yang et al. [9] | 2018 | ||
| Xiao et al. [25] | 2022 | TSPO | Zhang et al. [26] | 2022 | |
| Codrici et al. [1] | 2022 | IL-12B | Codrici et al. [1] | 2022 | CD163 |
| Zhang et al. [26] | 2022 | Kang et al. [11] | 2017 | ||
| Zhang et al. [26] | 2022 | IL-12A | Bianconi et al. [13] | 2022 | |
| Kang et al. [11] | 2017 | CD40 | Peng et al. [24] | 2022 | |
| Bianconi et al. [13] | 2022 | Jiang et al. [27] | 2022 | ||
| Hambardzumyan et al. [15] | 2016 | Zhang et al. [28] | 2022 | ||
| Xiao et al. [25] | 2022 | IL-1β | Hansen et al. [29] | 2022 | Timp1 |
| Jiang et al. [27] | 2022 | Umemura et al. [30] | 2008 | CD36 | |
| Umemura et al. [30] | 2008 | Xia et al. [31] | 2020 | 1IDO1 | |
| Xiao et al. [25] | 2022 | IL-6 | Umemura et al. [30] | 2008 | CCL17 |
| Curtale et al. [32] | 2019 | Curtale et al. [32] | 2019 | ||
| Liu et al. [33] | 2022 | CD80 | Sanders et al. [34] | 2021 | CCL4 |
| Bianconi et al. [13] | 2022 | CD74 | Peng et al. [24] | 2022 | VEGFA |
| Peng et al. [24] | 2022 | CD86 | Hansen et al. [29] | 2022 | |
| Xiao et al. [25] | 2022 | Xia et al. [31] | 2020 | EGF | |
| Curtale et al. [32] | 2019 | Codrici et al. [1] | 2022 | Fizz1 | |
| Kang et al. [11] | 2017 | CD197 | Yang et al. [9] | 2018 | |
| Zhang et al. [26] | 2022 | IL-23A | Bianconi et al. [13] | 2022 | |
| Liu et al. [33] | 2022 | Peng et al. [24] | 2022 | ||
| Codrici et al. [1] | 2022 | NOS2 | Codrici et al. [1] | 2022 | Arg-1 |
| Zhang et al. [26] | 2022 | Yang et al. [9] | 2018 | ||
| Zhang et al. [28] | 2022 | Kang et al. [11] | 2017 | ||
| Bianconi et al. [13] | 2022 | MHC-II | Bianconi et al. [13] | 2022 | |
| Xiao et al. [25] | 2022 | CXCL9 | Peng et al. [24] | 2022 | |
| Sanders et al. [34] | 2021 | Zhang et al. [26] | 2022 | ||
| Xiao et al. [25] | 2022 | CXCL10 | Jiang et al. [27] | 2022 | |
| Zhang et al. [26] | 2022 | Hansen et al. [29] | 2022 | ||
| Umemura et al. [30] | 2008 | Peng et al. [24] | 2022 | IL-10 | |
| Jiang et al. [27] | 2022 | TNF-α | Zhang et al. [26] | 2022 | |
| Umemura et al. [30] | 2008 | Hansen et al. [29] | 2022 | ||
| Zhang et al. [28] | 2022 | IRF-5 | Zhang et al. [26] | 2022 | RETNLB |
| Xiao et al. [25] | 2022 | CCL5 | Jiang et al. [27] | 2022 | TGF-β |
| Sanders et al. [34] | 2021 | Liu et al. [33] | 2022 | ||
| Codrici et al. [1] | 2022 | Ciita | Zhang et al. [28] | 2022 | MS4A4A |
| Sanders et al. [34] | 2021 | p-STAT1 | Zhang et al. [28] | 2022 | VSIG4 |
| Bianconi et al. [13] | 2022 | p-STAT3 | |||
| Yang et al. [9] | 2018 | Mgl-2 | |||
| Biswas et al. [35] | 2008 | ||||
| Codrici et al. [1] | 2022 | Chi3l3 | |||
| Codrici et al. [1] | 2022 | SOCS2 | |||
| Xia et al. [31] | 2020 | Ym1 | |||
| Biswas et al. [35] | 2008 | ||||
| Liu et al. [33] | 2022 | MSR1 | |||
| Yang et al. [9] | 2018 | CD11c | |||
| Codrici et al. [1] | 2022 | CD204 | |||
| Bianconi et al. [13] | 2022 | ||||
| Sanders et al. [34] | 2021 | ||||
| Kang et al. [11] | 2017 | CD206 | |||
| Bianconi et al. [13] | 2022 | ||||
| Peng et al. [24] | 2022 | ||||
| Jiang et al. [27] | 2022 | ||||
| Umemura et al. [30] | 2008 | ||||
| Peng et al. [24] | 2022 | CD68 | |||
| Codrici et al. [1] | 2022 | CCL2 | |||
| Xiao et al. [25] | 2022 | ||||
| Peng et al. [24] | 2022 | CCL18 | |||
| Xiao et al. [25] | 2022 | ||||
| Curtale et al. [32] | 2019 | ||||
| Xia et al. [31] | 2020 | CCL20 | |||
| Umemura et al. [30] | 2008 | CCL22 | |||
| Xia et al. [31] | 2020 | ||||
2. The Mechanism by Which M1- and M2-Polarized Macrophages Promote or Inhibit the Growth of Glioblastoma
2.1. M1 and M2 Polarization in Different Stages and Parts of Tumors
2.2. Galectin and AKT/GSK3β/IRF1/Gal-9/Tim3 Pathway
2.3. Chemerin/CMKLR1 Axis
2.4. MTA/A2B Receptor/M2 Pathway
2.5. HMGB1/RAGE/NF-κB/NLRP3/M1 Pathway
2.6. BACE1/IL-6R/SIL6R/IL-6/STAT3/M2 Pathway
2.7. The B2M/PIP5K1A/PI3K-AKT-MYC/TGF-β1/SMAD/M2 Pathway
2.8. GC/GR/FGF20/FGFR1/β-Catenin Pathway
2.9. The JMJD1C/mir-302a/H3K9/METTL3/SOCS2/M1 Pathway
2.10. The (Gal3BP/Gal)/CHI3L1/M2 Pathway
2.11. The MFG-E8/ITGB3/STAT3/M2 Pathway
2.12. The Exosome Pathway
2.13. Other Pathways
3. The Effect of M1 and M2 TAM on Glioma Progression
4. The Treatments for Gliomas
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Codrici, E.; Popescu, I.D.; Tanase, C.; Enciu, A.M. Friends with Benefits: Chemokines, Glioblastoma-Associated Microglia/Macrophages, and Tumor Microenvironment. Int. J. Mol. Sci. 2022, 23, 2509. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Lv, P.; Lou, Y. Alarm Signal S100-Related Signature Is Correlated with Tumor Microenvironment and Predicts Prognosis in Glioma. Dis. Markers 2022, 2022, 4968555. [Google Scholar] [CrossRef] [PubMed]
- Seyfrid, M.; Maich, W.T.; Shaikh, V.M.; Tatari, N.; Upreti, D.; Piyasena, D.; Subapanditha, M.; Savage, N.; McKenna, D.; Mikolajewicz, N.; et al. CD70 as an actionable immunotherapeutic target in recurrent glioblastoma and its microenvironment. J. Immunother. Cancer 2022, 10, e003289. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ping, Y.F.; Zhou, W.; He, Z.C.; Chen, C.; Bian, B.S.; Zhang, L.; Chen, L.; Lan, X.; Zhang, X.C.; et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 2017, 8, 15080. [Google Scholar] [CrossRef] [PubMed]
- Pancholi, S.; Tripathi, A.; Bhan, A.; Acharya, M.M.; Pillai, P. Emerging Concepts on the Role of Extracellular Vesicles and Its Cargo Contents in Glioblastoma-Microglial Crosstalk. Mol. Neurobiol. 2022, 59, 2822–2837. [Google Scholar] [CrossRef]
- Pan, Z.; Zhao, R.; Li, B.; Qi, Y.; Qiu, W.; Guo, Q.; Zhang, S.; Zhao, S.; Xu, H.; Li, M.; et al. EWSR1-induced circNEIL3 promotes glioma progression and exosome-mediated macrophage immunosuppressive polarization via stabilizing IGF2BP3. Mol. Cancer 2022, 21, 16. [Google Scholar] [CrossRef]
- Liu, T.; Zhu, C.; Chen, X.; Wu, J.; Guan, G.; Zou, C.; Shen, S.; Chen, L.; Cheng, P.; Cheng, W.; et al. Dual role of ARPC1B in regulating the network between tumor-associated macrophages and tumor cells in glioblastoma. Oncoimmunology 2022, 11, 2031499. [Google Scholar] [CrossRef]
- Chang, S.J.; Chao, C.T.; Kwan, A.L.; Chai, C.Y. The Diagnostic Significance of CXCL13 in M2 Tumor Immune Microenvironment of Human Astrocytoma. Pathol. Oncol. Res. 2022, 28, 1610230. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, S.; Liang, G.; Honggang, L.; Wu, H. Celastrol inhibits cancer metastasis by suppressing M2-like polarization of macrophages. Biochem. Biophys. Res. Commun. 2018, 503, 414–419. [Google Scholar] [CrossRef]
- Blitz, S.E.; Kappel, A.D.; Gessler, F.A.; Klinger, N.V.; Arnaout, O.; Lu, Y.; Peruzzi, P.P.; Smith, T.R.; Chiocca, E.A.; Friedman, G.K.; et al. Tumor-Associated Macrophages/Microglia in Glioblastoma Oncolytic Virotherapy: A Double-Edged Sword. Int. J. Mol. Sci. 2022, 23, 1808. [Google Scholar] [CrossRef]
- Kang, H.; Zhang, J.; Wang, B.; Liu, M.; Zhao, J.; Yang, M.; Li, Y. Puerarin inhibits M2 polarization and metastasis of tumor-associated macrophages from NSCLC xenograft model via inactivating MEK/ERK 1/2 pathway. Int. J. Oncol. 2017, 50, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Bianconi, A.; Aruta, G.; Rizzo, F.; Salvati, L.F.; Zeppa, P.; Garbossa, D.; Cofano, F. Systematic Review on Tumor Microenvironment in Glial Neoplasm: From Understanding Pathogenesis to Future Therapeutic Perspectives. Int. J. Mol. Sci. 2022, 23, 4166. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Jin, J.; Huang, X.; Wu, F.; Huang, H.; Zhan, R. EMP3 mediates glioblastoma-associated macrophage infiltration to drive T cell exclusion. J. Exp. Clin. Cancer Res. 2021, 40, 160. [Google Scholar] [CrossRef]
