
Exploiting DNA Replication Stress as a Therapeutic Strategy for Breast Cancer

Department of Systems Biology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
*
Authors to whom correspondence should be addressed.
Biomedicines 2022, 10(11), 2775; https://doi.org/10.3390/biomedicines10112775
Submission received: 13 September 2022
/
Revised: 19 October 2022
/
Accepted: 27 October 2022
/
Published: 1 November 2022
(This article belongs to the Special Issue Breast Cancer: From Biology to Therapeutic Opportunities)
Abstract
:Proliferating cells rely on DNA replication to ensure accurate genome duplication. Cancer cells, including breast cancer cells, exhibit elevated replication stress (RS) due to the uncontrolled oncogenic activation, loss of key tumor suppressors, and defects in the DNA repair machinery. This intrinsic vulnerability provides a great opportunity for therapeutic exploitation. An increasing number of drug candidates targeting RS in breast cancer are demonstrating promising efficacy in preclinical and early clinical trials. However, unresolved challenges lie in balancing the toxicity of these drugs while maintaining clinical efficacy. Furthermore, biomarkers of RS are urgently required to guide patient selection. In this review, we introduce the concept of targeting RS, detail the current therapies that target RS, and highlight the integration of RS with immunotherapies for breast cancer treatment. Additionally, we discuss the potential biomarkers to optimizing the efficacy of these therapies. Together, the continuous advances in our knowledge of targeting RS would benefit more patients with breast cancer.
1. Introduction
DNA replication in eukaryotic cells is a multifaceted process, which depends on the activation of numerous signaling pathways to accurately replicate the genome [1]. This process is constantly challenged by events of endogenous or exogenous origin that impede the rate and fidelity of DNA synthesis, thus affecting the integrity of genome. These events, collectively termed replication stress (RS), include DNA lesions such as DNA single-stranded or double-stranded breaks, unusual DNA secondary structures, RNA–DNA hybrids, deficiencies in nucleotide levels, oncogene activation, chromatin inaccessibility and limitation of essential replication factors [2]. In response to RS, cells elicit the DNA damage response (DDR) and subsequently inhibition of cell-cycle progression, which is known as the replication stress response (RSR). The purpose of RSR is to slow DNA synthesis and replication to allow time for DNA repair [3]. The RSR is primarily coordinated by two signaling cascades: ataxia telangiectasia and Rad3-related (ATR)–checkpoint kinase 1 (CHK1) pathway and ataxia telangiectasia mutated (ATM)–checkpoint kinase 2 (CHK2) pathway [4,5,6,7]. Cells rely on these coordinated pathways to prevent mitosis in the presence of DNA damage [8]. Defects in RSR allow for high levels of DNA damage and genomic instability that not only alter gene function, but also lead to continuous proliferation, ultimately provoking carcinogenesis and tumor progression [9,10,11,12,13].
Breast cancer is one of the most frequently diagnosed neoplasms worldwide, with one-third of these patients subsequently dying of this disease. In this heterogeneous disease, an important cause of replication stress is overexpression or constitutive activation of oncogenes, loss of key tumor suppressors, and defects in the DNA repair machinery [2]. Over the past decades, treatments for breast cancer have advanced considerably with effective therapies targeting the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor-2 (HER2). However, patients with triple-negative breast cancer (TNBC), i.e., estrogen receptor-negative, progesterone receptor-negative and HER2-negative, fail to benefit from these treatments. Notably, it was reported that TNBC has high levels of RS due to the activation of various oncogenes and germline BRCA mutations or “BRCAness” in the absence of BRCA mutations [14]. BRCAness is defined as a defect in double-strand break repair (DSBR) via homologous recombination repair (HRR) [15,16]. In the HRR process, recombinase Rad51 interacts with BRCA1 and BRCA2 to perform the search for homologous DNA sequences [16,17]. It has been shown that most hereditary breast cancers are associated with abnormal BRCA1 and BRCA2 genes [18]. Recently, Baldeyron et al. showed that TIPIN depletion results in apoptosis in breast cancer cells by enhancing replication stress [19]. In this context, breast cancer cells evolve various replication stress-resolving mechanisms to avoid toxic levels of replication stress for their survival. Thus, understanding the mechanisms that breast cancer cells have developed to deal with RS could provide promising therapeutic opportunities.
Due to the dependency of cancer cells on the replication stress response for survival, small molecular agents targeting DNA replication stress are under development and have shown antitumor activity in preclinical and clinical studies [20,21]. Cancer cells can be pushed toward apoptosis by introducing further DNA damage by targeting key kinases in RSR, or by using RS to stimulate innate immune response. In this review, we summarize recently gained insights into the mechanisms of the cellular response to RS, discuss the promising therapeutic strategies centered on targeting DNA replication stress in breast cancer, and highlight the potential biomarkers to optimizing the efficacy of these therapies.
