
AKT Signaling Modifies the Balance between Cell Proliferation and Migration in Neural Crest Cells from Patients Affected with Bosma Arhinia and Microphthalmia Syndrome

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Human iPSC Clones
2.3. Neural Crest Stem Cells Differentiation
2.4. Neural Crest-Like Cells Differentiation
2.5. Immunostaining
2.6. RNA Extraction, Quality Control and Library Preparation
2.7. RNA-Seq Data Processing and Differential Expression Analysis
2.8. Gene Ontology Analysis
2.9. Quantitative RT-PCR
2.10. RNA-Seq Data Analysis
2.11. Wound Healing Closure Scratch Test
2.12. Flow Cytometry Analysis
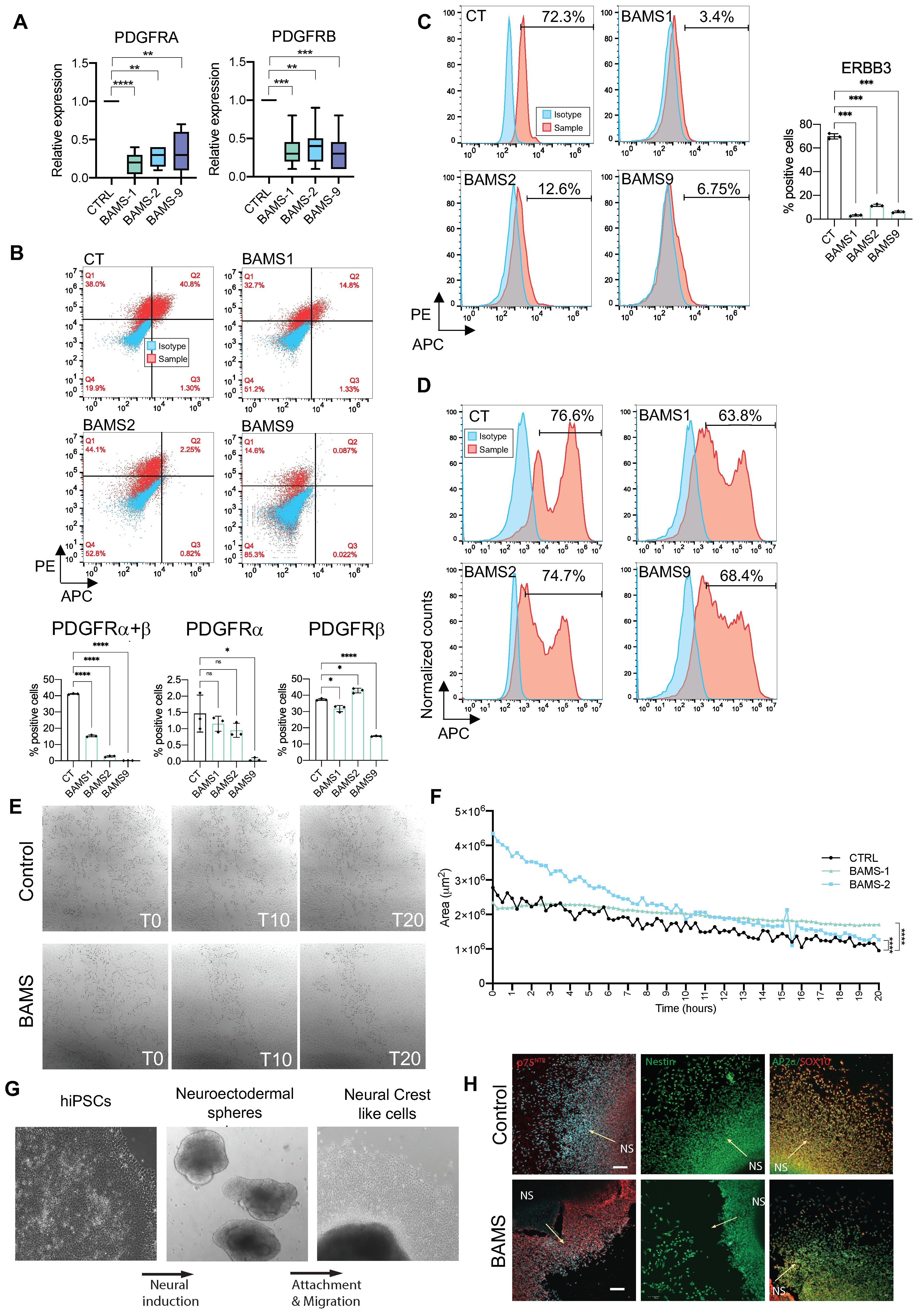
3. Results
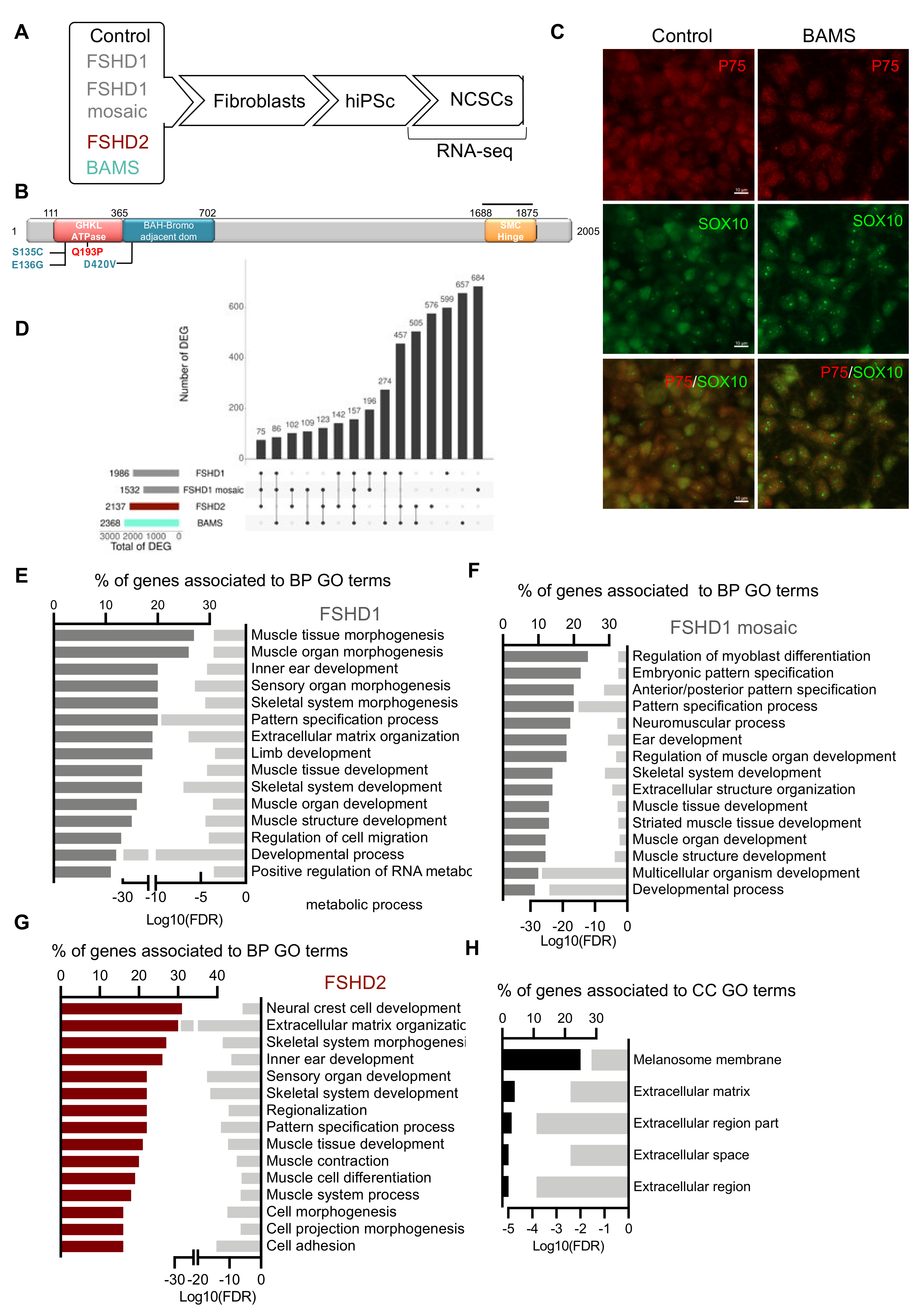
3.1. Experimental Design
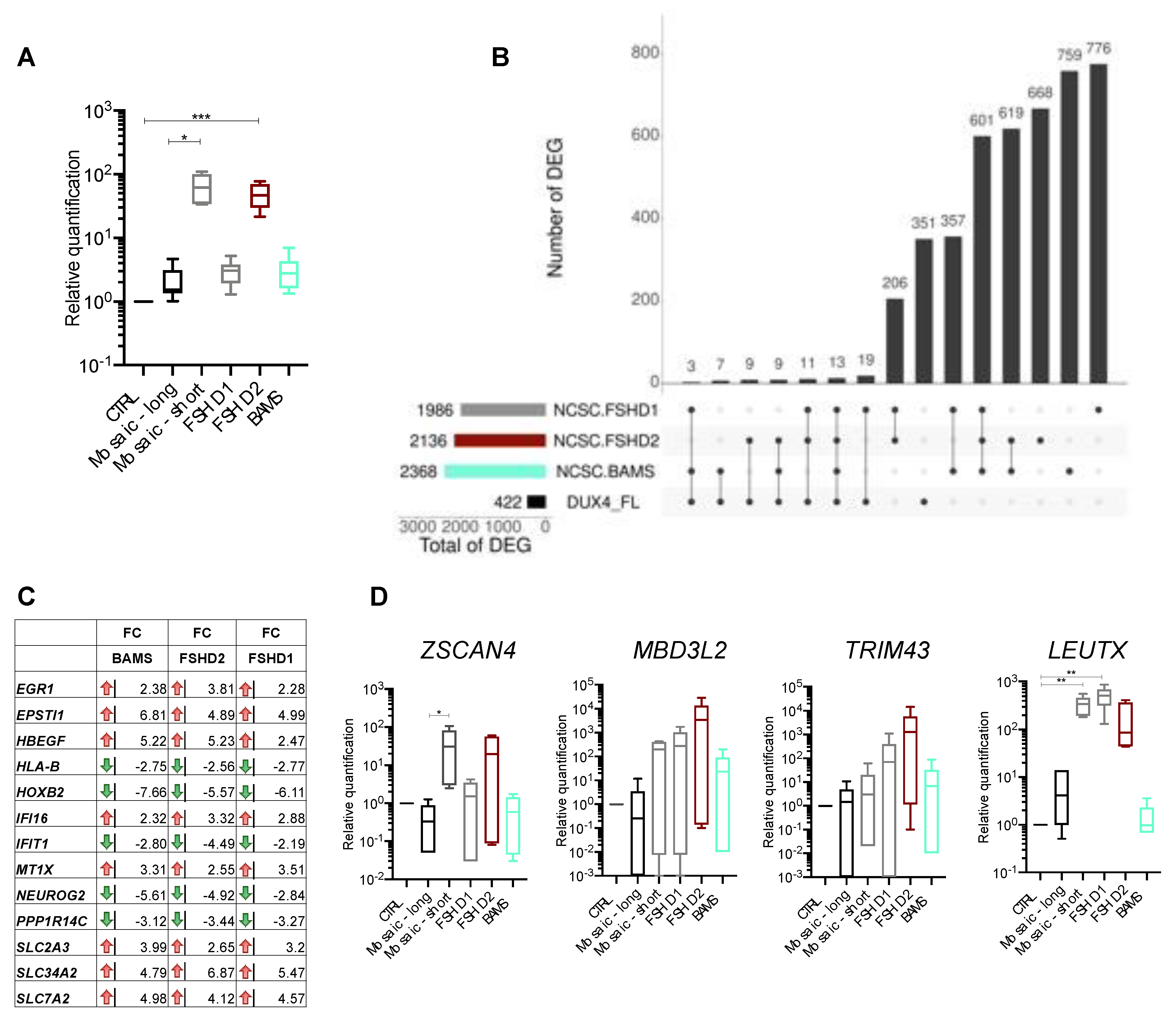
3.2. DUX4 and DUX4-Target Genes Are Detectable in FSHD and BAMS Neural Crest Cells
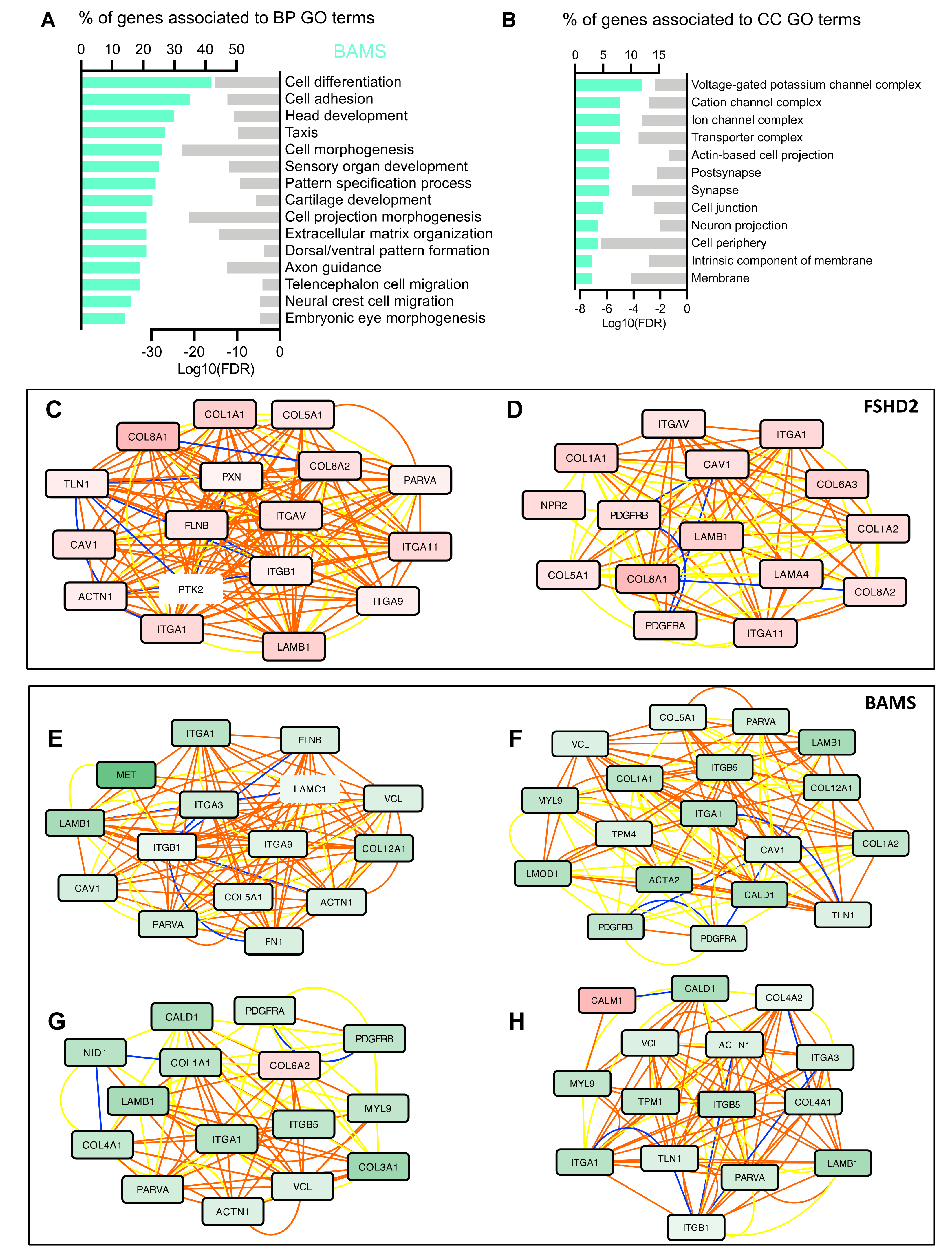
3.3. BAMS-Specific DEGs Are Associated with Cell Migration and Communication
3.4. Neural Crest Stem Cell (NCSC) Differentiation Reveals Impairment of Extracellular Matrix Synthesis
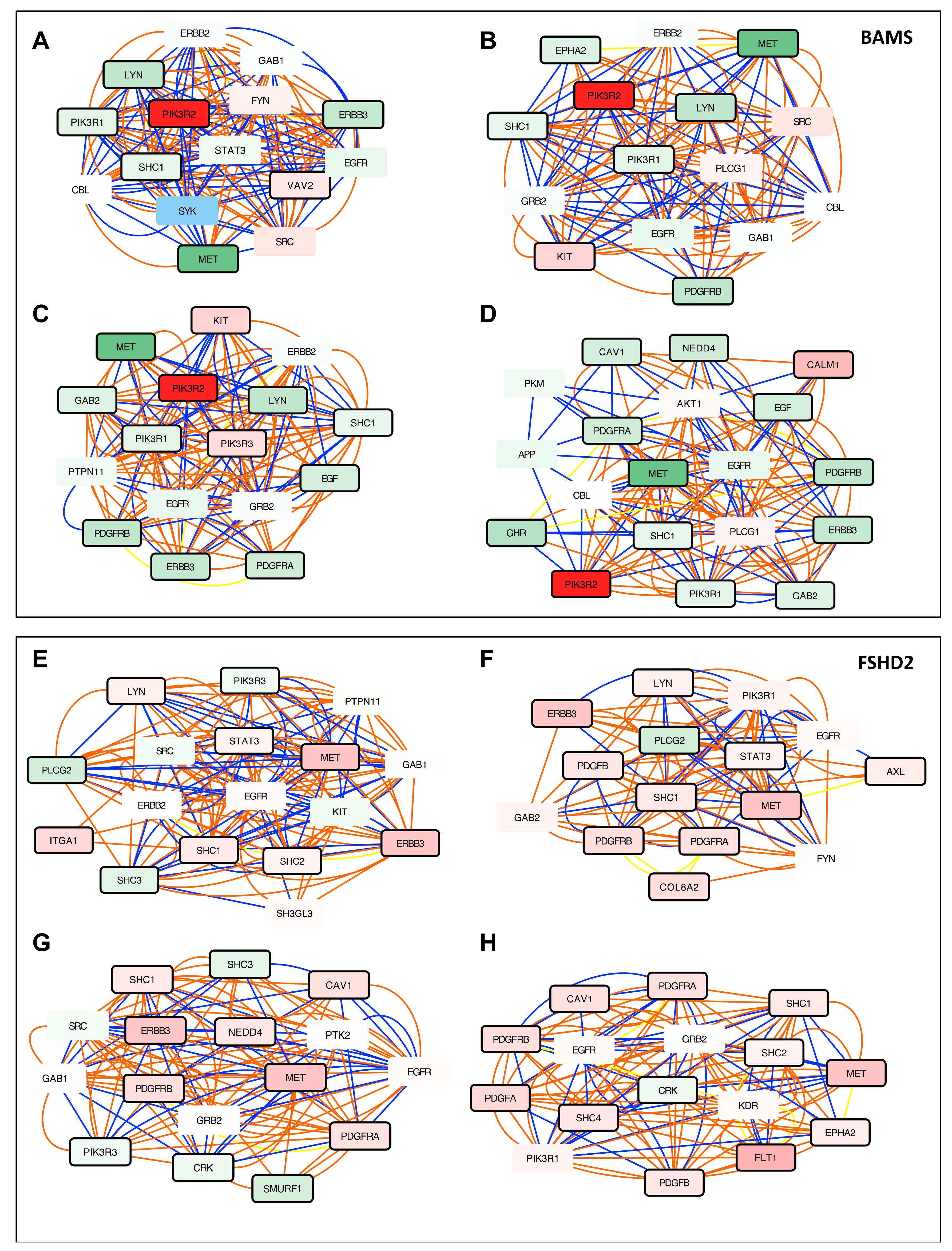
3.5. Altered PI3K/AKT Signaling is Specific to BAMS Cells
3.6. BAMS Neural Crest Cells Display a Defect in Cell Migration
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemmers, R.J.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [Green Version]
- Mostacciuolo, M.L.; Pastorello, E.; Vazza, G.; Miorin, M.; Angelini, C.; Tomelleri, G.; Galluzzi, G.; Trevisan, C.P. Facioscapulohumeral muscular dystrophy: Epidemiological and molecular study in a north-east Italian population sample. Clin. Genet. 2009, 75, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Padberg, G.W.; van Engelen, B.G. Facioscapulohumeral muscular dystrophy. Curr. Opin. Neurol. 2009, 22, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijmenga, C.; Sandkuijl, L.A.; Moerer, P.; van der Boorn, N.; Bodrug, S.E.; Ray, P.N.; Brouwer, O.F.; Murray, J.C.; van Ommen, G.J.; Padberg, G.W.; et al. Genetic linkage map of facioscapulohumeral muscular dystrophy and five polymorphic loci on chromosome 4q35-qter. Am. J. Hum. Genet. 1992, 51, 411–415. [Google Scholar] [PubMed]
