

Repeat Element Activation-Driven Inflammation: Role of NFκB and Implications in Normal Development and Cancer?
 , ,
, ,
Abstract
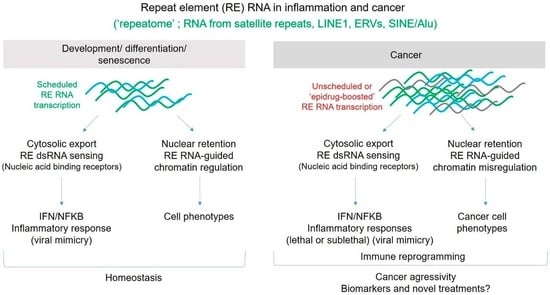
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Overview of Repeat Elements: Genomic Organization and Transcriptional Activity
2.1. Genomic Organization of Repeat Elements
2.2. Repeat Element Activity in Chromatin Organization, Gene Regulation and Innate Immune Signaling: State of the Art
3. Repeat Element Activation in Normal Development, Senescence and Cellular Differentiation: Emerging Paradigms
3.1. RE-Derived RNA in Development: Role in Chromatin Organization and Control of Gene Expression
3.2. Repeat Element Transcript-Activated Innate Immune Receptor Signaling, NFκB, Inflammation and Hematopoiesis
3.2.1. Inflammation and Hematopoiesis
3.2.2. RIG1 Receptors and Hematopoiesis
RIG-1-like Receptors
RIG Receptors and Hematopoiesis: Role of Repeat Element Transcripts and Interferon and NFκB Signaling
Transposable Element Transcript-Driven Activation of MDA5 for Enhanced Hematopoietic Regeneration after Chemotherapy
3.3. RE Activity in Senescence-Associated Inflammatory Signaling
4. Repeat Element Activation in Cancer: Functional Impact on Cancer Cell Phenotypes and Immune Signaling
4.1. Overview of Viral Mimicry in Solid Cancers and Acute Myeloid Leukemia
4.1.1. Pancreatic Ductal Adenocarcinoma (PDAC)
4.1.2. Colon Cancer
4.1.3. Ovarian Cancer
4.1.4. Melanoma
4.1.5. Triple Negative Breast Cancer (TNBC)
4.1.6. Acute Myeloid Leukemia (AML)
4.2. Therapeutic Intervention for Derepression of Repeat Elements in Cancer: A Novel Approach for Immuno-Oncology?
4.3. Repeat Element Activation in Oncology/Onco-Hematology: Biomarker Value?
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, S.; Allis, C.D.; Wang, G.G. The language of chromatin modification in human cancers. Nat. Rev. Cancer 2021, 21, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Ishak, C.A.; De Carvalho, D.D. Endogenous Retroelements and the Viral Mimicry Response in Cancer Therapy and Cellular Homeostasis. Cancer Discov. 2021, 11, 2707–2725. [Google Scholar] [CrossRef] [PubMed]
- Nicetto, D.; Zaret, K.S. Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Curr. Opin. Genet. Dev. 2019, 55, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640. [Google Scholar] [CrossRef]
- Kim, S.; Shendure, J. Mechanisms of Interplay between Transcription Factors and the 3D Genome. Mol. Cell 2019, 76, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Vourch, C.; Dufour, S.; Timcheva, K.; Seigneurin-Berny, D.; Verdel, A. HSF1-Activated Non-Coding Stress Response: Satellite lncRNAs and Beyond, an Emerging Story with a Complex Scenario. Genes 2022, 13, 597. [Google Scholar] [CrossRef]
- Percharde, M.; Bulut-Karslioglu, A.; Ramalho-Santos, M. Hypertranscription in Development, Stem Cells, and Regeneration. Dev. Cell 2017, 40, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Lefkopoulos, S.; Polyzou, A.; Derecka, M.; Bergo, V.; Clapes, T.; Cauchy, P.; Jerez-Longres, C.; Onishi-Seebacher, M.; Yin, N.; Martagon-Calderon, N.A.; et al. Repetitive Elements Trigger RIG-I-like Receptor Signaling that Regulates the Emergence of Hematopoietic Stem and Progenitor Cells. Immunity 2020, 53, 934–951. [Google Scholar] [CrossRef]
- Clapes, T.; Trompouki, E. Hematopoietic regeneration under the spell of epigenetic-epitranscriptomic factors and transposable elements. Curr. Opin. Hematol. 2020, 27, 264–272. [Google Scholar] [CrossRef]