2. Mechanisms of the Cellular Response to Replication Stress and Rationale in Breast Cancer Therapy
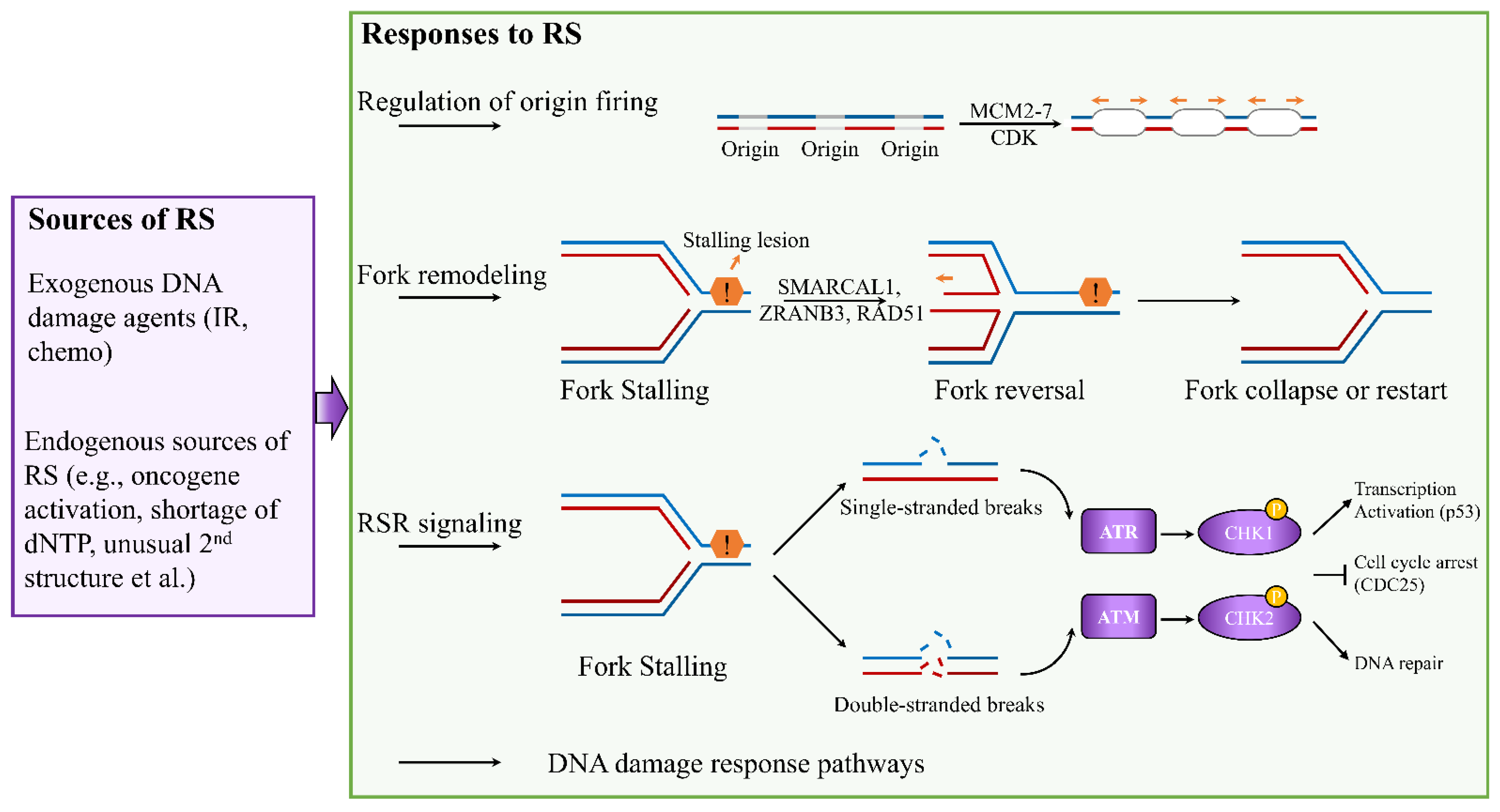
Accurate DNA replication is critical to ensure genomic integrity. The fidelity of this process is often challenged by RS, leading to altered replication fork progression and generation of DNA breaks [2,22]. Depending on the types of RS, cells activate four major responses to ensure accurate genome duplication: (1) regulation of origin firing, (2) remodeling of replication forks, (3) activation of replication stress response signaling, and (4) deployment of DNA damage response pathways (Figure 1) [23,24,25].
Eukaryotic genomes are duplicated exactly once during the S phases of each cell cycle. To control replication initiation, the replicative DNA helicase minichromosome maintenance complex 2–7 (MCM2–7) is loaded at replication origins during G1 phase, and activated only during the S phase [6]. This process is called licensing. Activation of licensed origins, which is known as origin firing, requires the activities of cyclin-dependent kinase (CDK) [6]. Deregulation of origin activation can generate replication stress in breast cancer. For example, high levels of MCM2 are associated with poor survival in patients with breast cancer [26,27]. Furthermore, Issac et al. have showed that increased protein expression of MCM2, MCM4, and MCM6 is associated with luminal B, HER2-positive, and TNBC [28]. These MCM proteins may serve as potential treatment targets for breast cancer patients. Unscheduled replication is another source of replication stress in breast cancer. This occurs when the timing of origin activation is altered, leading to DNA regions replicating more than once in one cell cycle or an increase in origin firing in early S phase [6]. Unscheduled replication occurs under the upregulation of DNA replication factor CDT1 and cell division control protein 6 (CDC6). High levels of CDT1 and CDC6 are associated with poorer survival in the breast cancer patients, suggesting that CDT1 and CDC6 are potential therapeutic targets for treatment of breast cancer [29]. To date, the mechanism that regulate origin firing remain largely unknown, and few drugs targeting these key proteins are in the clinical trials. Thus, better understanding the regulation of the origin firing can provide novel therapeutic strategies in the treatment of breast cancer.
Replication fork remodeling is a common response to RS, which involves unwinding of newly synthesized strands and annealing of parental strands. In this remodeling process, stalled forks are converted into four-way junctions to facilitate DNA damage repair [24]. Many key factors involved in reversed fork restart have been identified, including ZRANB3 and SMARCAL1 in reversed fork formation, and BRCA1 and BRCA2 in reversed fork protection [30,31]. RAD51, well-known for catalyzing strand invasion in homologous recombination (HR) repair of DNA double-strand breaks, also plays an important role in regulating replication fork reversal [23]. Furthermore, PARP1 has been linked to recruitment of MRE11 to stalled forks and regulation of fork restart to restore replication fork stability [32,33]. Similar to PARP1, RAD52 can promote the recruitment of MRE11 to stalled replication forks and fork degradation [34,35]. Additionally, DNA2, which functions in dsDNA break repair, has been shown to assist Werner syndrome helicase WRN in controlling HR-mediated restart of reversed replication forks [36,37]. Recently, Dibitetto et al. identified the essential role of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) in promoting fork reversal and preventing genome instability [38]. They found that DNA-PKcs inhibition in BRCA2-deficient breast cancer with acquired PARPi resistance efficiently restored drug sensitivity by impairing fork slowing. The identification of these key proteins in replication fork remodeling provides great opportunities to target RS in cancer therapy.