- Shaw, N.D.; Brand, H.; Kupchinsky, Z.A.; Bengani, H.; Plummer, L.; Jones, T.I.; Erdin, S.; Williamson, K.A.; Rainger, J.; Stortchevoi, A.; et al. SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and Bosma arhinia microphthalmia syndrome. Nat. Genet. 2017, 49, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Gordon, C.T.; Xue, S.; Yigit, G.; Filali, H.; Chen, K.; Rosin, N.; Yoshiura, K.I.; Oufadem, M.; Beck, T.J.; McGowan, R.; et al. De novo mutations in SMCHD1 cause Bosma arhinia microphthalmia syndrome and abrogate nasal development. Nat. Genet. 2017, 49, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Kinjo, K.; Nagasaki, K.; Muroya, K.; Suzuki, E.; Ishiwata, K.; Nakabayashi, K.; Hattori, A.; Nagao, K.; Nozawa, R.S.; Obuse, C.; et al. Rare variant of the epigenetic regulator SMCHD1 in a patient with pituitary hormone deficiency. Sci. Rep. 2020, 10, 10985. [Google Scholar] [CrossRef]
- Gurzau, A.D.; Chen, K.; Xue, S.; Dai, W.; Lucet, I.S.; Ly, T.T.N.; Reversade, B.; Blewitt, M.E.; Murphy, J.M. FSHD2- and BAMS-associated mutations confer opposing effects on SMCHD1 function. J. Biol. Chem. 2018, 293, 9841–9853. [Google Scholar] [CrossRef] [Green Version]
- Dion, C.; Roche, S.; Laberthonniere, C.; Broucqsault, N.; Mariot, V.; Xue, S.; Gurzau, A.D.; Nowak, A.; Gordon, C.T.; Gaillard, M.C.; et al. SMCHD1 is involved in de novo methylation of the DUX4-encoding D4Z4 macrosatellite. Nucleic Acids Res. 2019, 47, 2822–2839. [Google Scholar] [CrossRef] [Green Version]
- Mul, K.; Lemmers, R.; Kriek, M.; van der Vliet, P.J.; van den Boogaard, M.L.; Badrising, U.A.; Graham, J.M., Jr.; Lin, A.E.; Brand, H.; Moore, S.A.; et al. FSHD type 2 and Bosma arhinia microphthalmia syndrome: Two faces of the same mutation. Neurology 2018, 91, e562–e570. [Google Scholar] [CrossRef]