- Clapes, T.; Polyzou, A.; Prater, P.; Sagar; Morales-Hernandez, A.; Ferrarini, M.G.; Kehrer, N.; Lefkopoulos, S.; Bergo, V.; Hummel, B.; et al. Chemotherapy-induced transposable elements activate MDA5 to enhance haematopoietic regeneration. Nat. Cell Biol. 2021, 23, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, T.L.; Herzner, A.M.; Zhang, X.; Goyal, Y.; Watanabe, C.; Friedman, B.A.; Janakiraman, V.; Durinck, S.; Stinson, J.; Arnott, D.; et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol. 2017, 216, 3535–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deblois, G.; Tonekaboni, S.A.M.; Grillo, G.; Martinez, C.; Kao, Y.I.; Tai, F.; Ettayebi, I.; Fortier, A.M.; Savage, P.; Fedor, A.N.; et al. Epigenetic Switch-Induced Viral Mimicry Evasion in Chemotherapy-Resistant Breast Cancer. Cancer Discov. 2020, 10, 1312–1329. [Google Scholar] [CrossRef] [PubMed]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Levine, A.J.; Ting, D.T.; Greenbaum, B.D. P53 and the defenses against genome instability caused by transposons and repetitive elements. Bioessays 2016, 38, 508–513. [Google Scholar] [CrossRef]
- Zhu, Q.; Pao, G.M.; Huynh, A.M.; Suh, H.; Tonnu, N.; Nederlof, P.M.; Gage, F.H.; Verma, I.M. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 2011, 477, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Ishak, C.A.; Marshall, A.E.; Passos, D.T.; White, C.R.; Kim, S.J.; Cecchini, M.J.; Ferwati, S.; MacDonald, W.A.; Howlett, C.J.; Welch, I.D.; et al. An RB-EZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol. Cell 2016, 64, 1074–1087. [Google Scholar] [CrossRef] [Green Version]
- Puszyk, W.; Down, T.; Grimwade, D.; Chomienne, C.; Oakey, R.J.; Solomon, E.; Guidez, F. The epigenetic regulator PLZF represses L1 retrotransposition in germ and progenitor cells. EMBO J. 2013, 32, 1941–1952. [Google Scholar] [CrossRef]
- Altemose, N.; Logsdon, G.A.; Bzikadze, A.V.; Sidhwani, P.; Langley, S.A.; Caldas, G.V.; Hoyt, S.J.; Uralsky, L.; Ryabov, F.D.; Shew, C.J.; et al. Complete genomic and epigenetic maps of human centromeres. Science 2022, 376, eabl4178. [Google Scholar] [CrossRef]
- Altemose, N. A classical revival: Human satellite DNAs enter the genomics era. Semin. Cell Dev. Biol. 2022, 128, 2–14. [Google Scholar] [CrossRef]
- Hoyt, S.J.; Storer, J.M.; Hartley, G.A.; Grady, P.G.S.; Gershman, A.; de Lima, L.G.; Limouse, C.; Halabian, R.; Wojenski, L.; Rodriguez, M.; et al. From telomere to telomere: The transcriptional and epigenetic state of human repeat elements. Science 2022, 376, eabk3112. [Google Scholar] [CrossRef] [PubMed]
- Onishi-Seebacher, M.; Erikson, G.; Sawitzki, Z.; Ryan, D.; Greve, G.; Lubbert, M.; Jenuwein, T. Repeat to gene expression ratios in leukemic blast cells can stratify risk prediction in acute myeloid leukemia. BMC Med. Genom. 2021, 14, 166. [Google Scholar] [CrossRef]
- Burns, K.H. Transposable elements in cancer. Nat. Rev. Cancer 2017, 17, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Seluanov, A.; Mita, P.; McKerrow, W.; Fenyo, D.; Boeke, J.D.; Linker, S.B.; Gage, F.H.; Kreiling, J.A.; Petrashen, A.P.; et al. The role of retrotransposable elements in ageing and age-associated diseases. Nature 2021, 596, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.V.; Okamoto, I.; Casanova, M.; El Marjou, F.; Le Baccon, P.; Almouzni, G. A strand-specific burst in transcription of pericentric satellites is required for chromocenter formation and early mouse development. Dev. Cell 2010, 19, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.Y.; Shao, W.; Chang, L.; Yin, Y.; Li, T.; Zhang, H.; Hong, Y.; Percharde, M.; Guo, L.; Wu, Z.; et al. Genomic Repeats Categorize Genes with Distinct Functions for Orchestrated Regulation. Cell Rep. 2020, 30, 3296–3311. [Google Scholar] [CrossRef] [Green Version]