- Ma, W.; Zhang, K.; Bao, Z.; Jiang, T.; Zhang, Y. SAMD9 Is Relating With M2 Macrophage and Remarkable Malignancy Characters in Low-Grade Glioma. Front. Immunol. 2021, 12, 659659. [Google Scholar] [CrossRef]
- Chamseddine, A.N.; Assi, T.; Mir, O.; Chouaib, S. Modulating tumor-associated macrophages to enhance the efficacy of immune checkpoint inhibitors: A TAM-pting approach. Pharmacol. Ther. 2022, 231, 107986. [Google Scholar] [CrossRef]
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2021, 151, 3–12. [Google Scholar] [CrossRef]
- Lisi, L.; Stigliano, E.; Lauriola, L.; Navarra, P.; Dello Russo, C. Proinflammatory-activated glioma cells induce a switch in microglial polarization and activation status, from a predominant M2b phenotype to a mixture of M1 and M2a/B polarized cells. ASN Neuro 2014, 6, 171–183. [Google Scholar] [CrossRef]
- Arcuri, C.; Fioretti, B.; Bianchi, R.; Mecca, C.; Tubaro, C.; Beccari, T.; Franciolini, F.; Giambanco, I.; Donato, R. Microglia-glioma cross-talk: A two way approach to new strategies against glioma. Front. Biosci. (Landmark Ed.) 2017, 22, 268–309. [Google Scholar] [CrossRef]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.; Zhu, H.; Liu, D.; Chen, Z.; Zhang, X.; Guo, Z.; Dong, M.; Wan, L.; Zhang, P.; Liu, G.; et al. TGFBI secreted by tumor-associated macrophages promotes glioblastoma stem cell-driven tumor growth via integrin αvβ5-Src-Stat3 signaling. Theranostics 2022, 12, 4221–4236. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wang, Z.; Zhao, M.; Deng, Y.; Yang, M.; Su, G.; Yang, K.; Qian, C.; Hu, X.; Liu, Y.; et al. Single-Cell Transcriptomics Revealed Subtype-Specific Tumor Immune Microenvironments in Human Glioblastomas. Front. Immunol. 2022, 13, 914236. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, J.; Mi, W.; Zhang, Y.; Zhong, X.; Tan, G.; Li, F.; Li, X.; Xu, Y.; Zhang, Y. Comprehensive analysis of microglia gene and subpathway signatures for glioma prognosis and drug screening: Linking microglia to glioma. J. Transl. Med. 2022, 20, 277. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhao, J.; Xu, J.; Zhang, H.; Zhou, J.; Li, H.; Zhang, G.; Xu, K.; Jing, Z. Glioblastoma-associated microglia-derived exosomal circKIF18A promotes angiogenesis by targeting FOXC2. Oncogene 2022, 41, 3461–3473. [Google Scholar] [CrossRef]
- Zhang, F.; Cai, H.B.; Liu, H.Z.; Gao, S.; Wang, B.; Hu, Y.C.; Cheng, H.W.; Liu, J.X.; Gao, Y.; Hong, W.M. High Expression of CISD2 in Relation to Adverse Outcome and Abnormal Immune Cell Infiltration in Glioma. Dis. Markers 2022, 2022, 8133505. [Google Scholar] [CrossRef]
- Hansen, L.J.; Yang, R.; Roso, K.; Wang, W.; Chen, L.; Yang, Q.; Pirozzi, C.J.; He, Y. MTAP loss correlates with an immunosuppressive profile in GBM and its substrate MTA stimulates alternative macrophage polarization. Sci. Rep. 2022, 12, 4183. [Google Scholar] [CrossRef]
- Umemura, N.; Saio, M.; Suwa, T.; Kitoh, Y.; Bai, J.; Nonaka, K.; Ouyang, G.-F.; Okada, M.; Balazs, M.; Adany, R.; et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J. Leukoc. Biol. 2008, 83, 1136–1144. [Google Scholar] [CrossRef]
- Xia, Y.; Rao, L.; Yao, H.; Wang, Z.; Ning, P.; Chen, X. Engineering Macrophages for Cancer Immunotherapy and Drug Delivery. Adv. Mater. 2020, 32, e2002054. [Google Scholar] [CrossRef] [PubMed]
- Curtale, G.; Rubino, M.; Locati, M. MicroRNAs as Molecular Switches in Macrophage Activation. Front. Immunol. 2019, 10, 799. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shi, Y.; Wu, M.; Liu, J.; Wu, H.; Xu, C.; Chen, L. Hypoxia-induced polypoid giant cancer cells in glioma promote the transformation of tumor-associated macrophages to a tumor-supportive phenotype. CNS Neurosci. Ther. 2022, 28, 1326–1338. [Google Scholar] [CrossRef]
- Sanders, S.; Herpai, D.M.; Rodriguez, A.; Huang, Y.; Chou, J.; Hsu, F.-C.; Seals, D.; Mott, R.; Miller, L.D.; Debinski, W. The Presence and Potential Role of ALDH1A2 in the Glioblastoma Microenvironment. Cells 2021, 10, 2485. [Google Scholar] [CrossRef]
- Biswas, S.K.; Sica, A.; Lewis, C.E. Plasticity of macrophage function during tumor progression: Regulation by distinct molecular mechanisms. J. Immunol. 2008, 180, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, G.; Zhao, H. FDPS promotes glioma growth and macrophage recruitment by regulating CCL20 via Wnt/beta-catenin signalling pathway. J. Cell. Mol. Med. 2020, 24, 9055–9066. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Espinoza, F.I.; Walker, P.R. Untangling macrophage/microglia complexity in glioblastoma subtypes to elucidate the impact of CSF1R inhibition. Neuro Oncol. 2022, 24, 598–600. [Google Scholar] [CrossRef]
- Kim, A.R.; Choi, S.J.; Park, J.; Kwon, M.; Chowdhury, T.; Yu, H.J.; Kim, S.; Kang, H.; Kim, K.M.; Park, S.H.; et al. Spatial immune heterogeneity of hypoxia-induced exhausted features in high-grade glioma. Oncoimmunology 2022, 11, 2026019. [Google Scholar] [CrossRef]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Chen, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget 2016, 7, 80521–80542. [Google Scholar] [CrossRef] [PubMed]
- Kremsreiter, S.M.; Kroell, A.H.; Weinberger, K.; Boehm, H. Glycan-Lectin Interactions in Cancer and Viral Infections and How to Disrupt Them. Int. J. Mol. Sci. 2021, 22, 10577. [Google Scholar] [CrossRef] [PubMed]
- Pace, A.; Scirocchi, F.; Napoletano, C.; Zizzari, I.G.; D’Angelo, L.; Santoro, A.; Nuti, M.; Rahimi, H.; Rughetti, A. Glycan-Lectin Interactions as Novel Immunosuppression Drivers in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 6312. [Google Scholar] [CrossRef] [PubMed]
- RodrÍguez, E.; Schetters, S.T.T.; van Kooyk, Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat. Rev. Immunol. 2018, 18, 204–211. [Google Scholar] [CrossRef]
- Li, C.H.; Chang, Y.C.; Chan, M.H.; Yang, Y.F.; Liang, S.M.; Hsiao, M. Galectins in Cancer and the Microenvironment: Functional Roles, Therapeutic Developments, and Perspectives. Biomedicines 2021, 9, 1159. [Google Scholar] [CrossRef]
- Cedeno-Laurent, F.; Dimitroff, C.J. Galectins and their ligands: Negative regulators of anti-tumor immunity. Glycoconj. J. 2012, 29, 619–625. [Google Scholar] [CrossRef]
- Saussez, S.; Glinoer, D.; Chantrain, G.; Pattou, F.; Carnaille, B.; André, S.; Gabius, H.J.; Laurent, G. Serum galectin-1 and galectin-3 levels in benign and malignant nodular thyroid disease. Thyroid 2008, 18, 705–712. [Google Scholar] [CrossRef]
- Chen, Q.; Han, B.; Meng, X.; Duan, C.; Yang, C.; Wu, Z.; Magafurov, D.; Zhao, S.; Safin, S.; Jiang, C.; et al. Immunogenomic analysis reveals LGALS1 contributes to the immune heterogeneity and immunosuppression in glioma. Int. J. Cancer 2019, 145, 517–530. [Google Scholar] [CrossRef]
- Van Woensel, M.; Mathivet, T.; Wauthoz, N.; Rosiere, R.; Garg, A.D.; Agostinis, P.; Mathieu, V.; Kiss, R.; Lefranc, F.; Boon, L.; et al. Sensitization of glioblastoma tumor micro-environment to chemo- and immunotherapy by Galectin-1 intranasal knock-down strategy. Sci. Rep. 2017, 7, 1217. [Google Scholar] [CrossRef]