- Gendrel, A.V.; Tang, Y.A.; Suzuki, M.; Godwin, J.; Nesterova, T.B.; Greally, J.M.; Heard, E.; Brockdorff, N. Epigenetic functions of smchd1 repress gene clusters on the inactive X chromosome and on autosomes. Mol. Cell. Biol. 2013, 33, 3150–3165. [Google Scholar] [CrossRef] [Green Version]
- Jansz, N.; Keniry, A.; Trussart, M.; Bildsoe, H.; Beck, T.; Tonks, I.D.; Mould, A.W.; Hickey, P.; Breslin, K.; Iminitoff, M.; et al. Smchd1 regulates long-range chromatin interactions on the inactive X chromosome and at Hox clusters. Nat. Struct. Mol. Biol. 2018, 25, 766–777. [Google Scholar] [CrossRef]
- Jansz, N.; Chen, K.; Murphy, J.M.; Blewitt, M.E. The Epigenetic Regulator SMCHD1 in Development and Disease. Trends Genet. 2017, 33, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Blewitt, M.E.; Vickaryous, N.K.; Hemley, S.J.; Ashe, A.; Bruxner, T.J.; Preis, J.I.; Arkell, R.; Whitelaw, E. An N-ethyl-N-nitrosourea screen for genes involved in variegation in the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 7629–7634. [Google Scholar] [CrossRef] [Green Version]
- Mica, Y.; Lee, G.; Chambers, S.M.; Tomishima, M.J.; Studer, L. Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell Rep. 2013, 3, 1140–1152. [Google Scholar] [CrossRef] [Green Version]
- Bajpai, R.; Chen, D.A.; Rada-Iglesias, A.; Zhang, J.; Xiong, Y.; Helms, J.; Chang, C.P.; Zhao, Y.; Swigut, T.; Wysocka, J. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 2010, 463, 958–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Valdeolivas, A.; Tichit, L.; Navarro, C.; Perrin, S.; Odelin, G.; Levy, N.; Cau, P.; Remy, E.; Baudot, A. Random walk with restart on multiplex and heterogeneous biological networks. Bioinformatics 2019, 35, 497–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolland, T.; Tasan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef] [Green Version]
- Del-Toro, N.; Dumousseau, M.; Orchard, S.; Jimenez, R.C.; Galeota, E.; Launay, G.; Goll, J.; Breuer, K.; Ono, K.; Salwinski, L.; et al. A new reference implementation of the PSICQUIC web service. Nucleic Acids Res. 2013, 41, W601–W606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sales, G.; Calura, E.; Cavalieri, D.; Romualdi, C. Graphite—A Bioconductor package to convert pathway topology to gene network. BMC Bioinform. 