Fluorescent Tagged Vaccinia Virus Genome Allows Rapid and Efficient Measurement of Oncolytic Potential and Discovery of Oncolytic Modulators


 , ,
, ,
Abstract
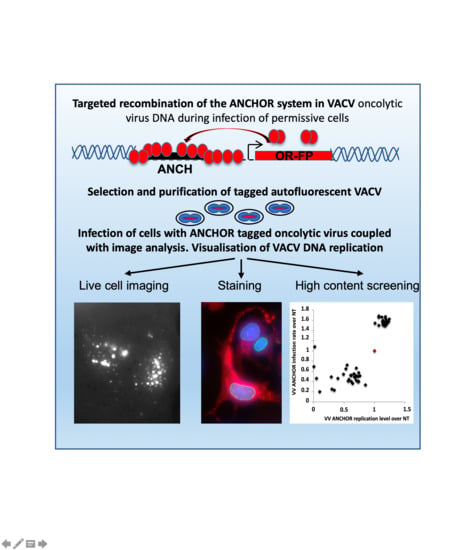
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Chemicals and Antibodies
2.3. Viruses and Plasmids
2.4. Visualization and Quantification of VACV ANCHOR
2.5. High-Content Screening Protocol
2.6. Cell Imaging and Immunofluorescence Staining
2.7. Statistical Analysis
3. Results
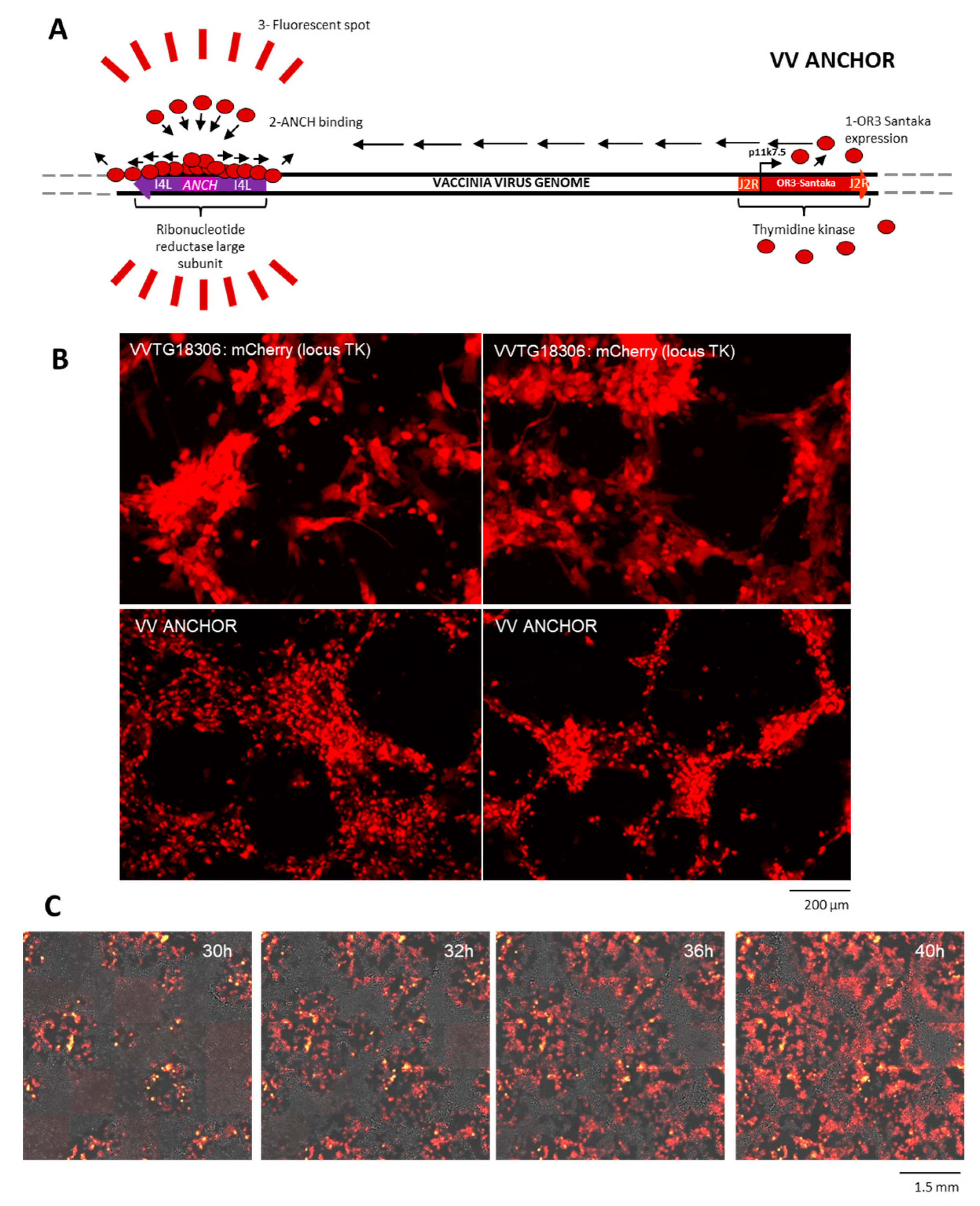
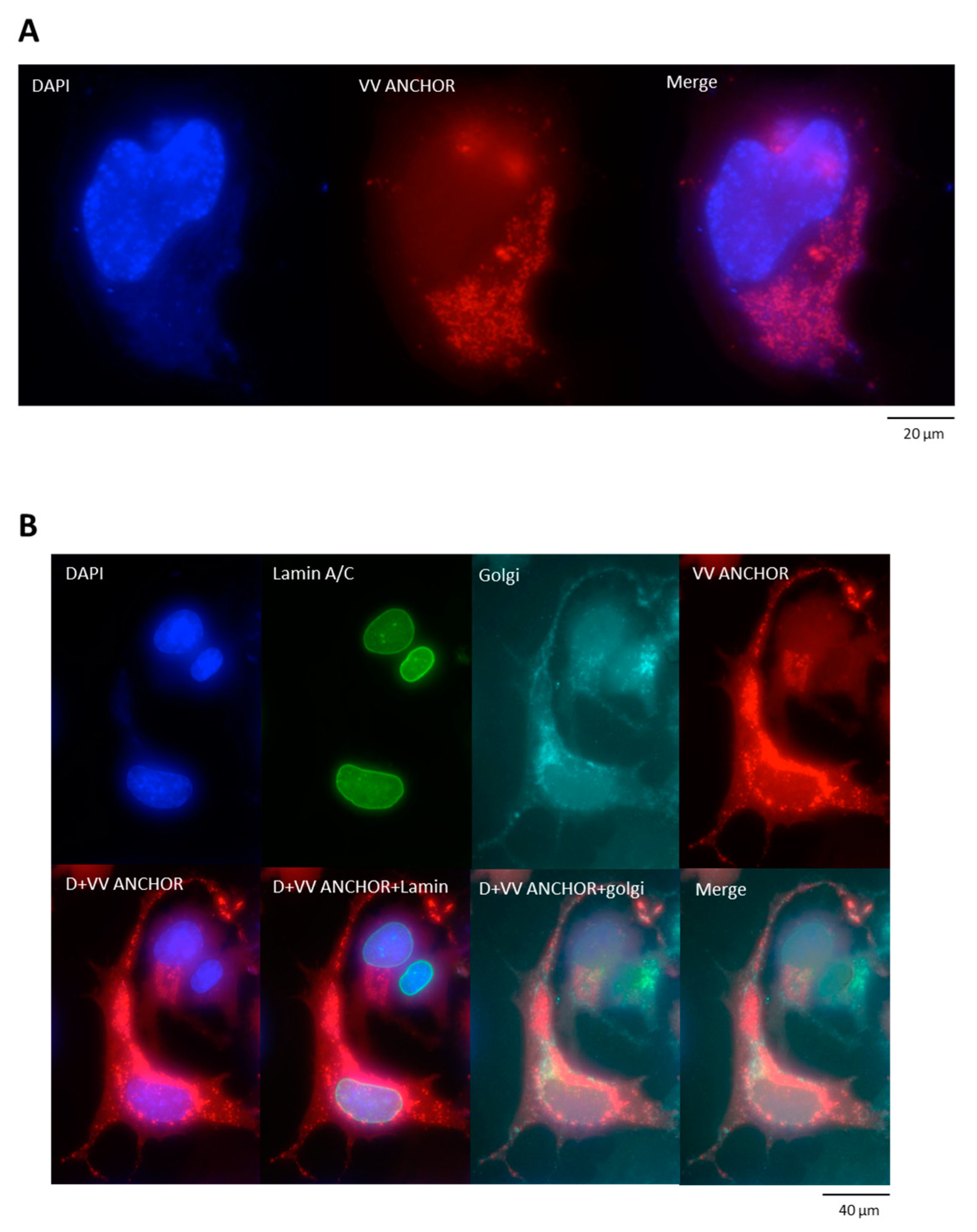
3.1. Generation of an Autofluorescent VACV
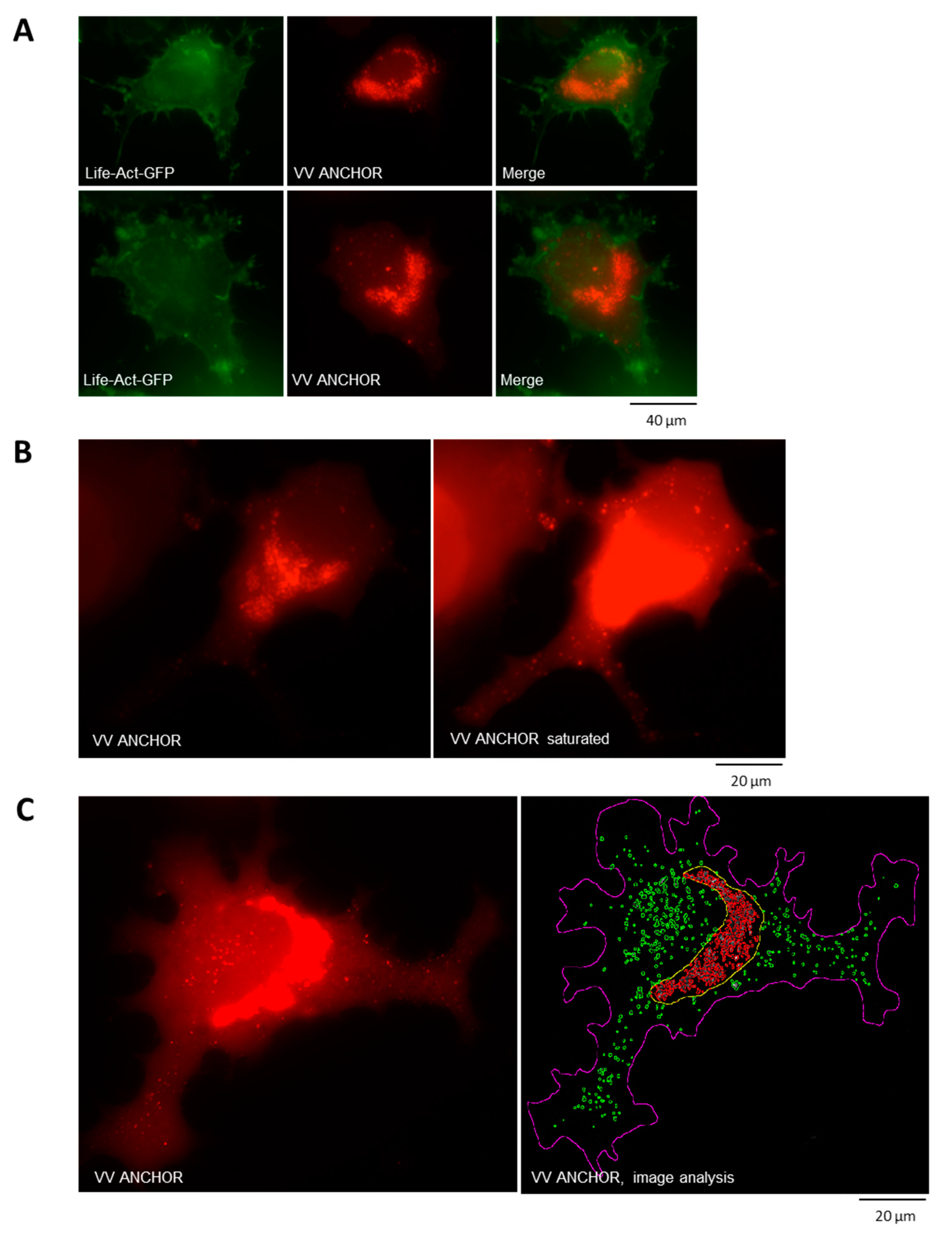
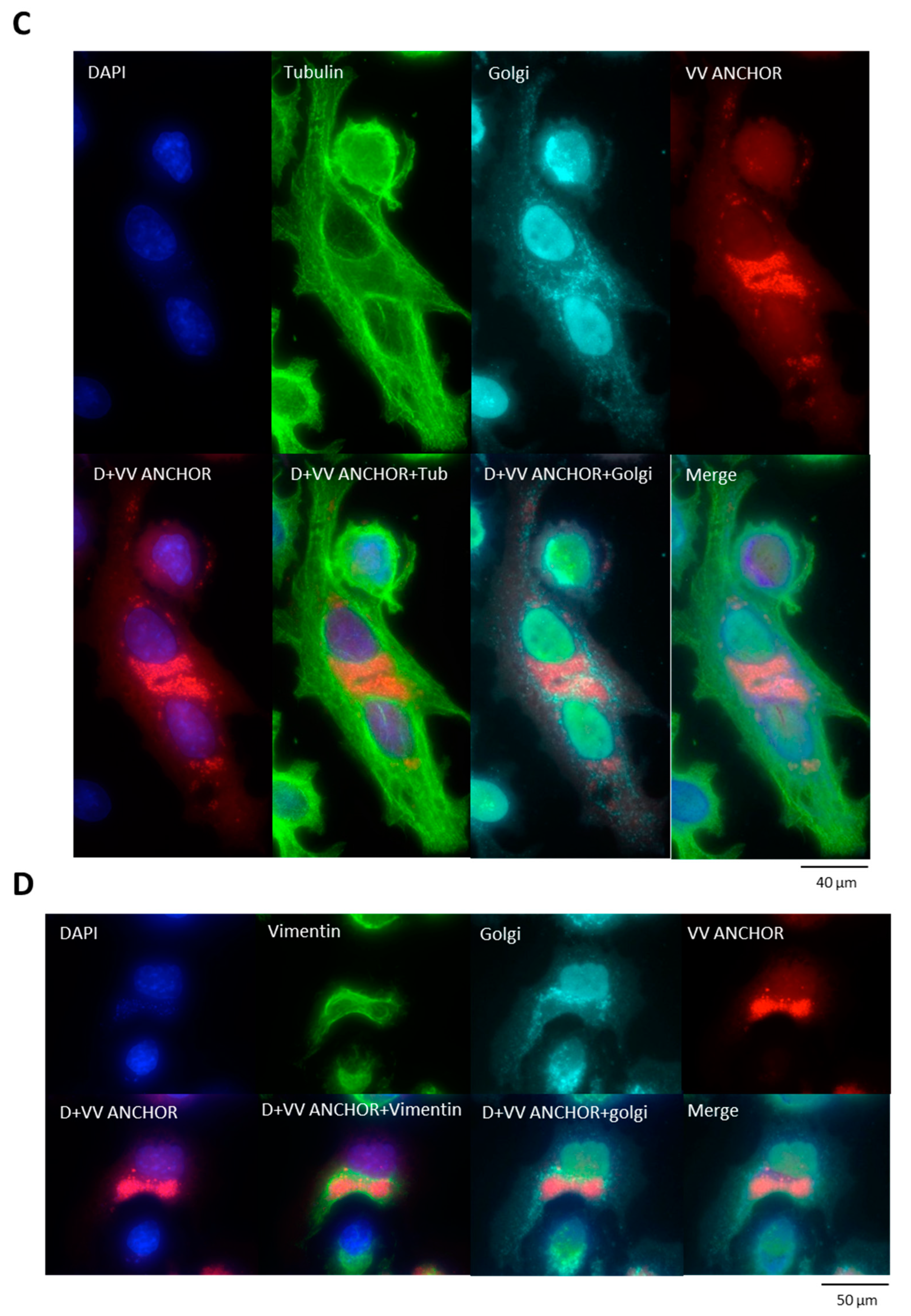
3.2. Biological Characterization of VACV ANCHOR
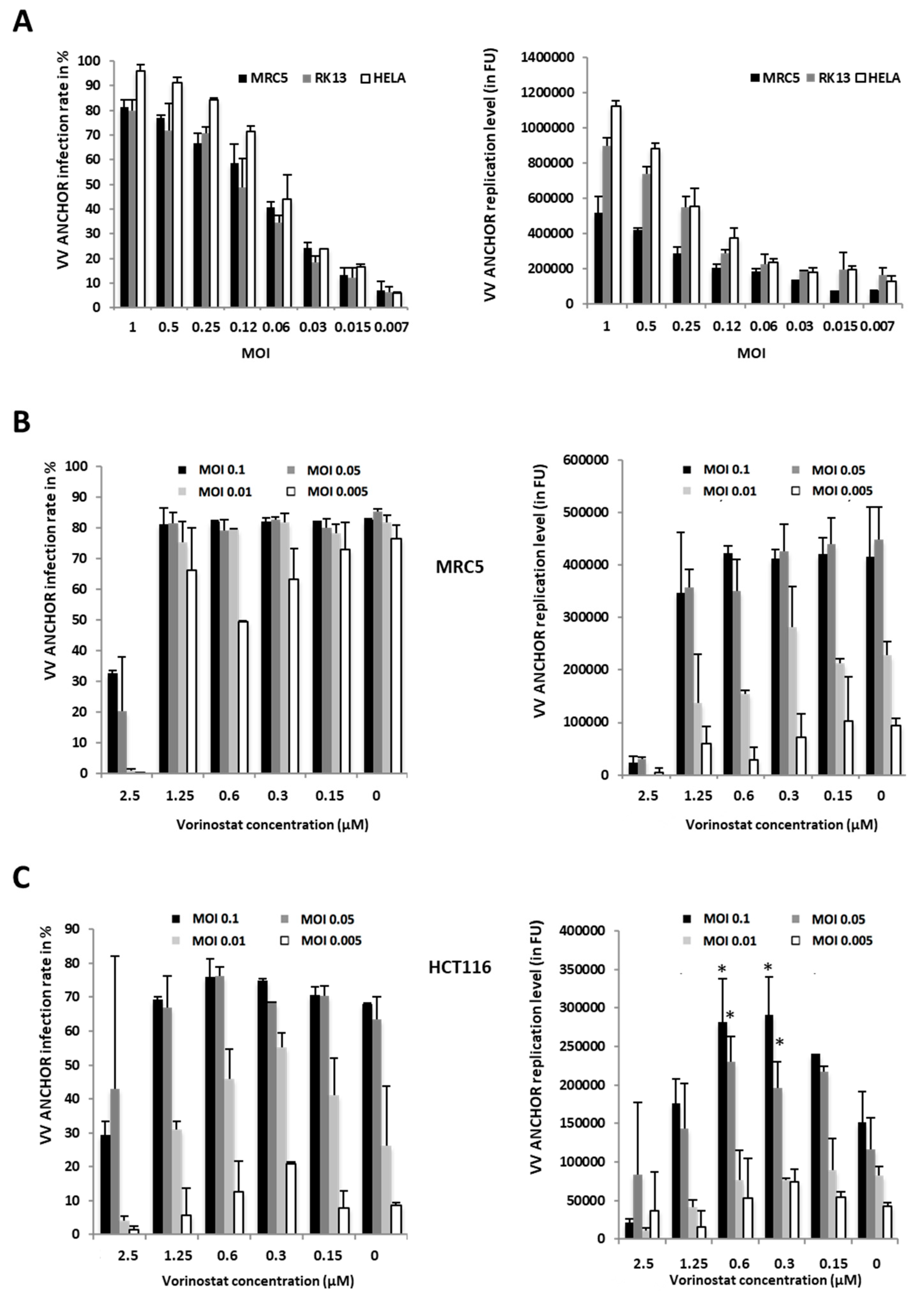
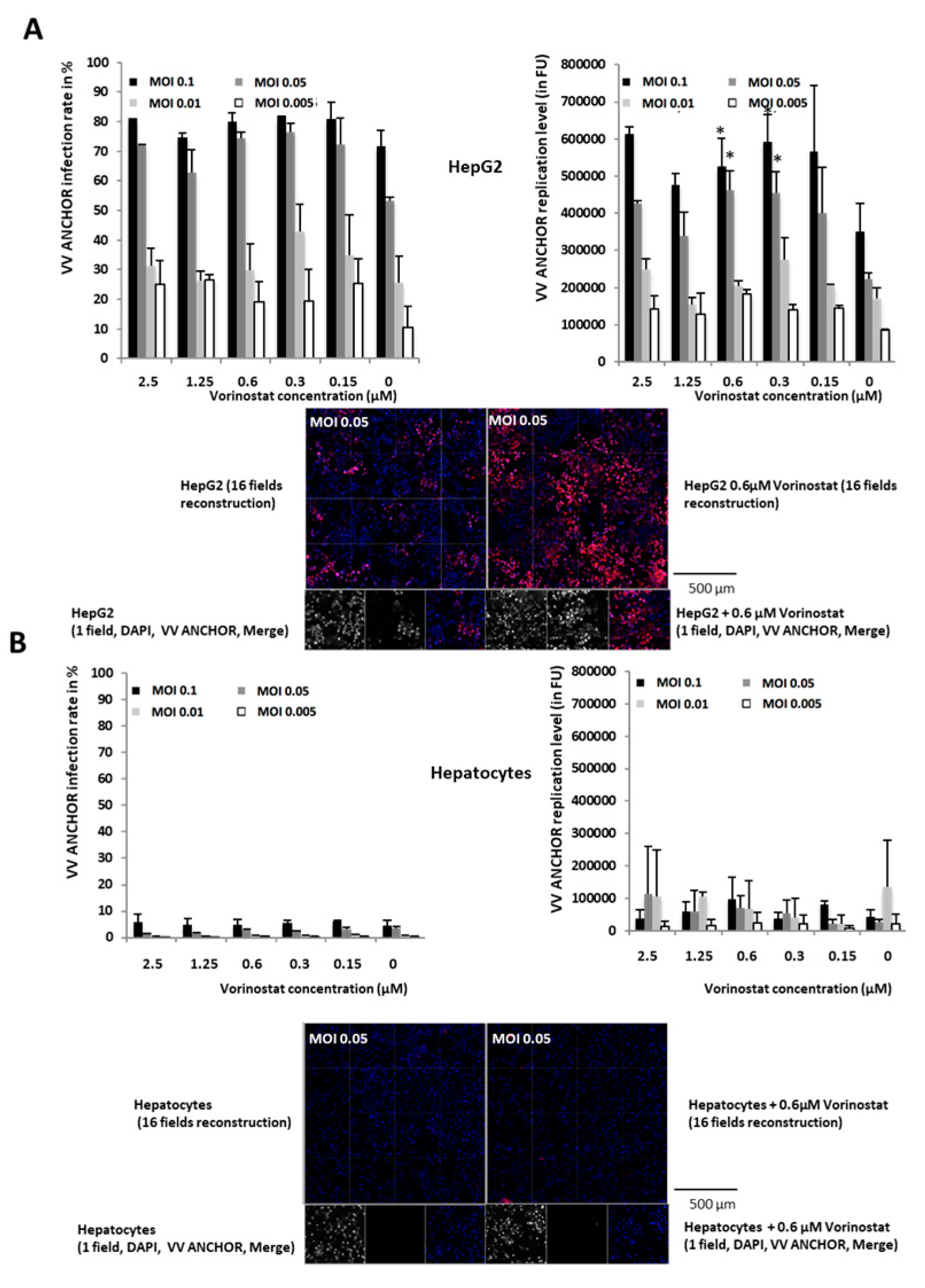
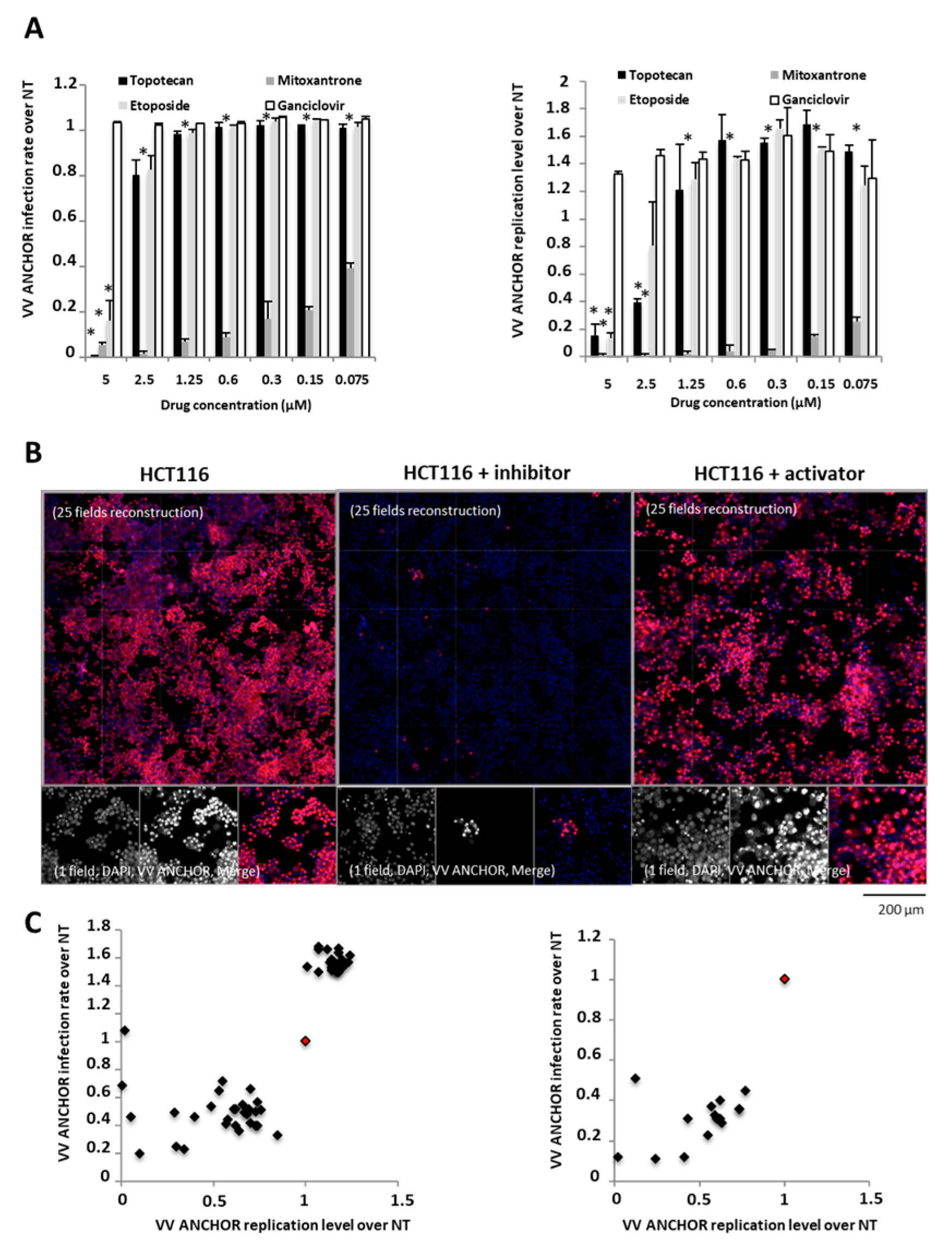
3.3. Identification of Modulators of Oncolytic Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaufman:, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Kirn, D.H.; Thorne, S.H. Targeted and armed oncolytic poxviruses: A novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer 2009, 9, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Nemunaitis, J. Fighting cancer with vaccinia virus: Teaching new tricks to an old dog. Mol. Ther. 2005, 11, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.M.; McFadden, G. Oncolytic Poxviruses. Annu. Rev. Virol. 2014, 1, 119–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, S.H. Immunotherapeutic potential of oncolytic vaccinia virus. Immunol. Res. 2011, 50, 286–293. [Google Scholar] [CrossRef]
- Parato, K.A.; Breitbach, C.J.; Le Boeuf, F.; Wang, J.; Storbeck, C.; Ilkow, C.; Diallo, J.S.; Falls, T.; Burns, J.; Garcia, V.; et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol. Ther. 2012, 20, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Fenner, F. A successful eradication campaign. Global eradication of smallpox. Rev. Infect. Dis. 1982, 4, 916–930. [Google Scholar] [CrossRef]
- Breitbach, C.J.; Burke, J.; Jonker, D.; Stephenson, J.; Haas, A.R.; Chow, L.Q.; Nieva, J.; Hwang, T.H.; Moon, A.; Patt, R.; et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 2011, 477, 99–102. [Google Scholar] [CrossRef]
- Hengstschlager, M.; Knofler, M.; Mullner, E.W.; Ogris, E.; Wintersberger, E.; Wawra, E. Different regulation of thymidine kinase during the cell cycle of normal versus DNA tumor virus-transformed cells. J. Biol. Chem. 1994, 269, 13836–13842. [Google Scholar]
- Puhlmann, M.; Brown, C.K.; Gnant, M.; Huang, J.; Libutti, S.K.; Alexander, H.R.; Bartlett, D.L. Vaccinia as a vector for tumor-directed gene therapy: Biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther. 2000, 7, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Foloppe, J.; Kintz, J.; Futin, N.; Findeli, A.; Cordier, P.; Schlesinger, Y.; Hoffmann, C.; Tosch, C.; Balloul, J.M.; Erbs, P. Targeted delivery of a suicide gene to human colorectal tumors by a conditionally replicating vaccinia virus. Gene Ther. 2008, 15, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Haddad, D. Genetically Engineered Vaccinia Viruses As Agents for Cancer Treatment, Imaging, and Transgene Delivery. Front. Oncol. 