- Solovyov, A.; Vabret, N.; Arora, K.S.; Snyder, A.; Funt, S.A.; Bajorin, D.F.; Rosenberg, J.E.; Bhardwaj, N.; Ting, D.T.; Greenbaum, B.D. Global Cancer Transcriptome Quantifies Repeat Element Polarization between Immunotherapy Responsive and T Cell Suppressive Classes. Cell Rep. 2018, 23, 512–521. [Google Scholar] [CrossRef]
- Ting, D.T.; Lipson, D.; Paul, S.; Brannigan, B.W.; Akhavanfard, S.; Coffman, E.J.; Contino, G.; Deshpande, V.; Iafrate, A.J.; Letovsky, S.; et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 2011, 331, 593–596. [Google Scholar] [CrossRef] [Green Version]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef] [Green Version]
- Porter, R.L.; Sun, S.; Flores, M.N.; Berzolla, E.; You, E.; Phillips, I.E.; Kc, N.; Desai, N.; Tai, E.C.; Szabolcs, A.; et al. Satellite repeat RNA expression in epithelial ovarian cancer associates with a tumor immunosuppressive phenotype. J. Clin. Investig. 2022, 132, e155931. [Google Scholar] [CrossRef]
- Kermi, C.; Lau, L.; Asadi Shahmirzadi, A.; Classon, M. Disrupting Mechanisms that Regulate Genomic Repeat Elements to Combat Cancer and Drug Resistance. Front. Cell Dev. Biol. 2022, 10, 826461. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.L.; Byron, M.; Carone, D.M.; Whitfield, T.W.; Pouliot, G.P.; Fischer, A.; Jones, P.; Lawrence, J.B. Demethylated HSATII DNA and HSATII RNA Foci Sequester PRC1 and MeCP2 into Cancer-Specific Nuclear Bodies. Cell Rep. 2017, 18, 2943–2956. [Google Scholar] [CrossRef] [PubMed]
- Fournier, A.; McLeer-Florin, A.; Lefebvre, C.; Duley, S.; Barki, L.; Ribeyron, J.; Alboukadel, K.; Hamaidia, S.; Granjon, A.; Gressin, R.; et al. 1q12 chromosome translocations form aberrant heterochromatic foci associated with changes in nuclear architecture and gene expression in B cell lymphoma. EMBO Mol. Med. 2010, 2, 159–171. [Google Scholar] [CrossRef]
- Tanne, A.; Muniz, L.R.; Puzio-Kuter, A.; Leonova, K.I.; Gudkov, A.V.; Ting, D.T.; Monasson, R.; Cocco, S.; Levine, A.J.; Bhardwaj, N.; et al. Distinguishing the immunostimulatory properties of noncoding RNAs expressed in cancer cells. Proc. Natl. Acad. Sci. USA 2015, 112, 15154–15159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vabret, N.; Bhardwaj, N.; Greenbaum, B.D. Sequence-Specific Sensing of Nucleic Acids. Trends Immunol. 2017, 38, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Rajurkar, M.; Parikh, A.R.; Solovyov, A.; You, E.; Kulkarni, A.S.; Chu, C.; Xu, K.H.; Jaicks, C.; Taylor, M.S.; Wu, C.; et al. Reverse Transcriptase Inhibition Disrupts Repeat Element Life Cycle in Colorectal Cancer. Cancer Discov. 2022, 12, 1462–1481. [Google Scholar] [CrossRef]
- Volkman, H.E.; Stetson, D.B. The enemy within: Endogenous retroelements and autoimmune disease. Nat. Immunol. 2014, 15, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Burton, A.; Brochard, V.; Galan, C.; Ruiz-Morales, E.R.; Rovira, Q.; Rodriguez-Terrones, D.; Kruse, K.; Le Gras, S.; Udayakumar, V.S.; Chin, H.G.; et al. Heterochromatin establishment during early mammalian development is regulated by pericentromeric RNA and characterized by non-repressive H3K9me3. Nat. Cell. Biol. 2020, 22, 767–778. [Google Scholar] [CrossRef]
- Nagasawa, T. Microenvironmental niches in the bone marrow required for B-cell development. Nat. Rev. Immunol. 2006, 6, 107–116. [Google Scholar] [CrossRef]
- Hosokawa, H.; Rothenberg, E.V. How transcription factors drive choice of the T cell fate. Nat. Rev. Immunol. 2021, 21, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Pucella, J.N.; Upadhaya, S.; Reizis, B. The Source and Dynamics of Adult Hematopoiesis: Insights from Lineage Tracing. Annu. Rev. Cell Dev. Biol. 2020, 36, 529–550. [Google Scholar] [CrossRef] [PubMed]
- Weijts, B.; Yvernogeau, L.; Robin, C. Recent Advances in Developmental Hematopoiesis: Diving Deeper With New Technologies. Front. Immunol. 2021, 12, 790379. [Google Scholar] [CrossRef] [PubMed]