2012, 13, 20. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, O.F.; Padberg, G.W.; Ruys, C.J.; Brand, R.; de Laat, J.A.; Grote, J.J. Hearing loss in facioscapulohumeral muscular dystrophy. Neurology 1991, 41, 1878–1881. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, R.J.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camano, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef] [Green Version]
- Snider, L.; Geng, L.N.; Lemmers, R.J.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6, e1001181. [Google Scholar] [CrossRef] [Green Version]
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 activates germline genes, retroelements, and immune mediators: Implications for facioscapulohumeral dystrophy. Dev. Cell 2012, 22, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Williams, K.; Kong, X.; Zeng, W.; Nguyen, N.V.; Ma, X.; Tawil, R.; Yokomori, K.; Mortazavi, A. Single-nucleus RNA-seq identifies divergent populations of FSHD2 myotube nuclei. PLoS Genet. 2020, 16, e1008754. [Google Scholar] [CrossRef]
- Banerji, C.R.S.; Panamarova, M.; Hebaishi, H.; White, R.B.; Relaix, F.; Severini, S.; Zammit, P.S. PAX7 target genes are globally repressed in facioscapulohumeral muscular dystrophy skeletal muscle. Nat. Commun. 2017, 8, 2152. [Google Scholar] [CrossRef] [Green Version]
- Raper, J.A.; Kapfhammer, J.P. The enrichment of a neuronal growth cone collapsing activity from embryonic chick brain. Neuron 1990, 4, 21–29. [Google Scholar] [CrossRef]
- Mitsogiannis, M.D.; Little, G.E.; Mitchell, K.J. Semaphorin-Plexin signaling influences early ventral telencephalic development and thalamocortical axon guidance. Neural Dev. 2017, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Z.; Guo, J.; Lu, Y.; Liu, W.; Fu, X.; Yao, T.; Zhou, Y.; Xu, H.A. Sema3E is required for migration of cranial neural crest cells in zebrafish: Implications for the pathogenesis of CHARGE syndrome. Int. J. Exp. Pathol. 2019, 100, 234–243. [Google Scholar] [CrossRef]