- Yuan, F.; Ming, H.; Wang, Y.; Yang, Y.; Yi, L.; Li, T.; Ma, H.; Tong, L.; Zhang, L.; Liu, P.; et al. Molecular and clinical characterization of Galectin-9 in glioma through 1,027 samples. J. Cell. Physiol. 2020, 235, 4326–4334. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Y.; He, Y.; Wang, X.; Fang, Q. Lipopolysaccharide mediates time-dependent macrophage M1/M2 polarization through the Tim-3/Galectin-9 signalling pathway. Exp. Cell Res. 2019, 376, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Oomizu, S.; Sakata, K.-m.; Sakata, A.; Arikawa, T.; Watanabe, K.; Ito, K.; Takeshita, K.; Niki, T.; Saita, N.; et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin. Immunol. 2008, 127, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- Zhou, F.; Shi, Q.; Fan, X.; Yu, R.; Wu, Z.; Wang, B.; Tian, W.; Yu, T.; Pan, M.; You, Y.; et al. Diverse Macrophages Constituted the Glioma Microenvironment and Influenced by PTEN Status. Front. Immunol. 2022, 13, 841404. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Pulido, R. PTEN: A yin-yang master regulator protein in health and disease. Methods 2015, 77–78, 3–10. [Google Scholar] [CrossRef]
- Ni, X.; Wu, W.; Sun, X.; Ma, J.; Yu, Z.; He, X.; Cheng, J.; Xu, P.; Liu, H.; Shang, T.; et al. Interrogating glioma-M2 macrophage interactions identifies Gal-9/Tim-3 as a viable target against PTEN-null glioblastoma. Sci. Adv. 2022, 8, eabl5165. [Google Scholar] [CrossRef]
- Treeck, O.; Buechler, C.; Ortmann, O. Chemerin and Cancer. Int. J. Mol. Sci. 2019, 20, 3750. [Google Scholar] [CrossRef]
- Rourke, J.L.; Dranse, H.J.; Sinal, C.J. Towards an integrative approach to understanding the role of chemerin in human health and disease. Obes. Rev. 2013, 14, 245–262. [Google Scholar] [CrossRef]
- Ben Dhaou, C.; Mandi, K.; Frye, M.; Acheampong, A.; Radi, A.; De Becker, B.; Antoine, M.; Baeyens, N.; Wittamer, V.; Parmentier, M. Chemerin regulates normal angiogenesis and hypoxia-driven neovascularization. Angiogenesis 2022, 25, 159–179. [Google Scholar] [CrossRef]
- Wu, J.; Shen, S.; Liu, T.; Ren, X.; Zhu, C.; Liang, Q.; Cui, X.; Chen, L.; Cheng, P.; Cheng, W.; et al. Chemerin enhances mesenchymal features of glioblastoma by establishing autocrine and paracrine networks in a CMKLR1-dependent manner. Oncogene 2022, 41, 3024–3036. [Google Scholar] [CrossRef]
- Sun, J.X.; Zhang, C.; Cheng, Z.B.; Tang, M.Y.; Liu, Y.Z.; Jiang, J.F.; Xiao, X.; Huang, L. Chemerin in atherosclerosis. Clin. Chim. Acta 2021, 520, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, T.; Huang, C.; Xu, M.; Xie, W.; Pei, Q.; Xie, X.; Wang, B.; Li, X. Chemerin located in bone marrow promotes osteogenic differentiation and bone formation via Akt/Gsk3beta/beta-catenin axis in mice. J. Cell. Physiol. 2021, 236, 6042–6054. [Google Scholar] [CrossRef] [PubMed]
- Helfer, G.; Wu, Q.F. Chemerin: A multifaceted adipokine involved in metabolic disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef] [PubMed]
- Rourke, J.L.; Dranse, H.J.; Sinal, C.J. CMKLR1 and GPR1 mediate chemerin signaling through the RhoA/ROCK pathway. Mol. Cell. Endocrinol. 2015, 417, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Goralski, K.B.; Jackson, A.E.; McKeown, B.T.; Sinal, C.J. More Than an Adipokine: The Complex Roles of Chemerin Signaling in Cancer. Int. J. Mol. Sci. 2019, 20, 4778. [Google Scholar] [CrossRef]
- Herová, M.; Schmid, M.; Gemperle, C.; Hersberger, M. ChemR23, the receptor for chemerin and resolvin E1, is expressed and functional on M1 but not on M2 macrophages. J. Immunol. 2015, 194, 2330–2337. [Google Scholar] [CrossRef]
- Dimitriadis, G.K.; Kaur, J.; Adya, R.; Miras, A.D.; Mattu, H.S.; Hattersley, J.G.; Kaltsas, G.; Tan, B.K.; Randeva, H.S. Chemerin induces endothelial cell inflammation: Activation of nuclear factor-kappa beta and monocyte-endothelial adhesion. Oncotarget 2018, 9, 16678–16690. [Google Scholar] [CrossRef]
- Wang, W.; Guo, J.; Wang, D. Promotion of chemerin in rat diabetic kidney disease through enhancement of TGF-β1/Smads/CTGF pathway. Am. J. Transl. Res. 2021, 13, 10206–10217. [Google Scholar]
- Shang, J.; Wang, L.; Zhang, Y.; Zhang, S.; Ning, L.; Zhao, J.; Cheng, G.; Liu, D.; Xiao, J.; Zhao, Z. Chemerin/ChemR23 axis promotes inflammation of glomerular endothelial cells in diabetic nephropathy. J. Cell. Mol. Med. 2019, 23, 3417–3428. [Google Scholar] [CrossRef]
- Ye, Y.; Fang, L.; Li, J.; Wu, H.; Tan, X.; Luo, H.; Li, X.; Huang, L. Chemerin/ChemR23 regulates cementoblast function and tooth resorption in mice via inflammatory factors. J. Periodontol. 2021, 92, 1470–1482. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef]
- Barekatain, Y.; Ackroyd, J.J.; Yan, V.C.; Khadka, S.; Wang, L.; Chen, K.C.; Poral, A.H.; Tran, T.; Georgiou, D.K.; Arthur, K.; et al. Homozygous MTAP deletion in primary human glioblastoma is not associated with elevation of methylthioadenosine. Nat. Commun. 2021, 12, 4228. [Google Scholar] [CrossRef]
- Moreno, B.; Hevia, H.; Santamaria, M.; Sepulcre, J.; Muñoz, J.; García-Trevijano, E.R.; Berasain, C.; Corrales, F.J.; Avila, M.A.; Villoslada, P. Methylthioadenosine reverses brain autoimmune disease. Ann. Neurol. 2006, 60, 323–334. [Google Scholar] [CrossRef]
- Yan, A.; Joachims, M.L.; Thompson, L.F.; Miller, A.D.; Canoll, P.D.; Bynoe, M.S. CD73 Promotes Glioblastoma Pathogenesis and Enhances Its Chemoresistance via A(2B) Adenosine Receptor Signaling. J. Neurosci. 2019, 39, 4387–4402. [Google Scholar] [CrossRef] [PubMed]
- Kitabatake, K.; Kaji, T.; Tsukimoto, M. Involvement of CD73 and A2B Receptor in Radiation-Induced DNA Damage Response and Cell Migration in Human Glioblastoma A172 Cells. Biol. Pharm. Bull. 2021, 44, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Yerneni, S.S.; Azambuja, J.H.; Gillespie, D.G.; Menshikova, E.V.; Jackson, E.K.; Whiteside, T.L. Tumor-derived exosomes promote angiogenesis via adenosine A(2B) receptor signaling. Angiogenesis 2020, 23, 599–610. [Google Scholar] [CrossRef]
- Ito, T.; Kaneko, T.; Yamanaka, Y.; Shigetani, Y.; Yoshiba, K.; Okiji, T. M2 macrophages participate in the biological tissue healing reaction to mineral trioxide aggregate. J. Endod. 2014, 40, 379–383. [Google Scholar] [CrossRef]
- Takei, E.; Shigetani, Y.; Yoshiba, K.; Hinata, G.; Yoshiba, N.; Okiji, T. Initial transient accumulation of M2 macrophage-associated molecule-expressing cells after pulpotomy with mineral trioxide aggregate in rat molars. J. Endod. 2014, 40, 1983–1988. [Google Scholar] [CrossRef]
- Li, Z.; Fu, W.-J.; Chen, X.-Q.; Wang, S.; Deng, R.-S.; Tang, X.-P.; Yang, K.-D.; Niu, Q.; Zhou, H.; Li, Q.-R.; et al. Autophagy-based unconventional secretion of HMGB1 in glioblastoma promotes chemosensitivity to temozolomide through macrophage M1-like polarization. J. Exp. Clin. Cancer Res. 2022, 41, 74. [Google Scholar] [CrossRef]