To cope with RS, cells can also rely on RSR cascade to arrest the cell cycle, protect stalled forks, and allow time to repair the replication fork (Figure 1). RS can perturb the coupling between the replicative helicase and polymerases, resulting in the uncoupling of leading- and lagging-strand synthesis, which generates double-strand breaks (DSBs) and/or single-strand DNA (ssDNA) gaps [6]. DSBs primarily trigger activation of ATM and DNA-dependent protein kinase (DNA-PK), whereas ssDNA coated with replication protein A (RPA) activates ATR via ATR-interacting protein (ATRIP) [39,40,41]. ATR and ATM are apical checkpoint kinases, which regulate the cellular response to replication fork blockage and DNA damage [42]. These kinases activate the effector checkpoint kinases CHK1 and CHK2, respectively, and they regulate the timing of replication origin firing independently of DNA damage [42]. Activation of the ATR–CHK1 pathway leads to cell-cycle arrest by inactivating the cell division cycle 25 (CDC25) phosphatase family or through WEE1 kinase [43,44]. This ATR–CHK1 signaling also promotes replication fork stabilization and restart [45]. Furthermore, H2AX, phosphorylated in an ATR-dependent manner in response to replication stress, recruits RAD51 at reversed forks to protect synthesized DNA degradation and assists with fork restart [46]. Both CHK1 and CHK2 can regulate transcription activation drive by p53 [6]. In addition to replication fork remodeling and RSR signaling, several DDR pathways are activated at stalled forks to allow for recovery from the RS [47,48]. Together, these steps are essential to ensure timely completion of DNA replication and maintain genome stability.
Failure to remove replication stressors due to the loss of replication stress response and repair proteins is a prominent feature of tumor cells. Because of this key feature that distinguishes cancer cells from normal cells, cancer cells harboring replication stress can be targeted through three major mechanisms (Figure 2). First, cancer cells can be pushed toward cell death by enhancing replication stress to induce replication catastrophe [21,49]. Second, cancer cells can be targeted by inhibiting the key kinases of the RSR cascade that cells depend upon to survive, such as ATR, ATM, CHK1, WEE1, and DNA-PK. Inhibition of these key mediators ultimately promotes the premature entry of tumor cells into mitosis, inducing mitotic catastrophe [7,49]. Third, replication stress due to chemotherapies or impaired DNA repair factors induces DNA release from the nucleus to cytoplasm. These DNA fragments trigger cytosolic DNA sensing and interferon signaling, which subsequently prime tumor for immune therapies [7,50,51,52]. The formation of micronuclei has been shown to be a key source of immunostimulatory DNA in cells lacking RNase H2 [53]. However, the release of ssDNA fragment directly from stalled replication forks in TREX1- or SAMHD1-deficient cells has also been shown to induce interferon signaling [51,52]. Several aspects of these mechanisms such as the interplay of proteins in response to RS to promote DNA fragments release from the nucleus into the cytoplasm have yet to be defined.
3. Therapeutic Strategies That Induce Replication Catastrophe in Breast Cancer
Since the persistence of replication stress is observed almost exclusively in cancer cells, enhancing replication stress can paradoxically lead to cell death by introducing further DNA damage in a catastrophic manner. Many traditional chemotherapeutic agents, acting by increasing the endogenous replication stress within breast cancer cells, have been developed and have shown antitumor activity (Table 1). For instance, gemcitabine, a deoxycytidine analogue, leads to a delay in replication fork progression by inhibiting ribonucleotide reductase and by competing with dCTP for incorporation into newly synthesized DNA [54]. Another nucleoside analogue 5-fluorouracil, frequently used to treat breast cancer, functions mainly by inhibition of thymidylate synthetase to reduce the amount of thymidine for DNA replication [55,56]. TAS1553, a small-molecule subunit interaction inhibitor of ribonucleotide reductase, has shown antiproliferative activity in breast cancer cells by dramatically reducing the intracellular dATP pool and causing DNA replication stress [57].
Unlike nucleoside analogues, which reduce dNTP pools to direct inhibition of DNA synthesis, alkylating agents and platinum-containing compounds increase the replication stress in breast cancer cells by directly modifying DNA through attacking the DNA bases forming covalent DNA adducts [83,84]. The formation of DNA adducts interferes with the progression of DNA polymerases, which results in delay replication fork progression. In addition to these RS inducers, topoisomerase inhibitors can increase replication stress by promoting R-loop formation, leading to global replication fork slowdown [85]. Another class of replication stress inducing agents that target breast cancer cells by directly inducing DNA damage is represented by poly(ADP-ribose) polymerase (PARP) inhibitors. PARP inhibitors are believed to interfere with the replication machinery and promote fork collapse by trapping PARP on DNA [86,87]. Additionally, PARP inhibitors cause an accumulation of single-strand breaks (SSBs) or Okazaki fragment processing [88,89]. More recently, PARP inhibitors were reported to increase the speed of fork elongation and amplify the replication stress in breast cancer cells [90].
Given that DNA polymerases play key roles in replication stress sensing upstream of ATR, inhibitors targeting DNA polymerases are a novel and attractive class of drugs that are in preclinical development. CD437, targeting POLA1 which encodes DNA polymerase α, has been shown to induce apoptosis in breast cancer cells but not in normal cells [67,91]. Additionally, a small-molecule inhibitor of DNA polymerase δ, zelpolib, displays superior antiproliferative properties in triple-negative breast cancer cells by inhibiting DNA replication and enhances the sensitivity of HR-proficient cells to PARP inhibitors [68]. Although these preclinical studies demonstrate the potential of these agents, further clinical evaluation is required to determine their efficiency.
In addition, understanding the collaboration among proteins that regulate replication fork remodeling provides novel therapeutic targets for capitalizing on endogenous replication stress in breast cancer (Table 1). DNA2 has been shown to play important roles in processing and restarting of reversed replication forks [36]. A selective small-molecule DNA2 inhibitor C5 has been reported to inhibit resection at stalled forks, as well as reduce recombination [69]. This compound is more potent in breast cancer cells defective in replication fork protection and sensitizes cells to traditional RS-inducing drugs. Recently, another DNA2 inhibitor NSC-105808 was identified to sensitize breast cancer cells to oncogene-induced replication stress [37]. However, these studies are still in their early stages. Whether these proteins involved in replication fork remodeling might be successfully developed as therapeutic targets, and what the therapeutic efficiency of these new drugs might remain to be investigated.