- Alto, L.T.; Terman, J.R. Semaphorins and their Signaling Mechanisms. In Semaphorin Signaling; Terman, J.R., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; Volume 1493, pp. 1–25. [Google Scholar]
- Aruga, J.; Yokota, N.; Mikoshiba, K. Human SLITRK family genes: Genomic organization and expression profiling in normal brain and brain tumor tissue. Gene 2003, 315, 87–94. [Google Scholar] [CrossRef]
- Tessier-Lavigne, M.; Goodman, C.S. The molecular biology of axon guidance. Science 1996, 274, 1123–1133. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Qian, X.; Zhu, M.; Li, A.; Fang, M.; Zhu, Y.; Zhang, J. miR1273g3p promotes proliferation, migration and invasion of LoVo cells via cannabinoid receptor 1 through activation of ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Mol. Med. Rep. 2018, 17, 4619–4626. [Google Scholar]
- Kuchay, S.; Duan, S.; Schenkein, E.; Peschiaroli, A.; Saraf, A.; Florens, L.; Washburn, M.P.; Pagano, M. FBXL2- and PTPL1-mediated degradation of p110-free p85beta regulatory subunit controls the PI(3)K signalling cascade. Nat. Cell Biol. 2013, 15, 472–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiszniak, S.; Kabbara, S.; Lumb, R.; Scherer, M.; Secker, G.; Harvey, N.; Kumar, S.; Schwarz, Q. The ubiquitin ligase Nedd4 regulates craniofacial development by promoting cranial neural crest cell survival and stem-cell like properties. Dev. Biol. 2013, 383, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Forni, P.E.; Wray, S. Neural crest and olfactory system: New prospective. Mol. Neurobiol. 2012, 46, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Prasad, M.S.; Charney, R.M.; Garcia-Castro, M.I. Specification and formation of the neural crest: Perspectives on lineage segregation. Genesis 2019, 57, e23276. [Google Scholar] [CrossRef] [Green Version]
- Novoa-del-Toro, E.-M.; Mezura-Montes, E.; Vignes, M.; Magdinier, F.; Tichit, L.; Baudot, A. Multi-Objective Genetic Algorithm to Find Active Modules from Multiplex Biological Networks. 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.05.25.114215v1 (accessed on 26 May 2020).