- Bassi, R.; Giussani, P.; Anelli, V.; Colleoni, T.; Pedrazzi, M.; Patrone, M.; Viani, P.; Sparatore, B.; Melloni, E.; Riboni, L. HMGB1 as an autocrine stimulus in human T98G glioblastoma cells: Role in cell growth and migration. J. Neurooncol. 2008, 87, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhang, Y.; Jiang, Y.; Wang, K.; Wang, X.; Zhou, D.; Wang, Y.; Yu, R.; Zhou, X. YAP promotes autophagy and progression of gliomas via upregulating HMGB1. J. Exp. Clin. Cancer Res. 2021, 40, 99. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Shrinet, K.; Kumar, A. HMGB1 protein as a novel target for cancer. Toxicol. Rep. 2019, 6, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Wang, S.; Shen, H.; Lin, J. Inhibitory Effect of miR-339-5p on Glioma through PTP4A1/HMGB1 Pathway. Dis. Markers 2022, 2022, 2231195. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Yan, Y.; Ma, C.; Chen, H.; Dong, Z.; Wang, Y.; Liu, Y.; Liu, M.; Yang, L. HMGB1 contributes to SASH1 methylation to attenuate astrocyte adhesion. Cell Death Dis. 2019, 10, 417. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, J.; Zhang, G.; Da, Q.; Chen, L.; Yu, S.; Zhou, Q.; Weng, Z.; Xin, Z.; Shi, L.; et al. HMGB1-Induced p62 Overexpression Promotes Snail-Mediated Epithelial-Mesenchymal Transition in Glioblastoma Cells via the Degradation of GSK-3beta. Theranostics 2019, 9, 1909–1922. [Google Scholar] [CrossRef]
- Curtin, J.F.; Liu, N.; Candolfi, M.; Xiong, W.; Assi, H.; Yagiz, K.; Edwards, M.R.; Michelsen, K.S.; Kroeger, K.M.; Liu, C.; et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009, 6, e10. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zhang, Q.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. HMGB1 in cancer: Good, bad, or both? Clin. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef]
- Zha, C.; Meng, X.; Li, L.; Mi, S.; Qian, D.; Li, Z.; Wu, P.; Hu, S.; Zhao, S.; Cai, J.; et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol. Med. 2020, 17, 154–168. [Google Scholar] [CrossRef]
- Zhang, I.Y.; Zhou, H.; Liu, H.; Zhang, L.; Gao, H.; Liu, S.; Song, Y.; Alizadeh, D.; Yin, H.H.; Pillai, R.; et al. Local and Systemic Immune Dysregulation Alters Glioma Growth in Hyperglycemic Mice. Clin. Cancer Res. 2020, 26, 2740–2753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, W.; Alizadeh, D.; Zhao, D.; Farrukh, O.; Lin, J.; Badie, S.A.; Badie, B. S100B attenuates microglia activation in gliomas: Possible role of STAT3 pathway. Glia 2011, 59, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Zhai, K.; Huang, Z.; Huang, Q.; Tao, W.; Fang, X.; Zhang, A.; Li, X.; Stark, G.R.; Hamilton, T.A.; Bao, S. Pharmacological inhibition of BACE1 suppresses glioblastoma growth by stimulating macrophage phagocytosis of tumor cells. Nat. Cancer 2021, 2, 1136–1151. [Google Scholar] [CrossRef] [PubMed]
- Farris, F.; Matafora, V.; Bachi, A. The emerging role of β-secretases in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 147. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Munzner, J.B.; Shuck, M.E.; Bienkowski, M.J. BACE2 functions as an alternative alpha-secretase in cells. J. Biol. Chem. 2001, 276, 34019–34027. [Google Scholar] [CrossRef]
- Yan, R.; Vassar, R. Targeting the beta secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319–329. [Google Scholar] [CrossRef]
- Marwarha, G.; Raza, S.; Meiers, C.; Ghribi, O. Leptin attenuates BACE1 expression and amyloid-beta genesis via the activation of SIRT1 signaling pathway. Biochim. Biophys. Acta 2014, 1842, 1587–1595. [Google Scholar] [CrossRef]
- Liu, C.; Wang, H.; Tang, L.; Huang, H.; Xu, M.; Lin, Y.; Zhou, L.; Ho, L.; Lu, J.; Ai, X. LncRNA BACE1-AS enhances the invasive and metastatic capacity of hepatocellular carcinoma cells through mediating miR-377-3p/CELF1 axis. Life Sci. 2021, 275, 119288. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, T.; Yang, L.; Na, X.; Qu, Y. LncRNA BACE1-AS promotes the progression of osteosarcoma through miR-762/SOX7 axis. Mol. Biol. Rep. 2022, 49, 5853–5862. [Google Scholar] [CrossRef]
- Peters, M.; Jacobs, S.; Ehlers, M.; Vollmer, P.; Müllberg, J.; Wolf, E.; Brem, G.; Meyer zum Büschenfelde, K.H.; Rose-John, S. The function of the soluble interleukin 6 (IL-6) receptor in vivo: Sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. J. Exp. Med. 1996, 183, 1399–1406. [Google Scholar] [CrossRef]
- Arosa, F.A.; Santos, S.G.; Powis, S.J. Open conformers: The hidden face of MHC-I molecules. Trends Immunol. 2007, 28, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [PubMed]
- Abele, R.; Tampé, R. The ABCs of immunology: Structure and function of TAP, the transporter associated with antigen processing. Physiology 2004, 19, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Zhao, Y.H.; Zhang, Q.; Wei, W.; Tian, S.F.; Li, C.; Yao, J.; Wang, Z.F.; Li, Z.Q. Impact of beta-2 microglobulin expression on the survival of glioma patients via modulating the tumor immune microenvironment. CNS Neurosci. Ther. 2021, 27, 951–962. [Google Scholar] [CrossRef]
- Sarwar, M.; Syed Khaja, A.S.; Aleskandarany, M.; Karlsson, R.; Althobiti, M.; Ødum, N.; Mongan, N.P.; Dizeyi, N.; Johnson, H.; Green, A.R.; et al. The role of PIP5K1α/pAKT and targeted inhibition of growth of subtypes of breast cancer using PIP5K1α inhibitor. Oncogene 2019, 38, 375–389. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Q.; Li, L.; Chen, K.; Yang, J.; Dixit, D.; Gimple, R.C.; Ci, S.; Lu, C.; Hu, L.; et al. beta2-Microglobulin Maintains Glioblastoma Stem Cells and Induces M2-like Polarization of Tumor-Associated Macrophages. Cancer Res. 2022, 82, 3321–3334. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Wang, Y. The role of PIP5K1A in cancer development and progression. Med. Oncol. 2022, 39, 151. [Google Scholar] [CrossRef]
- Peng, J.M.; Lin, S.H.; Yu, M.C.; Hsieh, S.Y. CLIC1 recruits PIP5K1A/C to induce cell-matrix adhesions for tumor metastasis. J. Clin. Investig. 2021, 131, e133525. [Google Scholar] [CrossRef] [PubMed]
- Feng, N.; Guo, Z.; Wu, X.; Tian, Y.; Li, Y.; Geng, Y.; Yu, Y. Circ_PIP5K1A regulates cisplatin resistance and malignant progression in non-small cell lung cancer cells and xenograft murine model via depending on miR-493-5p/ROCK1 axis. Respir. Res. 2021, 22, 248. [Google Scholar] [CrossRef]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.J.; Perry, S.R.; Tonon, G.; Chu, G.C.; Ding, Z.; et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008, 455, 1129–1133. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Li, Z.; Wu, Q.; Lathia, J.D.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. c-Myc is required for maintenance of glioma cancer stem cells. PLoS ONE 2008, 3, e3769. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. FGF signaling network in the gastrointestinal tract (review). Int. J. Oncol. 2006, 29, 163–168. [Google Scholar] [CrossRef]
- Chamorro, M.N.; Schwartz, D.R.; Vonica, A.; Brivanlou, A.H.; Cho, K.R.; Varmus, H.E. FGF-20 and DKK1 are transcriptional targets of beta-catenin and FGF-20 is implicated in cancer and development. EMBO J. 2005, 24, 73–84. [Google Scholar] [CrossRef]
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013, 14, 166–180. [Google Scholar] [CrossRef]
- Yang, Y.; Ye, Y.C.; Chen, Y.; Zhao, J.L.; Gao, C.C.; Han, H.; Liu, W.C.; Qin, H.Y. Crosstalk between hepatic tumor cells and macrophages via Wnt/beta-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018, 9, 793. [Google Scholar] [CrossRef]
- Deng, F.; Yan, J.; Lu, J.; Luo, M.; Xia, P.; Liu, S.; Wang, X.; Zhi, F.; Liu, D. M2 Macrophage-Derived Exosomal miR-590-3p Attenuates DSS-Induced Mucosal Damage and Promotes Epithelial Repair via the LATS1/YAP/ beta-Catenin Signalling Axis. J. Crohn’s Colitis 2021, 15, 665–677. [Google Scholar] [CrossRef]
- Cosin-Roger, J.; Ortiz-Masià, M.D.; Barrachina, M.D. Macrophages as an Emerging Source of Wnt Ligands: Relevance in Mucosal Integrity. Front. Immunol. 2019, 10, 2297. [Google Scholar] [CrossRef]