While these replication stress inducers target breast cancer directly, their efficacy is limited due to their associated toxicities and the rapid emergence of resistance. Thus, synergistic effects of two different replication stress inducers have been investigated. For example, combinations of PARPi and other replication stress inducers, such as cisplatin [92,93], carboplatin [94,95], temozolomide (NCT05128734), and topoisomerase inhibitors [96,97] have been proven effective in early clinical trials. Despite the moderate success, more effective therapies are still needed for breast cancers which are resistant to these RS inducers. In addition to the combination of different replication stress inducers, targeting the replication stress response signaling pathways is emerging as a promising strategy.
4. Targeting Replication Stress Response Signaling
If breast cancer cells with high levels of replicative lesions do not initiate cell death, they likely rely on RSR to provide sufficient time to deal with such lesions. The main RSR mediators for inducing cell-cycle delay or protecting stalled forks could, therefore, be promising targets (Table 1). Given the central role of ATR in preventing replication fork collapse, ATR inhibition initiates widespread DNA synthesis from dormant replication origins, generating ssDNA to exhaust the cellular pools of RPA [98,99]. ATR inhibitors (ATRi), such as AZD6738, BAY1895344, and M6620, have shown antitumor activity in preclinical and clinical studies [100]. Additional agent RP-3500 targeting ATR in breast cancer is currently under clinical phase 1 trial [72]. CHK1, the key downstream effector protein of the ATR response, plays an important role in triggering the S-phase checkpoint upon replication stress and preventing premature entry into mitosis. Currently, two CHK1 inhibitors, prexasertib [101] and GDC-0575 [74], have been reported to show preliminary antitumor activity in patients with breast cancer.
In addition to these two key players, the RSR also involves other important kinases, including WEE1, DNA-PK, and ATM, to arrest the cell cycle, protect stalled forks, and allow time for replication fork repair. For instance, the activation of ATR–CHK1 signaling can delay cell-cycle progression through WEE1 kinase [43]. A small-molecular inhibitor of WEE1 kinase, AZD1775, can increase unscheduled origin firings, leading to replication fork stalling and driving HER2-positive or TNBC breast cells into unscheduled mitosis [102,103]. Another WEE1 inhibitor ZN-c3 is undergoing evaluation in clinical trials for treatment of TNBC or metastatic breast cancer (NCT04158336, NCT05368506). The DNA-dependent protein kinase (DNA-PK), which has been well characterized in nonhomologous end-joining, has recently been shown to be required for efficient replication restart of stalled forks and ATR signaling [104,105]. Peposertib (M3814), an orally available DNA-PK inhibitor, is in clinical development and has shown modest efficacy in TNBC breast cancer [80]. AZD7648, a highly specific and potent DNA-PK inhibitor, is now in phase 1 clinical trial testing in patients with advanced cancers including breast cancer [81]. Additionally, ATM has been identified to play a replication stress-specific role in preserving replication fork integrity and maintaining DNA replication [106]. Currently, ATM inhibitors are under development and have been shown to be potent radiosensitizers in preclinical studies [82]. Among these ATM inhibitors, AZD0156 is currently being evaluated in phase 1 studies (NCT02588105). It is important to point out that the efficacy of these inhibitors as monotherapies depends on their ability to exploit the intrinsically high levels of replication stress within tumor cells and the ability to identify potential biomarkers to enhance sensitivity.
Limitations associated with the use of these inhibitors, such as excessive toxicity at effective doses and resistance to the treatment through compensatory pathways, promote the development of combination therapies. ATR and CHK1 inhibitors synergize with compounds that induce replication stress in breast cancers, including nucleoside analogues, platinum-based agents, and PARP inhibitors [107,108]. Additionally, a preclinical study showed that DNA-PK inhibitor AZD7648 enhances the efficiency of doxorubicin and PARP inhibitors in breast cancer cell lines and TNBC patient-derived xenograft models [109]. Clinical trials evaluating the antitumor efficacy of combining DNA-PK inhibitor and CHK1 inhibitor in breast cancer are ongoing (NCT04032080 and NCT02124148). Recently, a phase 2 clinical trial demonstrated that the WEE1 inhibitor adavosertib combined with cisplatin improved clinical outcomes for patients with metastatic TNBC [110]. Interestingly, dual inhibition of WEE1 and ATR sensitizes TNBC cells to cisplatin and PARP inhibitors [111], extending the catalog of possible combinations with other targeted inhibitors that could be implemented.
Despite these encouraging findings, challenges remain in determining the optimal therapeutic strategies, both monotherapy and combination therapy, to balance dose-dependent toxicity and antitumor efficiency. Another promising therapeutic strategy is the combination of agents targeting RS with immunotherapeutic agents, which share nonoverlapping toxicity profiles with agents targeting RS.
5. Emerging Combination Strategies of Immunotherapy with Agents Targeting RS
Immunotherapy, especially immune checkpoint blockade (ICB), is emerging as a new treatment modality in breast cancer. Monotherapy using antibodies against programmed death-1 (PD-1) and programmed death ligand-1 (PD-L1) reported objective response rates (ORRs) of around 10% to 20% in patients with metastatic breast cancer [112,113]. The lack of response of patients with breast cancer to immunotherapy has directed research toward novel combination therapeutic strategies aimed at transforming a higher proportion of non-responders into responders.