- Sittewelle, M.; Monsoro-Burq, A.H. AKT signaling displays multifaceted functions in neural crest development. Dev. Biol. 2018, 444 (Suppl. 1), S144–S155. [Google Scholar] [CrossRef]
- Carmona-Fontaine, C.; Matthews, H.K.; Kuriyama, S.; Moreno, M.; Dunn, G.A.; Parsons, M.; Stern, C.D.; Mayor, R. Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature 2008, 456, 957–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuchel, R.; Berg, A.; Tallquist, M.; Ahlen, K.; Reed, R.K.; Rubin, K.; Claesson-Welsh, L.; Heldin, C.H.; Soriano, P. Platelet-derived growth factor beta receptor regulates interstitial fluid homeostasis through phosphatidylinositol-3’ kinase signaling. Proc. Natl. Acad. Sci. USA 1999, 96, 11410–11415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallejo-Diaz, J.; Chagoyen, M.; Olazabal-Moran, M.; Gonzalez-Garcia, A.; Carrera, A.C. The Opposing Roles of PIK3R1/p85α and PIK3R2/p85β in Cancer. Trends Cancer 2019, 5, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantauzzo, K.A.; Soriano, P. PDGFRβ regulates craniofacial development through homodimers and functional heterodimers with PDGFRα. Genes Dev. 2016, 30, 2443–2458. [Google Scholar] [CrossRef] [Green Version]
- Schneller, M.; Vuori, K.; Ruoslahti, E. αvβ3 integrin associates with activated insulin and PDGFβ receptors and potentiates the biological activity of PDGF. EMBO J. 1997, 16, 5600–5607. [Google Scholar] [CrossRef] [Green Version]
- Morrison-Graham, K.; Schatteman, G.C.; Bork, T.; Bowen-Pope, D.F.; Weston, J.A. A PDGF receptor mutation in the mouse (Patch) perturbs the development of a non-neuronal subset of neural crest-derived cells. Development 1992, 115, 133–142. [Google Scholar] [CrossRef]
- Orr-Urtreger, A.; Lonai, P. Platelet-derived growth factor-A and its receptor are expressed in separate, but adjacent cell layers of the mouse embryo. Development 1992, 115, 1045–1058. [Google Scholar] [CrossRef]