- Zhao, S.J.; Kong, F.Q.; Jie, J.; Li, Q.; Liu, H.; Xu, A.D.; Yang, Y.Q.; Jiang, B.; Wang, D.D.; Zhou, Z.Q.; et al. Macrophage MSR1 promotes BMSC osteogenic differentiation and M2-like polarization by activating PI3K/AKT/GSK3beta/beta-catenin pathway. Theranostics 2020, 10, 17–35. [Google Scholar] [CrossRef]
- Sarode, P.; Zheng, X.; Giotopoulou, G.A.; Weigert, A.; Kuenne, C.; Günther, S.; Friedrich, A.; Gattenlöhner, S.; Stiewe, T.; Brüne, B.; et al. Reprogramming of tumor-associated macrophages by targeting β-catenin/FOSL2/ARID5A signaling: A potential treatment of lung cancer. Sci. Adv. 2020, 6, eaaz6105. [Google Scholar] [CrossRef] [PubMed]
- Matias, D.; Dubois, L.G.; Pontes, B.; Rosario, L.; Ferrer, V.P.; Balca-Silva, J.; Fonseca, A.C.C.; Macharia, L.W.; Romao, L.; TCLS, E.S.; et al. GBM-Derived Wnt3a Induces M2-Like Phenotype in Microglial Cells Through Wnt/beta-Catenin Signaling. Mol. Neurobiol. 2019, 56, 1517–1530. [Google Scholar] [CrossRef]
- Cai, X.; Tao, W.; Li, L. Glioma cell-derived FGF20 suppresses macrophage function by activating beta-catenin. Cell. Signal. 2022, 89, 110181. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Lee, J.H.; Jiang, H.; Wang, C.; Wang, S.; Zheng, Z.; Shao, F.; Xu, D.; Xia, Y.; Li, J.; et al. β-Catenin induces transcriptional expression of PD-L1 to promote glioblastoma immune evasion. J. Exp. Med. 2020, 217, e20191115. [Google Scholar] [CrossRef]
- Osuka, S.; Zhu, D.; Zhang, Z.; Li, C.; Stackhouse, C.T.; Sampetrean, O.; Olson, J.J.; Gillespie, G.Y.; Saya, H.; Willey, C.D.; et al. N-cadherin upregulation mediates adaptive radioresistance in glioblastoma. J. Clin. Investig. 2021, 131, e136098. [Google Scholar] [CrossRef]
- Vallee, A.; Guillevin, R.; Vallee, J.N. Vasculogenesis and angiogenesis initiation under normoxic conditions through Wnt/beta-catenin pathway in gliomas. Rev. Neurosci. 2018, 29, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Kim, S.S.; Choi, E.; Oh, Y.T.; Lin, W.; Kim, T.H.; Sa, J.K.; Hong, J.H.; Park, S.H.; Kwon, H.J.; et al. ARS2/MAGL signaling in glioblastoma stem cells promotes self-renewal and M2-like polarization of tumor-associated macrophages. Nat. Commun. 2020, 11, 2978. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bogler, O.; et al. m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.e6. [Google Scholar] [CrossRef]
- Das Gupta, K.; Shakespear, M.R.; Iyer, A.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases in monocyte/macrophage development, activation and metabolism: Refining HDAC targets for inflammatory and infectious diseases. Clin. Transl. Immunol. 2016, 5, e62. [Google Scholar] [CrossRef]
- Ishii, M.; Wen, H.; Corsa, C.A.; Liu, T.; Coelho, A.L.; Allen, R.M.; Carson, W.F.t.; Cavassani, K.A.; Li, X.; Lukacs, N.W.; et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009, 114, 3244–3254. [Google Scholar] [CrossRef]
- Satoh, T.; Takeuchi, O.; Vandenbon, A.; Yasuda, K.; Tanaka, Y.; Kumagai, Y.; Miyake, T.; Matsushita, K.; Okazaki, T.; Saitoh, T.; et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010, 11, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Park, J.W.; Drissi, H.; Wang, X.; Xu, R.H. Epigenetic regulation of miR-302 by JMJD1C inhibits neural differentiation of human embryonic stem cells. J. Biol. Chem. 2014, 289, 2384–2395. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Yang, X.; Wu, S.; Ding, X.; Zhu, H.; Long, X.; Wang, Y.; Zhai, S.; Chen, Y.; Che, N.; et al. Jmjd1c demethylates STAT3 to restrain plasma cell differentiation and rheumatoid arthritis. Nat. Immunol. 2022, 23, 1342–1354. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Chen, M.; Eng, R.; DeJong, J.; Sinha, A.U.; Rahnamay, N.F.; Koche, R.; Al-Shahrour, F.; Minehart, J.C.; Chen, C.W.; et al. MLL-AF9- and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J. Clin. Investig. 2016, 126, 997–1011. [Google Scholar] [CrossRef]
- Izaguirre-Carbonell, J.; Christiansen, L.; Burns, R.; Schmitz, J.; Li, C.; Mokry, R.L.; Bluemn, T.; Zheng, Y.; Shen, J.; Carlson, K.S.; et al. Critical role of Jumonji domain of JMJD1C in MLL-rearranged leukemia. Blood Adv. 2019, 3, 1499–1511. [Google Scholar] [CrossRef]
- Xu, X.; Wang, L.; Hu, L.; Dirks, W.G.; Zhao, Y.; Wei, Z.; Chen, D.; Li, Z.; Wang, Z.; Han, Y.; et al. Small molecular modulators of JMJD1C preferentially inhibit growth of leukemia cells. Int. J. Cancer 2020, 146, 400–412. [Google Scholar] [CrossRef]
- Yoshihama, Y.; LaBella, K.A.; Kim, E.; Bertolet, L.; Colic, M.; Li, J.; Shang, X.; Wu, C.J.; Spring, D.J.; Wang, Y.A.; et al. AR-negative prostate cancer is vulnerable to loss of JMJD1C demethylase. Proc. Natl. Acad. Sci. USA 2021, 118, e2026324118. [Google Scholar] [CrossRef]
- Yu, S.; Li, Y.; Zhao, H.; Wang, Q.; Chen, P. The Histone Demethylase JMJD1C Regulates CAMKK2-AMPK Signaling to Participate in Cardiac Hypertrophy. Front. Physiol. 2020, 11, 539. [Google Scholar] [CrossRef]
- Zhong, C.; Tao, B.; Yang, F.; Xia, K.; Yang, X.; Chen, L.; Peng, T.; Xia, X.; Li, X.; Peng, L. Histone demethylase JMJD1C promotes the polarization of M1 macrophages to prevent glioma by upregulating miR-302a. Clin. Transl. Med. 2021, 11, e424. [Google Scholar] [CrossRef]
- Chai, R.-C.; Chang, Y.-Z.; Chang, X.; Pang, B.; An, S.Y.; Zhang, K.-N.; Chang, Y.-H.; Jiang, T.; Wang, Y.-Z. YTHDF2 facilitates UBXN1 mRNA decay by recognizing METTL3-mediated m6A modification to activate NF-κB and promote the malignant progression of glioma. J. Hematol. Oncol. 2021, 14, 109. [Google Scholar] [CrossRef]
- Tassinari, V.; Cesarini, V.; Tomaselli, S.; Ianniello, Z.; Silvestris, D.A.; Ginistrelli, L.C.; Martini, M.; De Angelis, B.; De Luca, G.; Vitiani, L.R.; et al. ADAR1 is a new target of METTL3 and plays a pro-oncogenic role in glioblastoma by an editing-independent mechanism. Genome Biol. 2021, 22, 51. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.G.; et al. m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef]
- Han, J.; Du, S.; Wu, C.; Qiu, M.; Su, L.; Zhao, Z.; Cheng, S.; Tao, W. METTL3 participates in glioma development by regulating the methylation level of COL4A1. J. BUON 2021, 26, 1556–1562. [Google Scholar] [PubMed]
- Martini, M.; Pallini, R.; Luongo, G.; Cenci, T.; Lucantoni, C.; Larocca, L.M. Prognostic relevance of SOCS3 hypermethylation in patients with glioblastoma multiforme. Int. J. Cancer 2008, 123, 2955–2960. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Huang, C.; Ma, T.T.; Bian, E.B.; He, Y.; Zhang, L.; Li, J. SOCS1 hypermethylation mediated by DNMT1 is associated with lipopolysaccharide-induced inflammatory cytokines in macrophages. Toxicol. Lett. 2014, 225, 488–497. [Google Scholar] [CrossRef]
- Zhou, H.; Miki, R.; Eeva, M.; Fike, F.M.; Seligson, D.; Yang, L.; Yoshimura, A.; Teitell, M.A.; Jamieson, C.A.; Cacalano, N.A. Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin. Cancer Res. 2007, 13, 2344–2353. [Google Scholar] [CrossRef]
- Shervington, A.; Patel, R. Silencing DNA methyltransferase (DNMT) enhances glioma chemosensitivity. Oligonucleotides 2008, 18, 365–374. [Google Scholar] [CrossRef]