Preclinical studies have shown the link between immune response and pathways involved in RS. Accumulating evidence indicates that RS triggers the immune response through the accumulation of cytosolic DNA derived from the nucleus and the activation of DNA sensing pathways [21,114]. For instance, S-phase-specific DNA damage in breast cancer was identified to be associated with increased T-cell infiltration and PD-L1 expression in a STING-dependent manner [115]. Additionally, Diamond et al. showed that irradiated breast cancer cells transferred cytosolic dsDNA to dendritic cells and stimulated dendritic cell upregulation of costimulatory molecules and STING-dependent activation of IFN signaling [116]. Recently, a preclinical study reported that induction of RSR defects by CHK1/2 inhibition improved ICB response in murine breast cancer models [117]. Furthermore, other recent studies have illustrated how key RSR members including ATR, CHK1, and DNA-PK regulate immune response in breast cancer [118,119,120]. These studies have, thus, suggested that targeting RS would amplify the response of patients with breast cancer to ICB.
On the basis of these promising data in preclinical settings, combination strategies integrating drugs targeting RS with immunotherapies have advanced to clinical trials (Table 2). For example, the TOPACIO/KEYNOTE-162 trial reported that the anti-PD-1 plus PARP inhibitor combination therapy exhibited a good tolerability profile, with the ORR of 45% being higher in BRCA1/2 mutant TNBC patients than in nonmutant ones [121]. Meanwhile, the MEDIOLA trial showed that a combination of PARPi and anti-PD-L1 led to enhanced antitumor activity in one of the four cohorts for patients with advanced-stage BRCA1/2-mutant breast cancers [122]. Additionally, several ongoing trials are evaluating combinations of ATRi, DNA-PKi, or WEE1i with ICB therapy in breast cancer [123,124,125]. These studies would support the use of replication stress as a predictive marker for immunotherapy efficacy.
Despite these robust associations, a conflicting study reported that inhibition of RSR checkpoint kinases such as ATM, ATR, or CHK1 suppressed PD-L1 upregulation [126]. In addition, Burleign et al. identified DNA-PK as a DNA sensor that plays an essential role in the activation a STING-independent DNA sensing pathway [127]. Inhibition of DNA-PK led to the complete inhibition of this STING-independent DNA sensing pathway. In this context, blunting of this DNA sensing pathway by DNA-PK inhibition may be counterproductive to the activation of innate immunity. Together, these studies demonstrate that the mechanisms underlying the interplay between replication stress response and tumor immunogenicity have not yet been completely elucidated, and further research is required to provide insight into how these responses can be modulated optimally.
6. Predictive Biomarkers for Therapies Targeting Replication Stress
Optimal design of therapeutic strategies targeting RS requires reliable predictive biomarkers that can help to select, before the initiation of treatments, patients with breast cancer who would be most likely to benefit. Currently, there are a few biomarkers that have been identified to predict the response of patients with breast cancer to drugs enhancing RS. For example, Birkbak et al. reported that the expression levels of the BLM and FANCI genes are potential biomarkers that predict response of TNBC to platinum-based therapy [128]. Another recent report also suggested that the expression level of EZH2 can be used to identify patients with breast cancer who may benefit from platinum-based therapy [129]. They found that EZH2 inhibition enhanced the response of BRCA1-deficient breast tumors to platinum drugs. These two studies identified different biomarkers for platinum-based therapy in patients with breast cancer, suggesting that a single-gene function dependency usually represents a limited number of cases. Biomarkers identified in preclinical studies for predicting response of patients with breast cancer to RS-inducing agents need to be validated with clinical biopsy samples before being considered as useful biomarkers.
On the basis of positive outcomes in clinical trials, PARP inhibitors olaparib and talazoparib are approved as monotherapies for the treatment of patients with germline BRCA-mutated, HER2-negative advanced or metastatic breast cancer [130,131,132]. However, PARPi resistance has proved to be a major problem in the clinic [133]. The mechanisms underlying this resistance have been characterized, and restoration of replication fork stability is one of major mechanisms for PARPi resistance [133]. Collectively, these studies suggest that, owing to the complexity of tumor microenvironment, single biomarkers are insufficient to accurately predict clinical outcomes with therapies targeting RS, and a combination of genetic deficiencies will be required to deliver a sufficient degree of sensitivity to drugs targeting RS.
In addition to these biomarkers for predicting response to RS-inducing agents, other genomic aberrations have been identified as potential markers for evaluating the response of breast cancer patients to drugs targeting RSR signaling. In a preclinical study, ARID1A deficiency was found to sensitize breast cancer cells to ATR inhibitor both in vivo and in vitro by causing topoisomerase 2A and cell-cycle defects [134]. This study indicates that ARID1 defects may serve as a biomarker of single-agent ATR inhibitor response. Additionally, inhibition of RAD51 has been demonstrated to increase dependency on ATR-CHK1-mediated RSR signaling, and subsequent inhibition of ATR or CHK1 results in preferentially killing of HR-deficient breast cancer cells [135]. It is important to point out that these biomarkers identified in preclinical models need to be validated using clinical samples. A functional genetic screen showed that silencing of Fanconi anemia and HR genes resulted in increased replication stress and nucleotide depletion after WEE1 inhibition, culminating in unscheduled mitotic entry [136]. On the basis of this preclinical model, a clinical trial is ongoing using alterations in homologous recombination repair-related genes as selective biomarkers for patients with advanced breast cancer (NCT03330847) [137]. Currently, there are few biomarkers that effectively predict the response of patients with breast cancer to RSR inhibitors.
Unlike studies using a sole predictive biomarker, McGrail et al. developed a gene signature which can predicts RSR defects (RSRDs) in patient breast hyperplasias and cell lines [5]. Importantly, they found the RSRD gene signature can accurately identify patients with non-hypermutated cancer across seven tumor types, including breast cancer, who may benefit from ICB [117]. Although this study has not yet been validated using clinical samples, such an RSRD signature may provide broader means of patient selection and could become relevant as potential biomarkers if validated in the clinic.
One key question remains to be addressed, when identifying biomarkers that indicate a dependency on the RSR and a likely response to inhibitors targeting key RSR kinases, is whether the clinical biopsy taken at initial diagnosis still indicates the level of RSR dependence in the tumor to be treated. Therefore, genetic analysis of clinical samples obtained at different timepoints, i.e., pre- and post-treatment tumor samples, may represent an important approach for understanding the dynamic changes of response to agents targeting RSR, and an additional goal would be the identification of more reliable predictive biomarkers that can help to select patients prior to therapy.