- Twamley-Stein, G.M.; Pepperkok, R.; Ansorge, W.; Courtneidge, S.A. The Src family tyrosine kinases are required for platelet-derived growth factor-mediated signal transduction in NIH 3T3 cells. Proc. Natl. Acad. Sci. USA 1993, 90, 7696–7700. [Google Scholar] [CrossRef] [Green Version]
- Tallquist, M.D.; Soriano, P. Cell autonomous requirement for PDGFRα in populations of cranial and cardiac neural crest cells. Development 2003, 130, 507–518. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Soriano, P. A critical role for PDGFRα signaling in medial nasal process development. PLoS Genet. 2013, 9, e1003851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsinovi, D.; Giannetti, K.; Cericola, A.; Naef, V.; Ori, M. PDGF-B: The missing piece in the mosaic of PDGF family role in craniofacial development. Dev. Dyn. 2019, 248, 603–612. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, N.; Liu, J.S.; Richarte, A.M.; Eskiocak, B.; Lovely, C.B.; Tallquist, M.D.; Eberhart, J.K. Pdgfra and Pdgfrb genetically interact during craniofacial development. Dev. Dyn. 2016, 245, 641–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattanasopha, S.; Tongkobpetch, S.; Srichomthong, C.; Siriwan, P.; Suphapeetiporn, K.; Shotelersuk, V. PDGFRa mutations in humans with isolated cleft palate. Eur. J. Hum. Genet. 2012, 20, 1058–1062. [Google Scholar] [CrossRef] [Green Version]
- Takenouchi, T.; Yamaguchi, Y.; Tanikawa, A.; Kosaki, R.; Okano, H.; Kosaki, K. Novel overgrowth syndrome phenotype due to recurrent de novo PDGFRB mutation. J. Pediatr. 2015, 166, 483–486. [Google Scholar] [CrossRef]
- Nakamura, T.; Gulick, J.; Colbert, M.C.; Robbins, J. Protein tyrosine phosphatase activity in the neural crest is essential for normal heart and skull development. Proc. Natl. Acad. Sci. USA 2009, 106, 11270–11275. [Google Scholar] [CrossRef] [Green Version]
- Takayama, H.; La Rochelle, W.J.; Anver, M.; Bockman, D.E.; Merlino, G. Scatter factor/hepatocyte growth factor as a regulator of skeletal muscle and neural crest development. Proc. Natl. Acad. Sci. USA 1996, 93, 5866–5871. [Google Scholar] [CrossRef] [Green Version]