- Rajendran, G.; Shanmuganandam, K.; Bendre, A.; Muzumdar, D.; Goel, A.; Shiras, A. Epigenetic regulation of DNA methyltransferases: DNMT1 and DNMT3B in gliomas. J. Neurooncol. 2011, 104, 483–494. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, J.H.; Chie, E.K.; Young, P.D.; Kim, I.A.; Kim, I.H. DNMT (DNA methyltransferase) inhibitors radiosensitize human cancer cells by suppressing DNA repair activity. Radiat. Oncol. 2012, 7, 39. [Google Scholar] [CrossRef]
- Cheray, M.; Pacaud, R.; Nadaradjane, A.; Oliver, L.; Vallette, F.M.; Cartron, P.F. Specific Inhibition of DNMT3A/ISGF3gamma Interaction Increases the Temozolomide Efficiency to Reduce Tumor Growth. Theranostics 2016, 6, 1988–1999. [Google Scholar] [CrossRef]
- Heidari, A.; Sharif, P.M.; Rezaei, N. The Association between Tumor-associated Macrophages and Glioblastoma: A Potential Target for Therapy. Curr. Pharm. Des. 2021, 27, 4650–4662. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Liu, D.; Yu, L.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574. [Google Scholar] [CrossRef]
- Zhao, T.; Zeng, J.; Xu, Y.; Su, Z.; Chong, Y.; Ling, T.; Xu, H.; Shi, H.; Zhu, M.; Mo, Q.; et al. Chitinase-3 like-protein-1 promotes glioma progression via the NF-kappaB signaling pathway and tumor microenvironment reprogramming. Theranostics 2022, 12, 6989–7008. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Da Silva, C.A.; Dela Cruz, C.S.; Ahangari, F.; Ma, B.; Kang, M.J.; He, C.H.; Takyar, S.; Elias, J.A. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu. Rev. Physiol. 2011, 73, 479–501. [Google Scholar] [CrossRef]
- Zhao, T.; Su, Z.; Li, Y.; Zhang, X.; You, Q. Chitinase-3 like-protein-1 function and its role in diseases. Signal Transduct. Target. Ther. 2020, 5, 201. [Google Scholar] [CrossRef] [PubMed]
- Steponaitis, G.; Skiriute, D.; Kazlauskas, A.; Golubickaite, I.; Stakaitis, R.; Tamasauskas, A.; Vaitkiene, P. High CHI3L1 expression is associated with glioma patient survival. Diagn. Pathol. 2016, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.M.; Lee, Y.K.; Ryu, J.; Jeong, J.Y.; Choi, J.; Eun, K.M.; Shin, H.Y.; Kim, D.G.; Hwang, E.M.; Yoo, J.C.; et al. CHI3L1 (YKL-40) is expressed in human gliomas and regulates the invasion, growth and survival of glioma cells. Int. J. Cancer 2011, 128, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Jiang, Y.; Li, Z.; Wu, L.; Santiago, U.; Zou, H.; Cai, C.; Sharma, V.; Guan, Y.; McCarl, L.H.; et al. Chitinase-3-like 1 protein complexes modulate macrophage-mediated immune suppression in glioblastoma. J. Clin. Investig. 2021, 131, e147552. [Google Scholar] [CrossRef]
- Aziz, M.; Jacob, A.; Matsuda, A.; Wang, P. Review: Milk fat globule-EGF factor 8 expression, function and plausible signal transduction in resolving inflammation. Apoptosis 2011, 16, 1077–1086. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, L.; Pan, J.; Luo, B.; Zeng, H.; Shao, Y.; Zhang, H.; Guan, H.; Guo, D.; Zeng, C.; et al. MFG-E8 regulated by miR-99b-5p protects against osteoarthritis by targeting chondrocyte senescence and macrophage reprogramming via the NF-kappaB pathway. Cell Death Dis. 2021, 12, 533. [Google Scholar] [CrossRef]
- Yamada, K.; Uchiyama, A.; Uehara, A.; Perera, B.; Ogino, S.; Yokoyama, Y.; Takeuchi, Y.; Udey, M.C.; Ishikawa, O.; Motegi, S. MFG-E8 Drives Melanoma Growth by Stimulating Mesenchymal Stromal Cell-Induced Angiogenesis and M2 Polarization of Tumor-Associated Macrophages. Cancer Res. 2016, 76, 4283–4292. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhao, L.; Jia, Z.; Bi, J.; Wei, Q.; Song, X.; Jiang, L.; Lin, S.; Wei, M. MFG-E8 overexpression is associated with poor prognosis in breast cancer patients. Pathol. Res. Pract. 2019, 215, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Xu, L.; Sun, X.; Zhang, K.; Shen, H.; Tian, Y.; Sun, F.; Li, Y. MFG-E8 overexpression promotes colorectal cancer progression via AKT/MMPs signalling. Tumour Biol. 2017, 39, 1010428317707881. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Hanayama, R.; Kawane, K. Autoimmunity and the clearance of dead cells. Cell 2010, 140, 619–630. [Google Scholar] [CrossRef]
- Soki, F.N.; Koh, A.J.; Jones, J.D.; Kim, Y.W.; Dai, J.; Keller, E.T.; Pienta, K.J.; Atabai, K.; Roca, H.; McCauley, L.K. Polarization of prostate cancer-associated macrophages is induced by milk fat globule-EGF factor 8 (MFG-E8)-mediated efferocytosis. J. Biol. Chem. 2014, 289, 24560–24572. [Google Scholar] [CrossRef]
- Goldberg, G.S.; Bechberger, J.F.; Tajima, Y.; Merritt, M.; Omori, Y.; Gawinowicz, M.A.; Narayanan, R.; Tan, Y.; Sanai, Y.; Yamasaki, H.; et al. Connexin43 suppresses MFG-E8 while inducing contact growth inhibition of glioma cells. Cancer Res. 2000, 60, 6018–6026. [Google Scholar]
- Wu, J.; Yang, H.; Cheng, J.; Zhang, L.; Ke, Y.; Zhu, Y.; Wang, C.; Zhang, X.; Zhen, X.; Zheng, L.T. Knockdown of milk-fat globule EGF factor-8 suppresses glioma progression in GL261 glioma cells by repressing microglial M2 polarization. J. Cell. Physiol. 2020, 235, 8679–8690. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, Y.; Li, H.; Xu, L.; You, H.; Liu, Y.; Liu, Z.; Liu, X.; Zheng, D.; Bie, J.; et al. Exosomal microRNAs induce tumor-associated macrophages via PPARgamma during tumor progression in SHH medulloblastoma. Cancer Lett. 2022, 535, 215630. [Google Scholar] [CrossRef]
- Simon, T.; Jackson, E.; Giamas, G. Breaking through the glioblastoma micro-environment via extracellular vesicles. Oncogene 2020, 39, 4477–4490. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef]
- Dou, X.; Hua, Y.; Chen, Z.; Chao, F.; Li, M. Extracellular vesicles containing PD-L1 contribute to CD8+ T-cell immune suppression and predict poor outcomes in small cell lung cancer. Clin. Exp. Immunol. 2022, 207, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Wang, S.; Guo, X.; Wang, J.; Zhang, Z.; Qiu, W.; Gao, X.; Chen, Z.; Xu, J.; Zhao, R.; et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-kappaB pathways. Oncogene 2020, 39, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; You, J.Y.; Paek, K.; Park, J.; Kang, S.J.; Han, E.H.; Choi, N.; Chung, S.; Rhee, W.J.; Kim, J.A. Inhibition of tumor progression and M2 microglial polarization by extracellular vesicle-mediated microRNA-124 in a 3D microfluidic glioblastoma microenvironment. Theranostics 2021, 11, 9687–9704. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Nunez, R.T.; Louafi, F.; Sanchez-Elsner, T. The Interleukin 13 (IL-13) Pathway in Human Macrophages Is Modulated by MicroRNA-155 via Direct Targeting of Interleukin 13 Receptor α1 (IL13Rα1). J. Biol. Chem. 2011, 286, 1786–1794. [Google Scholar] [CrossRef]
- Ying, H.; Kang, Y.; Zhang, H.; Zhao, D.; Xia, J.; Lu, Z.; Wang, H.; Xu, F.; Shi, L. MiR-127 modulates macrophage polarization and promotes lung inflammation and injury by activating the JNK pathway. J. Immunol. 2015, 194, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Li, X. MicroRNA-32 targeting PTEN enhances M2 macrophage polarization in the glioma microenvironment and further promotes the progression of glioma. Mol. Cell. Biochem. 2019, 460, 67–79. [Google Scholar] [CrossRef]
- Xu, S.; Wei, J.; Wang, F.; Kong, L.Y.; Ling, X.Y.; Nduom, E.; Gabrusiewicz, K.; Doucette, T.; Yang, Y.; Yaghi, N.K.; et al. Effect of miR-142-3p on the M2 macrophage and therapeutic efficacy against murine glioblastoma. J. Natl. Cancer Inst. 2014, 106, dju162. [Google Scholar] [CrossRef]