7. Conclusions and Future Perspectives
In this review, we highlighted the advances in understanding the mechanism of the cellular response to replication stress and development of promising drug candidates targeting RS. Despite these significant advancements, challenges remain in discerning the optimal therapeutic strategies. For instance, the applicability and efficacy of these drugs that enhance endogenous RS or target RSR is limited by their associated toxicities and the high rate of drug resistance [20,21,138]. As noted, these therapies can induce apoptosis in normal cells within the gut epithelium and bone marrow, leading to undesirable and intolerable side-effects [139,140].
In fact, clinical studies assessing the potential of drugs targeting RSR pathways in breast cancer, both as monotherapy and as combination therapy described above, are still in the very early stages; the antitumor activity of these drugs has not yet been completely elucidated. Additionally, whether the combination of these drugs with immunotherapeutic agents is as effective and less toxic than the combination with DDR agents needs to be addressed.
Another important limitation is the current technologies available to personalize and define patient-specific RS in real time in the clinic. Potential predictive biomarkers have been identified to select patients who are most likely to benefit from certain therapies. However, only a few biomarkers are currently FDA-approved for clinic use. Other emerging biomarker candidates have shown their unique advantages, as well as limitations, in both preclinical and clinical studies [7,43]. Indeed, novel technologies are needed to assist precision medicine approaches. Genome-wide screening using CRISPR/Cas9 technology performed in more physiologically relevant settings, such as in 3D organoid cultures or in vivo mouse models, will likely identify novel clinically relevant targets in DNA replication and RSR.
Targeting replication stress has shown promise in breast cancer treatment. Future research in this area would likely provide insightful clues for identification of novel molecular targets in DNA replication and RSR, for finding the optimal multimodality therapy, as well as for discovering additional biomarkers that can collectively predict RS phenotype and maximize clinical benefit.
Author Contributions
Conceptualization, J.Z. and S.-Y.L.; writing—original draft preparation, J.Z.; writing—review and editing, J.Z., D.W.C. and S.-Y.L.; figure and table design: J.Z.; funding acquisition, S.-Y.L. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the NCI (R01CA247862).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [Green Version]
- McGrail, D.J.; Lin, C.C.; Dai, H.; Mo, W.; Li, Y.; Stephan, C.; Davies, P.; Lu, Z.; Mills, G.B.; Lee, J.S.; et al. Defective Replication Stress Response Is Inherently Linked to the Cancer Stem Cell Phenotype. Cell Rep. 2018, 23, 2095–2106. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Pham, M.M.; Tan, D.S.P.; Yap, T.A. Targeting the replication stress response through synthetic lethal strategies in cancer medicine. Trends Cancer 2021, 7, 930–957. [Google Scholar] [CrossRef]
- Dobbelstein, M.; Sørensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Dapic, V.; Carvalho, M.A.; Monteiro, A.N.A. Breast Cancer Susceptibility and the DNA Damage Response. Cancer Control 2005, 12, 127–136. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborn, A.J.; Elledge, S.J.; Zou, L. Checking on the fork: The DNA-replication stress-response pathway. Trends Cell Biol. 2002, 12, 509–516. [Google Scholar] [CrossRef]
- Nazareth, D.; Jones, M.J.; Gabrielli, B. Everything in Moderation: Lessons Learned by Exploiting Moderate Replication Stress in Cancer. Cancers 2019, 11, 1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajamanickam, S.; Park, J.; Bates, K.; Timilsina, S.; Eedunuri, V.; Onyeagucha, B.; Subbarayalu, P.; Abdelfattah, N.; Jung, K.; Favours, E.; et al. Abstract P6-06-04: Targeting replication stress in triple negative breast cancer treatment regimen: An emerging approach. Cancer Res. 2018, 78, P6-06-04. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar] [CrossRef]
- Parmigiani, G.; Berry, D.A.; Aguilar, O. Determining Carrier Probabilities for Breast Cancer–Susceptibility Genes BRCA1 and BRCA2. Am. J. Hum. Genet. 1998, 62, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Baldeyron, C.; Brisson, A.; Tesson, B.; Némati, F.; Koundrioukoff, S.; Saliba, E.; De Koning, L.; Martel, E.; Ye, M.; Rigaill, G.; et al. TIPIN depletion leads to apoptosis in breast cancer cells. Mol. Oncol. 2015, 9, 1580–1598. [Google Scholar] [CrossRef]
- Jo, U.; Murai, Y.; Takebe, N.; Thomas, A.; Pommier, Y. Precision Oncology with Drugs Targeting the Replication Stress, ATR, and Schlafen 11. Cancers 2021, 13, 4601. [Google Scholar] [CrossRef]
- Ubhi, T.; Brown, G.W. Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res. 2019, 79, 1730–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Técher, H.; Koundrioukoff, S.; Nicolas, A.; Debatisse, M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 2017, 18, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Krishnamoorthy, A.; Zhao, R.; Cortez, D. Two replication fork remodeling pathways generate nuclease substrates for distinct fork protection factors. Sci. Adv. 2020, 6, eabc3598. [Google Scholar] [CrossRef]
- Quinet, A.; Lemaçon, D.; Vindigni, A. Replication Fork Reversal: Players and Guardians. Mol. Cell 2017, 68, 830–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, S.; Zou, L. Hallmarks of DNA replication stress. Mol. Cell 2022, 82, 2298–2314. [Google Scholar] [CrossRef]
- Abe, S.; Yamamoto, K.; Kurata, M.; Abe-Suzuki, S.; Horii, R.; Akiyama, F.; Kitagawa, M. Targeting MCM2 function as a novel strategy for the treatment of highly malignant breast tumors. Oncotarget 2015, 6, 34892–34909. [Google Scholar] [CrossRef] [Green Version]