- Sugden, W.W.; North, T.E. Making Blood from the Vessel: Extrinsic and Environmental Cues Guiding the Endothelial-to-Hematopoietic Transition. Life 2021, 11, 1027. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Crosier, P.; Crosier, K. Inflammatory cytokines provide both infection-responsive and developmental signals for blood development: Lessons from the zebrafish. Mol. Immunol. 2016, 69, 113–122. [Google Scholar] [CrossRef]
- Espin-Palazon, R.; Traver, D. The NF-kappaB family: Key players during embryonic development and HSC emergence. Exp. Hematol. 2016, 44, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Clapes, T.; Lefkopoulos, S.; Trompouki, E. Stress and Non-Stress Roles of Inflammatory Signals during HSC Emergence and Maintenance. Front. Immunol. 2016, 7, 487. [Google Scholar] [CrossRef] [Green Version]
- Sawamiphak, S.; Kontarakis, Z.; Stainier, D.Y. Interferon gamma signaling positively regulates hematopoietic stem cell emergence. Dev. Cell 2014, 31, 640–653. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Esain, V.; Teng, L.; Xu, J.; Kwan, W.; Frost, I.M.; Yzaguirre, A.D.; Cai, X.; Cortes, M.; Maijenburg, M.W.; et al. Inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell production. Genes Dev. 2014, 28, 2597–2612. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Zhang, C.; Wang, L.; Zhang, P.; Ma, D.; Lv, J.; Liu, F. Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood 2015, 125, 1098–1106. [Google Scholar] [CrossRef]
- Espin-Palazon, R.; Stachura, D.L.; Campbell, C.A.; Garcia-Moreno, D.; Del Cid, N.; Kim, A.D.; Candel, S.; Meseguer, J.; Mulero, V.; Traver, D. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell 2014, 159, 1070–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rotig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ukadike, K.C.; Mustelin, T. Implications of Endogenous Retroelements in the Etiopathogenesis of Systemic Lupus Erythematosus. J. Clin. Med. 2021, 10, 856. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, H.; Wu, J.; Cai, Z.Y.; Li, B.; Ni, H.; Qiu, X.; Chen, H.; Liu, W.; Yang, Z.H.; et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 2020, 580, 386–390. [Google Scholar] [CrossRef]
- Bersani, F.; Lee, E.; Kharchenko, P.V.; Xu, A.W.; Liu, M.; Xega, K.; MacKenzie, O.C.; Brannigan, B.W.; Wittner, B.S.; Jung, H.; et al. Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15148–15153. [Google Scholar] [CrossRef] [Green Version]
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021, 595, 309–314. [Google Scholar] [CrossRef]
- Mehdipour, P.; Marhon, S.A.; Ettayebi, I.; Chakravarthy, A.; Hosseini, A.; Wang, Y.; de Castro, F.A.; Yau, H.L.; Ishak, C.; Abelson, S.; et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 2020, 588, 169–173. [Google Scholar] [CrossRef]
- Wylie, A.; Jones, A.E.; D’Brot, A.; Lu, W.J.; Kurtz, P.; Moran, J.V.; Rakheja, D.; Chen, K.S.; Hammer, R.E.; Comerford, S.A.; et al. p53 genes function to restrain mobile elements. Genes Dev. 2016, 30, 64–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonova, K.I.; Brodsky, L.; Lipchick, B.; Pal, M.; Novototskaya, L.; Chenchik, A.A.; Sen, G.C.; Komarova, E.A.; Gudkov, A.V. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc. Natl. Acad. Sci. USA 2013, 110, E89–E98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinet, E.; Gu, Z.; Imbusch, C.D.; Giese, N.A.; Buscher, M.; Safavi, M.; Weisenburger, S.; Klein, C.; Vogel, V.; Falcone, M.; et al. Aggressive PDACs Show Hypomethylation of Repetitive Elements and the Execution of an Intrinsic IFN Program Linked to a Ductal Cell of Origin. Cancer Discov. 2021, 11, 638–659. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.L.; Magnus, N.K.C.; Thapar, V.; Morris, R.; Szabolcs, A.; Neyaz, A.; Kulkarni, A.S.; Tai, E.; Chougule, A.; Hillis, A.; et al. Epithelial to mesenchymal plasticity and differential response to therapies in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 26835–26845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.e27. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.; Sajed, D.; Arora, K.S.; Solovyov, A.; Rajurkar, M.; Bledsoe, J.R.; Sil, S.; Amri, R.; Tai, E.; MacKenzie, O.C.; et al. Diverse repetitive element RNA expression defines epigenetic and immunologic features of colon cancer. JCI Insight 2017, 2, e91078. [Google Scholar] [CrossRef] [Green Version]
- Nogalski, M.T.; Solovyov, A.; Kulkarni, A.S.; Desai, N.; Oberstein, A.; Levine, A.J.; Ting, D.T.; Shenk, T.; Greenbaum, B.D. A tumor-specific endogenous repetitive element is induced by herpesviruses. Nat. Commun. 2019, 10, 90. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, S. Breaking satellite silence: Human satellite II RNA expression in ovarian cancer. J. Clin. Investig. 2022, 132, e161981. [Google Scholar] [CrossRef]
- Badal, B.; Solovyov, A.; Di Cecilia, S.; Chan, J.M.; Chang, L.W.; Iqbal, R.; Aydin, I.T.; Rajan, G.S.; Chen, C.; Abbate, F.; et al. Transcriptional dissection of melanoma identifies a high-risk subtype underlying TP53 family genes and epigenome deregulation. JCI Insight 2017, 2, e92102. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Nie, D.Y.; Ba-Alawi, W.; Ji, Y.; Zhang, Z.; Cruickshank, J.; Haight, J.; Ciamponi, F.E.; Chen, J.; Duan, S.; et al. PRMT inhibition induces a viral mimicry response in triple-negative breast cancer. Nat. Chem. Biol. 2022, 18, 821–830. [Google Scholar] [CrossRef]
- Colombo, A.R.; Zubair, A.; Thiagarajan, D.; Nuzhdin, S.; Triche, T.J.; Ramsingh, G. Suppression of Transposable Elements in Leukemic Stem Cells. Sci. Rep. 2017, 7, 7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Singh, M.; Sanz Santos, G.; Guerlavais, V.; Carvajal, L.A.; Aivado, M.; Zhan, Y.; Oliveira, M.M.S.; Westerberg, L.S.; Annis, D.A.; et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021, 11, 3090–3105. [Google Scholar] [CrossRef] [PubMed]
- Limagne, E.; Nuttin, L.; Thibaudin, M.; Jacquin, E.; Aucagne, R.; Bon, M.; Revy, S.; Barnestein, R.; Ballot, E.; Truntzer, C.; et al. MEK inhibition overcomes chemoimmunotherapy resistance by inducing CXCL10 in cancer cells. Cancer Cell 2022, 40, 136–152. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.; Lobb, R.J. The evolving translational potential of small extracellular vesicles in cancer. Nat. Rev. Cancer 2020, 20, 697–709. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dumetier, B.; Sauter, C.; Hajmirza, A.; Pernon, B.; Aucagne, R.; Fournier, C.; Row, C.; Guidez, F.; Rossi, C.; Lepage, C.; et al. Repeat Element Activation-Driven Inflammation: Role of NFκB and Implications in Normal Development and Cancer? Biomedicines 2022, 10, 3101. https://doi.org/10.3390/biomedicines10123101
Dumetier B, Sauter C, Hajmirza A, Pernon B, Aucagne R, Fournier C, Row C, Guidez F, Rossi C, Lepage C, et al. Repeat Element Activation-Driven Inflammation: Role of NFκB and Implications in Normal Development and Cancer? Biomedicines. 2022; 10(12):3101. https://doi.org/10.3390/biomedicines10123101
Chicago/Turabian StyleDumetier, Baptiste, Camille Sauter, Azadeh Hajmirza, Baptiste Pernon, Romain Aucagne, Cyril Fournier, Céline Row, Fabien Guidez, Cédric Rossi, Côme Lepage, and et al. 2022. "Repeat Element Activation-Driven Inflammation: Role of NFκB and Implications in Normal Development and Cancer?" Biomedicines 10, no. 12: 3101. https://doi.org/10.3390/biomedicines10123101