2017, 7, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foloppe, J.; Kempf, J.; Futin, N.; Kintz, J.; Cordier, P.; Pichon, C.; Findeli, A.; Vorburger, F.; Quemeneur, E.; Erbs, P. The Enhanced Tumor Specificity of TG6002, an Armed Oncolytic Vaccinia Virus Deleted in Two Genes Involved in Nucleotide Metabolism. Mol. Ther. Oncolytics 2019, 14, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, H.; Gallardo, F.; Dalvai, M.; Tanguy-le-Gac, N.; Lane, D.; Bystricky, K. DNA dynamics during early double-strand break processing revealed by non-intrusive imaging of living cells. PLoS Genet. 2014, 10, e1004187. [Google Scholar] [CrossRef] [Green Version]
- Germier, T.; Kocanova, S.; Walther, N.; Bancaud, A.; Shaban, H.A.; Sellou, H.; Politi, A.Z.; Ellenberg, J.; Gallardo, F.; Bystricky, K. Real-Time Imaging of a Single Gene Reveals Transcription-Initiated Local Confinement. Biophys. J. 2017, 113, 1383–1394. [Google Scholar] [CrossRef] [Green Version]
- Mariame, B.; Kappler-Gratias, S.; Kappler, M.; Balor, S.; Gallardo, F.; Bystricky, K. Real-Time Visualization and Quantification of Human Cytomegalovirus Replication in Living Cells Using the ANCHOR DNA Labeling Technology. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, T.; Quentin-Froignant, C.; Carlon-Andres, I.; Lagadec, F.; Rayne, F.; Ragues, J.; Kehlenbach, R.H.; Zhang, W.; Ehrhardt, A.; Bystricky, K.; et al. In Vivo Labelling of Adenovirus DNA Identifies Chromatin Anchoring and Biphasic Genome Replication. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Rodriguez, G.; Gazi, A.; Monel, B.; Frabetti, S.; Scoca, V.; Mueller, F.; Schwartz, O.; Krijnse-Locker, J.; Charneau, P.; Di Nunzio, F. Remodeling of the Core Leads HIV-1 Preintegration Complex into the Nucleus of Human Lymphocytes. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Hinsberger, A.; Graillot, B.; Blachere Lopez, C.; Juliant, S.; Cerutti, M.; King, L.A.; Possee, R.D.; Gallardo, F.; Lopez Ferber, M. Tracing Baculovirus AcMNPV Infection Using a Real-Time Method Based on ANCHOR(TM) DNA Labeling Technology. Viruses 2020, 12, 50. [Google Scholar] [CrossRef] [Green Version]
- Erbs, P.; Findeli, A.; Kintz, J.; Cordier, P.; Hoffmann, C.; Geist, M.; Balloul, J.M. Modified vaccinia virus Ankara as a vector for suicide gene therapy. Cancer Gene Ther. 2008, 15, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Risco, C.; Rodriguez, J.R.; Lopez-Iglesias, C.; Carrascosa, J.L.; Esteban, M.; Rodriguez, D. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 2002, 76, 1839–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacTavish, H.; Diallo, J.S.; Huang, B.; Stanford, M.; Le Boeuf, F.; De Silva, N.; Cox, J.; Simmons, J.G.; Guimond, T.; Falls, T.; et al. Enhancement of vaccinia virus based oncolysis with histone deacetylase inhibitors. PLoS ONE 2010, 5, e14462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Chemaly, R.F.; Hill, J.A.; Voigt, S.; Peggs, K.S. In vitro comparison of currently available and investigational antiviral agents against pathogenic human double-stranded DNA viruses: A systematic literature review. Antiviral Res. 2019, 163, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2016, 36, 248–299. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Dai, P.; Ciro, A.; Smee, D.F.; Djaballah, H.; Shuman, S. Identification of novel antipoxviral agents: Mitoxantrone inhibits vaccinia virus replication by blocking virion assembly. J. Virol. 2007, 81, 13392–13402. [Google Scholar] [CrossRef] [Green Version]
- Moss, B. Poxvirus DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Dales, S.; Siminovitch, L. The development of vaccinia virus in Earle’s L strain cells as examined by electron microscopy. J. Biophys. Biochem. Cytol. 1961, 10, 475–503. [Google Scholar] [CrossRef] [Green Version]
- Harford, C.G.; Hamlin, A.; Rieders, E. Electron microscopic autoradiography of DNA synthesis in cells infected with vaccinia virus. Exp. Cell Res. 1966, 42, 50–57. [Google Scholar] [CrossRef]
- Katsafanas, G.C.; Moss, B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2007, 2, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolonen, N.; Doglio, L.; Schleich, S.; Krijnse Locker, J. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol. Biol. Cell 2001, 12, 2031–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszkowski, P.; Noyce, R.S.; Evans, D.H. Live-Cell Imaging of Vaccinia Virus Recombination. PLoS Pathog. 2016, 12, e1005824. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallardo, F.; Schmitt, D.; Brandely, R.; Brua, C.; Silvestre, N.; Findeli, A.; Foloppe, J.; Top, S.; Kappler-Gratias, S.; Quentin-Froignant, C.; et al. Fluorescent Tagged Vaccinia Virus Genome Allows Rapid and Efficient Measurement of Oncolytic Potential and Discovery of Oncolytic Modulators. Biomedicines 2020, 8, 543. https://doi.org/10.3390/biomedicines8120543
Gallardo F, Schmitt D, Brandely R, Brua C, Silvestre N, Findeli A, Foloppe J, Top S, Kappler-Gratias S, Quentin-Froignant C, et al. Fluorescent Tagged Vaccinia Virus Genome Allows Rapid and Efficient Measurement of Oncolytic Potential and Discovery of Oncolytic Modulators. Biomedicines. 2020; 8(12):543. https://doi.org/10.3390/biomedicines8120543
Chicago/Turabian StyleGallardo, Franck, Doris Schmitt, Renée Brandely, Catherine Brua, Nathalie Silvestre, Annie Findeli, Johann Foloppe, Sokunthea Top, Sandrine Kappler-Gratias, Charlotte Quentin-Froignant, and et al. 2020. "Fluorescent Tagged Vaccinia Virus Genome Allows Rapid and Efficient Measurement of Oncolytic Potential and Discovery of Oncolytic Modulators" Biomedicines 8, no. 12: 543. https://doi.org/10.3390/biomedicines8120543