- Juríková, M.; Danihel, Ľ.; Polák, Š.; Varga, I. Ki67, PCNA, and MCM proteins: Markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 2016, 118, 544–552. [Google Scholar] [CrossRef]
- Issac, M.S.M.; Yousef, E.; Tahir, M.R.; Gaboury, L.A. MCM2, MCM4, and MCM6 in Breast Cancer: Clinical Utility in Diagnosis and Prognosis. Neoplasia 2019, 21, 1015–1035. [Google Scholar] [CrossRef]
- Mahadevappa, R.; Neves, H.; Yuen, S.M.; Bai, Y.; McCrudden, C.M.; Yuen, H.F.; Wen, Q.; Zhang, S.D.; Kwok, H.F. The prognostic significance of Cdc6 and Cdt1 in breast cancer. Sci. Rep. 2017, 7, 985. [Google Scholar] [CrossRef] [Green Version]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Técher, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881.e7. [Google Scholar] [CrossRef]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.-W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Ray Chaudhuri, A.; Callen, E.; Pang, Y.; Biswas, K.; Klarmann, K.D.; Martin, B.K.; Burkett, S.; Cleveland, L.; Stauffer, S.; et al. Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat. Commun. 2016, 7, 12425. [Google Scholar] [CrossRef]
- Wu, X. Replication Stress Response Links RAD52 to Protecting Common Fragile Sites. Cancers 2019, 11, 1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Peng, X.; Daley, J.; Yang, L.; Shen, J.; Nguyen, N.; Bae, G.; Niu, H.; Peng, Y.; Hsieh, H.J.; et al. Inhibition of DNA2 nuclease as a therapeutic strategy targeting replication stress in cancer cells. Oncogenesis 2017, 6, e319. [Google Scholar] [CrossRef] [Green Version]
- Dibitetto, D.; Marshall, S.; Sanchi, A.; Liptay, M.; Badar, J.; Lopes, M.; Rottenberg, S.; Smolka, M.B. DNA-PKcs promotes fork reversal and chemoresistance. Mol. Cell 2022, 82, 3932–3942.e6. [Google Scholar] [CrossRef]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [Green Version]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Ashley, A.K.; Shrivastav, M.; Nie, J.; Amerin, C.; Troksa, K.; Glanzer, J.G.; Liu, S.; Opiyo, S.O.; Dimitrova, D.D.; Le, P.; et al. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair 2014, 21, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Shechter, D.; Costanzo, V.; Gautier, J. ATR and ATM regulate the timing of DNA replication origin firing. Nat. Cell Biol. 2004, 6, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Falck, J.; Lukas, C.; Syljuâsen, R.G.; Welcker, M.; Bartek, J.; Lukas, J. Rapid destruction of human Cdc25A in response to DNA damage. Science 2000, 288, 1425–1429. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef] [Green Version]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef]
- Cortez, D. Replication-Coupled DNA Repair. Mol. Cell 2019, 74, 866–876. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Cortez, D.; Lopes, M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 2020, 21, 633–651. [Google Scholar] [CrossRef]
- Schoonen, P.M.; Guerrero Llobet, S.; van Vugt, M. Replication stress: Driver and therapeutic target in genomically instable cancers. Adv. Protein Chem. Struct. Biol. 2019, 115, 157–201. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Shih, D.J.H.; Lin, S.-Y. Role of DNA repair defects in predicting immunotherapy response. Biomark. Res. 2020, 8, 23. [Google Scholar] [CrossRef]
- Coquel, F.; Silva, M.-J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.-L.; et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017, 31, 353–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merriman, R.L.; Hertel, L.W.; Schultz, R.M.; Houghton, P.J.; Houghton, J.A.; Rutherford, P.G.; Tanzer, L.R.; Boder, G.B.; Grindey, G.B. Comparison of the antitumor activity of gemcitabine and ara-C in a panel of human breast, colon, lung and pancreatic xenograft models. Investig. New Drugs 1996, 14, 243–247. [Google Scholar] [CrossRef]
- Cameron, D.A.; Gabra, H.; Leonard, R.C. Continuous 5-fluorouracil in the treatment of breast cancer. Br. J. Cancer 1994, 70, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Ponce-Cusi, R.; Calaf, G.M. Apoptotic activity of 5-fluorouracil in breast cancer cells transformed by low doses of ionizing α-particle radiation. Int. J. Oncol. 2016, 48, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Ueno, H.; Hoshino, T.; Yano, W.; Tsukioka, S.; Suzuki, T.; Hara, S.; Ogino, Y.; Chong, K.T.; Suzuki, T.; Tsuji, S.; et al. TAS1553, a small molecule subunit interaction inhibitor of ribonucleotide reductase, exhibits antitumor activity by causing DNA replication stress. Commun. Biol. 2022, 5, 571. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Butler, C.; Sprowls, S.; Szalai, G.; Arsiwala, T.; Saralkar, P.; Straight, B.; Hatcher, S.; Tyree, E.; Yost, M.; Kohler, W.J.; et al. Hypomethylating Agent Azacitidine Is Effective in Treating Brain Metastasis Triple-Negative Breast Cancer Through Regulation of DNA Methylation of Keratin 18 Gene. Transl. Oncol. 2020, 13, 100775. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Merkher, Y.; Chen, L.; Liu, N.; Leonov, S.; Chen, Y. Recent advances in therapeutic strategies for triple-negative breast cancer. J. Hematol. Oncol. 2022, 15, 121. [Google Scholar] [CrossRef]
- Byrski, T.; Dent, R.; Blecharz, P.; Foszczynska-Kloda, M.; Gronwald, J.; Huzarski, T.; Cybulski, C.; Marczyk, E.; Chrzan, R.; Eisen, A.; et al. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast Cancer Res. 2012, 14, R110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trudeau, M.E.; Crump, M.; Charpentier, D.; Yelle, L.; Bordeleau, L.; Matthews, S.; Eisenhauer, E. Temozolomide in metastatic breast cancer (MBC): A phase II trial of the National Cancer Institute of Canada - Clinical Trials Group (NCIC-CTG). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2006, 17, 952–956. [Google Scholar] [CrossRef]