- Bier, A.; Hong, X.; Cazacu, S.; Goldstein, H.; Rand, D.; Xiang, C.; Jiang, W.; Ben-Asher, H.W.; Attia, M.; Brodie, A.; et al. miR-504 modulates the stemness and mesenchymal transition of glioma stem cells and their interaction with microglia via delivery by extracellular vesicles. Cell Death Dis. 2020, 11, 899. [Google Scholar] [CrossRef]
- Xu, Y.; Liao, C.; Liu, R.; Liu, J.; Chen, Z.; Zhao, H.; Li, Z.; Chen, L.; Wu, C.; Tan, H.; et al. IRGM promotes glioma M2 macrophage polarization through p62/TRAF6/NF-κB pathway mediated IL-8 production. Cell Biol. Int. 2019, 43, 125–135. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, W.; Sun, X.; Dang, R.; Zhou, R.; Bai, H.; Ben, J.; Zhu, X.; Zhang, Y.; Yang, Q.; et al. Class A1 scavenger receptor modulates glioma progression by regulating M2-like tumor-associated macrophage polarization. Oncotarget 2016, 7, 50099–50116. [Google Scholar] [CrossRef]
- Kreatsoulas, D.; Bolyard, C.; Wu, B.X.; Cam, H.; Giglio, P.; Li, Z. Translational landscape of glioblastoma immunotherapy for physicians: Guiding clinical practice with basic scientific evidence. J. Hematol. Oncol. 2022, 15, 80. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Sun, F.; Zhao, P.; Liang, G.; Sun, X.; Zeng, L.; Huang, Y. Brain-targeting biomimetic nanoparticles for codelivery of celastrol and LY2157299 for reversing glioma immunosuppression. Int. J. Pharm. 2022, 619, 121709. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Moustakas, A. Role of Smads in TGFbeta signaling. Cell Tissue Res. 2012, 347, 21–36. [Google Scholar] [CrossRef]
- Hu, Z.; Qu, S. EVA1C Is a Potential Prognostic Biomarker and Correlated With Immune Infiltration Levels in WHO Grade II/III Glioma. Front. Immunol. 2021, 12, 683572. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Frei, K.; Gramatzki, D.; Tritschler, I.; Schroeder, J.J.; Espinoza, L.; Rushing, E.J.; Weller, M. Transforming growth factor-β pathway activity in glioblastoma. Oncotarget 2015, 6, 5963–5977. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Liu, Z.; Kuang, W.; Zhou, Q.; Zhang, Y. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int. J. Mol. Med. 2018, 42, 3395–3403. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. The regulation of TGFbeta signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef]
- Thapa, N.; Lee, B.H.; Kim, I.S. TGFBIp/betaig-h3 protein: A versatile matrix molecule induced by TGF-beta. Int. J. Biochem. Cell Biol. 2007, 39, 2183–2194. [Google Scholar] [CrossRef]
- Lang, K.; Kahveci, S.; Bonberg, N.; Wichert, K.; Behrens, T.; Hovanec, J.; Roghmann, F.; Noldus, J.; Tam, Y.C.; Tannapfel, A.; et al. TGFBI Protein Is Increased in the Urine of Patients with High-Grade Urothelial Carcinomas, and Promotes Cell Proliferation and Migration. Int. J. Mol. Sci. 2019, 20, 4483. [Google Scholar] [CrossRef] [PubMed]
- Ween, M.P.; Oehler, M.K.; Ricciardelli, C. Transforming growth Factor-Beta-Induced Protein (TGFBI)/(betaig-H3): A matrix protein with dual functions in ovarian cancer. Int. J. Mol. Sci. 2012, 13, 10461–10477. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Cai, H.; Chen, Y.; Hu, B.; Luo, H.; Wu, Y.; Wu, J. The role of TGFBI (betaig-H3) in gastrointestinal tract tumorigenesis. Mol. Cancer 2015, 14, 64. [Google Scholar] [CrossRef]
- Tumbarello, D.A.; Temple, J.; Brenton, J.D. ß3 integrin modulates transforming growth factor beta induced (TGFBI) function and paclitaxel response in ovarian cancer cells. Mol. Cancer 2012, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef]
- Bournazou, E.; Bromberg, J. Targeting the tumor microenvironment: JAK-STAT3 signaling. JAKSTAT 2013, 2, e23828. [Google Scholar] [CrossRef]
- Chaudhary, R.; Morris, R.J.; Steinson, E. The multifactorial roles of microglia and macrophages in the maintenance and progression of glioblastoma. J. Neuroimmunol. 2021, 357, 577633. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Kros, J.M.; Cheng, C.; Mustafa, D. The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro Oncol. 2017, 19, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, X.; Zhang, Y.; Wang, H.; Rong, X.; Peng, J.; He, L.; Peng, Y. An miR-340-5p-macrophage feedback loop modulates the progression and tumor microenvironment of glioblastoma multiforme. Oncogene 2019, 38, 7399–7415. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Chu, C.; Zhou, W.; Huang, Z.; Zhai, K.; Fang, X.; Huang, Q.; Zhang, A.; Wang, X.; Yu, X.; et al. Dual Role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat. Commun. 2020, 11, 3015. [Google Scholar] [CrossRef]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef]
- Perrotta, C.; Cervia, D.; Di Renzo, I.; Moscheni, C.; Bassi, M.T.; Campana, L.; Martelli, C.; Catalani, E.; Giovarelli, M.; Zecchini, S.; et al. Nitric Oxide Generated by Tumor-Associated Macrophages Is Responsible for Cancer Resistance to Cisplatin and Correlated With Syntaxin 4 and Acid Sphingomyelinase Inhibition. Front. Immunol. 2018, 9, 1186. [Google Scholar] [CrossRef]
- Perrotta, C.; Bizzozero, L.; Cazzato, D.; Morlacchi, S.; Assi, E.; Simbari, F.; Zhang, Y.; Gulbins, E.; Bassi, M.T.; Rosa, P.; et al. Syntaxin 4 is required for acid sphingomyelinase activity and apoptotic function. J. Biol. Chem. 2010, 285, 40240–40251. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, G.; Chu, H.; Wang, X.; Xiong, L.; Cai, G.; Liu, R.; Gao, H.; Tao, B.; Li, W.; et al. Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis. Mol. Cell 2018, 71, 201–215.e7. [Google Scholar] [CrossRef]
- Zhu, C.; Chrifi, I.; Mustafa, D.; van der Weiden, M.; Leenen, P.J.M.; Duncker, D.J.; Kros, J.M.; Cheng, C. CECR1-mediated cross talk between macrophages and vascular mural cells promotes neovascularization in malignant glioma. Oncogene 2017, 36, 5356–5368. [Google Scholar] [CrossRef]
- Zhu, C.; Mustafa, D.; Zheng, P.P.; van der Weiden, M.; Sacchetti, A.; Brandt, M.; Chrifi, I.; Tempel, D.; Leenen, P.J.M.; Duncker, D.J.; et al. Activation of CECR1 in M2-like TAMs promotes paracrine stimulation-mediated glial tumor progression. Neuro Oncol. 2017, 19, 648–659. [Google Scholar] [CrossRef]
- Mustafa, D.A.; Dekker, L.J.; Stingl, C.; Kremer, A.; Stoop, M.; Sillevis Smitt, P.A.; Kros, J.M.; Luider, T.M. A proteome comparison between physiological angiogenesis and angiogenesis in glioblastoma. Mol. Cell. Proteom. 2012, 11, M111.008466. [Google Scholar] [CrossRef]
- Liu, A.Y.; Zheng, H.; Ouyang, G. Periostin, a multifunctional matricellular protein in inflammatory and tumor microenvironments. Matrix Biol. 2014, 37, 150–156. [Google Scholar] [CrossRef]
- Morisse, M.C.; Jouannet, S.; Dominguez-Villar, M.; Sanson, M.; Idbaih, A. Interactions between tumor-associated macrophages and tumor cells in glioblastoma: Unraveling promising targeted therapies. Expert Rev. Neurother. 2018, 18, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, Y.; Sun, J.; Chen, W.; Zhao, L.; Ma, C.; Wang, Q.; Sun, J.; Huang, B.; Zhang, Y.; et al. M2-like tumor-associated macrophages drive vasculogenic mimicry through amplification of IL-6 expression in glioma cells. Oncotarget 2017, 8, 819–832. [Google Scholar] [CrossRef]
- Qi, L.; Yu, H.; Zhang, Y.; Zhao, D.; Lv, P.; Zhong, Y.; Xu, Y. IL-10 secreted by M2 macrophage promoted tumorigenesis through interaction with JAK2 in glioma. Oncotarget 2016, 7, 71673–71685. [Google Scholar] [CrossRef]