- Gammill, L.S.; Roffers-Agarwal, J. Division of labor during trunk neural crest development. Dev. Biol. 2010, 344, 555–565. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis | Genotype | SMCHD1 Status | Age | Gender | |
|---|---|---|---|---|---|
| AG08498 | Healthy | >10 | No Mutation | 1 | Male |
| 12759 | FSHD1 | 7RU | No mutation | 51 | Female |
| 17706 | FSHD1 | 2RU | No mutation | 56 | Female |
| Mosaic | 25% | ||||
| 14586 | FSHD2 | >10 | c.573A > C°; p.Q193P | 67 | Male |
| 11440 | FSHD2 | >10 | c.2338 + 4A > G; p.S754 * | 37 | Male |
| BAMS-1 | BAMS | N/A | c.407A > G | 5 | Male |
| p.E136G | |||||
| BAMS-2 | BAMS | N/A | c.403A > T | 28 | Female |
| p.S135C | |||||
| BAMS-9 | BAMS | N/A | c.1259A > T | 3 | Male |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laberthonnière, C.; Novoa-del-Toro, E.M.; Chevalier, R.; Broucqsault, N.; Rao, V.V.; Trani, J.P.; Nguyen, K.; Xue, S.; Reversade, B.; Robin, J.D.; et al. AKT Signaling Modifies the Balance between Cell Proliferation and Migration in Neural Crest Cells from Patients Affected with Bosma Arhinia and Microphthalmia Syndrome. Biomedicines 2021, 9, 751. https://doi.org/10.3390/biomedicines9070751
Laberthonnière C, Novoa-del-Toro EM, Chevalier R, Broucqsault N, Rao VV, Trani JP, Nguyen K, Xue S, Reversade B, Robin JD, et al. AKT Signaling Modifies the Balance between Cell Proliferation and Migration in Neural Crest Cells from Patients Affected with Bosma Arhinia and Microphthalmia Syndrome. Biomedicines. 2021; 9(7):751. https://doi.org/10.3390/biomedicines9070751
Chicago/Turabian StyleLaberthonnière, Camille, Elva Maria Novoa-del-Toro, Raphaël Chevalier, Natacha Broucqsault, Vanitha Venkoba Rao, Jean Philippe Trani, Karine Nguyen, Shifeng Xue, Bruno Reversade, Jérôme D. Robin, and et al. 2021. "AKT Signaling Modifies the Balance between Cell Proliferation and Migration in Neural Crest Cells from Patients Affected with Bosma Arhinia and Microphthalmia Syndrome" Biomedicines 9, no. 7: 751. https://doi.org/10.3390/biomedicines9070751