- Song, L.; Luan, B.; Xu, Q.; Shi, R.; Wang, X. microRNA-155-3p delivered by M2 macrophages-derived exosomes enhances the progression of medulloblastoma through regulation of WDR82. J. Transl. Med. 2022, 20, 13. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Wang, Z.; Cheng, Y.; Ma, C.; Zhong, Y.; Xiao, Y.; Gao, X.; Li, Z. M2 macrophage-derived exosomal microRNAs inhibit cell migration and invasion in gliomas through PI3K/AKT/mTOR signaling pathway. J. Transl. Med. 2021, 19, 99. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guan, J.; Jiang, Z.; Cheng, S.; Hou, W.; Yao, J.; Wang, Z. Microglial Exosome miR-7239-3p Promotes Glioma Progression by Regulating Circadian Genes. Neurosci. Bull. 2021, 37, 497–510. [Google Scholar] [CrossRef]
- Chao, M.P.; Weissman, I.L.; Majeti, R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 2012, 24, 225–232. [Google Scholar] [CrossRef]
- Matlung, H.L.; Szilagyi, K.; Barclay, N.A.; van den Berg, T.K. The CD47-SIRPalpha signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef]
- Soto-Pantoja, D.R.; Kaur, S.; Roberts, D.D. CD47 signaling pathways controlling cellular differentiation and responses to stress. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 212–230. [Google Scholar] [CrossRef] [PubMed]
- Tsai, R.K.; Discher, D.E. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 2008, 180, 989–1003. [Google Scholar] [CrossRef] [PubMed]
- Bener, G.; Félix, A.J.; Sanchez de Diego, C.; Pascual Fabregat, I.; Ciudad, C.J.; Noe, V. Silencing of CD47 and SIRPalpha by Polypurine reverse Hoogsteen hairpins to promote MCF-7 breast cancer cells death by PMA-differentiated THP-1 cells. BMC Immunol. 2016, 17, 32. [Google Scholar] [CrossRef] [PubMed]
- Veillette, A.; Chen, J. SIRPalpha-CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef]
- Zhang, M.; Hutter, G.; Kahn, S.A.; Azad, T.D.; Gholamin, S.; Xu, C.Y.; Liu, J.; Achrol, A.S.; Richard, C.; Sommerkamp, P.; et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PLoS ONE 2016, 11, e0153550. [Google Scholar] [CrossRef]
- Ma, D.; Liu, S.; Lal, B.; Wei, S.; Wang, S.; Zhan, D.; Zhang, H.; Lee, R.S.; Gao, P.; Lopez-Bertoni, H.; et al. Extracellular Matrix Protein Tenascin C Increases Phagocytosis Mediated by CD47 Loss of Function in Glioblastoma. Cancer Res. 2019, 79, 2697–2708. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef]
- Fierro, J., Jr.; DiPasquale, J.; Perez, J.; Chin, B.; Chokpapone, Y.; Tran, A.M.; Holden, A.; Factoriza, C.; Sivagnanakumar, N.; Aguilar, R.; et al. Dual-sgRNA CRISPR/Cas9 knockout of PD-L1 in human U87 glioblastoma tumor cells inhibits proliferation, invasion, and tumor-associated macrophage polarization. Sci. Rep. 2022, 12, 2417. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, H.; Chen, B.; Liu, X.; Zhang, S.; Zong, Z.; Gao, M. PD-L1-Mediated Immunosuppression in Glioblastoma Is Associated With the Infiltration and M2-Polarization of Tumor-Associated Macrophages. Front. Immunol. 2020, 11, 588552. [Google Scholar] [CrossRef]
- Rao, G.; Latha, K.; Ott, M.; Sabbagh, A.; Marisetty, A.; Ling, X.; Zamler, D.; Doucette, T.A.; Yang, Y.; Kong, L.Y.; et al. Anti-PD-1 Induces M1 Polarization in the Glioma Microenvironment and Exerts Therapeutic Efficacy in the Absence of CD8 Cytotoxic T Cells. Clin. Cancer Res. 2020, 26, 4699–4712. [Google Scholar] [CrossRef]
- Cao, Y.; Ding, S.; Zeng, L.; Miao, J.; Wang, K.; Chen, G.; Li, C.; Zhou, J.; Bian, X.W.; Tian, G. Reeducating Tumor-Associated Macrophages Using CpG@Au Nanocomposites to Modulate Immunosuppressive Microenvironment for Improved Radio-Immunotherapy. ACS Appl. Mater. Interfaces 2021, 13, 53504–53518. [Google Scholar] [CrossRef]
- Ghoochani, A.; Schwarz, M.A.; Yakubov, E.; Engelhorn, T.; Doerfler, A.; Buchfelder, M.; Bucala, R.; Savaskan, N.E.; Eyupoglu, I.Y. MIF-CD74 signaling impedes microglial M1 polarization and facilitates brain tumorigenesis. Oncogene 2016, 35, 6246–6261. [Google Scholar] [CrossRef] [PubMed]
- Zeiner, P.S.; Preusse, C.; Blank, A.E.; Zachskorn, C.; Baumgarten, P.; Caspary, L.; Braczynski, A.K.; Weissenberger, J.; Bratzke, H.; Reiss, S.; et al. MIF Receptor CD74 is Restricted to Microglia/Macrophages, Associated with a M1-Polarized Immune Milieu and Prolonged Patient Survival in Gliomas. Brain Pathol. 2015, 25, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, Y.; Sun, X.; Zhao, X.; Ma, Y.; Wang, Y.; Zhang, X. AEG-1 silencing attenuates M2-polarization of glioma-associated microglia/macrophages and sensitizes glioma cells to temozolomide. Sci. Rep. 2021, 11, 17348. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, J.; Zheng, H.; Li, C.; Xiong, J.; Wang, W.; Bao, H.; Jin, H.; Liang, P. Modulating lncRNA SNHG15/CDK6/miR-627 circuit by palbociclib, overcomes temozolomide resistance and reduces M2-polarization of glioma associated microglia in glioblastoma multiforme. J. Exp. Clin. Cancer Res. 2019, 38, 380. [Google Scholar] [CrossRef]
- Hernandez-SanMiguel, E.; Gargini, R.; Cejalvo, T.; Segura-Collar, B.; Nunez-Hervada, P.; Hortiguela, R.; Sepulveda-Sanchez, J.M.; Hernandez-Lain, A.; Perez-Nunez, A.; Sanz, E.; et al. Ocoxin Modulates Cancer Stem Cells and M2 Macrophage Polarization in Glioblastoma. Oxid. Med. Cell. Longev. 2019, 2019, 9719730. [Google Scholar] [CrossRef]
- Xue, N.; Zhou, Q.; Ji, M.; Jin, J.; Lai, F.; Chen, J.; Zhang, M.; Jia, J.; Yang, H.; Zhang, J.; et al. Chlorogenic acid inhibits glioblastoma growth through repolarizating macrophage from M2 to M1 phenotype. Sci. Rep. 2017, 7, 39011. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Wang, C.; Chen, X.; Duan, C.; Zhang, X.; Zhang, J.; Chai, H.; Tang, T.; Chen, H.; Yue, J.; et al. Dopamine induces growth inhibition and vascular normalization through reprogramming M2-polarized macrophages in rat C6 glioma. Toxicol. Appl. Pharmacol. 2015, 286, 112–123. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Komohara, Y.; Kudo, R.; Tsurushima, K.; Ohnishi, K.; Ikeda, T.; Takeya, M. Oleanolic acid inhibits macrophage differentiation into the M2 phenotype and glioblastoma cell proliferation by suppressing the activation of STAT3. Oncol. Rep. 2011, 26, 1533–1537. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Komohara, Y.; Ikeda, T.; Takeya, M. Corosolic acid inhibits glioblastoma cell proliferation by suppressing the activation of signal transducer and activator of transcription-3 and nuclear factor-kappa B in tumor cells and tumor-associated macrophages. Cancer Sci. 2011, 102, 206–211. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, J.; Xu, B.; Ren, J.; Liu, Z.; Cai, L.; Zhang, X.; Wang, W.; Li, S.; Jin, L.; Ding, L. The Importance of M1-and M2-Polarized Macrophages in Glioma and as Potential Treatment Targets. Brain Sci. 2023, 13, 1269. https://doi.org/10.3390/brainsci13091269
Ren J, Xu B, Ren J, Liu Z, Cai L, Zhang X, Wang W, Li S, Jin L, Ding L. The Importance of M1-and M2-Polarized Macrophages in Glioma and as Potential Treatment Targets. Brain Sciences. 2023; 13(9):1269. https://doi.org/10.3390/brainsci13091269
Chicago/Turabian StyleRen, Jiangbin, Bangjie Xu, Jianghao Ren, Zhichao Liu, Lingyu Cai, Xiaotian Zhang, Weijie Wang, Shaoxun Li, Luhao Jin, and Lianshu Ding. 2023. "The Importance of M1-and M2-Polarized Macrophages in Glioma and as Potential Treatment Targets" Brain Sciences 13, no. 9: 1269. https://doi.org/10.3390/brainsci13091269