- Zimmer, A.S.; Steinberg, S.M.; Smart, D.D.; Gilbert, M.R.; Armstrong, T.S.; Burton, E.; Houston, N.; Biassou, N.; Gril, B.; Brastianos, P.K.; et al. Temozolomide in secondary prevention of HER2-positive breast cancer brain metastases. Future Oncol. (Lond. Engl.) 2020, 16, 899–909. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, S.; Ao, D. Targeting DNA repair pathway in cancer: Mechanisms and clinical application. MedComm 2021, 2, 654–691. [Google Scholar] [CrossRef]
- Geyer, C.E.; Sikov, W.M.; Huober, J.; Rugo, H.S.; Wolmark, N.; O’Shaughnessy, J.; Maag, D.; Untch, M.; Golshan, M.; Lorenzo, J.P.; et al. Long-term efficacy and safety of addition of carboplatin with or without veliparib to standard neoadjuvant chemotherapy in triple-negative breast cancer: 4-year follow-up data from BrighTNess, a randomized phase III trial. Ann. Oncol. 2022, 33, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Goralski, M.; Capota, E.; Padrick, S.B.; Kim, J.; Xie, Y.; Nijhawan, D. The antitumor toxin CD437 is a direct inhibitor of DNA polymerase α. Nat. Chem. Biol. 2016, 12, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, B.; Zhang, S.; Zhao, H.; Darzynkiewicz, Z.; Lee, E.Y.C.; Lee, M.; Zhang, Z. Discovery of a novel DNA polymerase inhibitor and characterization of its antiproliferative properties. Cancer Biol. Ther. 2019, 20, 474–486. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhou, M.; Li, Z.; Li, H.; Polaczek, P.; Dai, H.; Wu, Q.; Liu, C.; Karanja, K.K.; Popuri, V.; et al. A Selective Small Molecule DNA2 Inhibitor for Sensitization of Human Cancer Cells to Chemotherapy. EBioMedicine 2016, 6, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef]
- Toma, M.; Sullivan-Reed, K.; Śliwiński, T.; Skorski, T. RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies. Cancers 2019, 11, 1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roulston, A.; Zimmermann, M.; Papp, R.; Skeldon, A.; Pellerin, C.; Dumas-Bérube, É.; Dumais, V.; Dorich, S.; Fader, L.D.; Fournier, S.; et al. RP-3500: A Novel, Potent, and Selective ATR Inhibitor that is Effective in Preclinical Models as a Monotherapy and in Combination with PARP Inhibitors. Mol. Cancer Ther. 2022, 21, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Barnieh, F.M.; Loadman, P.M.; Falconer, R.A. Progress towards a clinically-successful ATR inhibitor for cancer therapy. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100017. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Infante, J.R.; Shapiro, G.I.; Moore, K.N.; LoRusso, P.M.; Hamilton, E.; Cousin, S.; Toulmonde, M.; Postel-Vinay, S.; Tolaney, S.; et al. Phase I study of the checkpoint kinase 1 inhibitor GDC-0575 in combination with gemcitabine in patients with refractory solid tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Ellis, M.J.; Petroni, G.R.; Guo, Z.; Cai, S.R.; Ryan, C.E.; Craig Lockhart, A.; Naughton, M.J.; Pluard, T.J.; Brenin, C.M.; et al. A phase II study of UCN-01 in combination with irinotecan in patients with metastatic triple negative breast cancer. Breast Cancer Res. Treat. 2013, 137, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Leijen, S.; Geel, R.M.J.M.V.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.A.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.; Mamdani, H.; Chalasani, P.; Meric-Bernstam, F.; Gazdoiu, M.; Makris, L.; Pultar, P.; Voliotis, D. Abstract CT016: Clinical activity of single-agent ZN-c3, an oral WEE1 inhibitor, in a phase 1 dose-escalation trial in patients with advanced solid tumors. Cancer Res. 2021, 81, CT016. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Sharma, M.; Dummer, R.; Imedio, E.R.; Yge, M.-L.; Micallef, S.; Bellon, A.; Stathis, A. A phase 1 dose-finding and dose-expansion study evaluating the safety, tolerability, pharmacokinetics, and efficacy of a highly selective WEE1 inhibitor (Debio 0123) in adult patients with advanced solid tumors. J. Clin. Oncol. 2022, 40, TPS2702. [Google Scholar] [CrossRef]
- van Bussel, M.T.J.; Awada, A.; de Jonge, M.J.A.; Mau-Sørensen, M.; Nielsen, D.; Schöffski, P.; Verheul, H.M.W.; Sarholz, B.; Berghoff, K.; El Bawab, S.; et al. A first-in-man phase 1 study of the DNA-dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. Br. J. Cancer 2021, 124, 728–735. [Google Scholar] [CrossRef]
- Yap, T.A.; Chen, Y.; Butler, L.H.; Lao-Sirieix, S.-H.; Cadogan, E.; Oplustil O’onnor, L.; Vishwanathan, K.; Kim, Y.; Landers, D.; Dean, E. Abstract CT248: AZD7648: A Phase I/IIa first-in-human trial of a novel, potent and selective DNA-PK inhibitor in patients with advanced malignancies. Cancer Res. 2020, 80, CT248. [Google Scholar] [CrossRef]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer Ther. 2020, 19, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [Green Version]
- Promonet, A.; Padioleau, I.; Liu, Y.; Sanz, L.; Biernacka, A.; Schmitz, A.-L.; Skrzypczak, M.; Sarrazin, A.; Mettling, C.; Rowicka, M.; et al. Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites. Nat. Commun. 2020, 11, 3940. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst.) 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Michelena, J.; Lezaja, A.; Teloni, F.; Schmid, T.; Imhof, R.; Altmeyer, M. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat. Commun. 2018, 9, 2678. [Google Scholar] [CrossRef] [Green Version]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 2018, 71, 319–331.e3. [Google Scholar] [CrossRef] [Green Version]
- Cong, K.; Peng, M.; Kousholt, A.N.; Lee, W.T.C.; Lee, S.; Nayak, S.; Krais, J.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Calvo, J.; et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 2021, 81, 3128–3144.e7. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef]
- Shao, Z.M.; Dawson, M.I.; Li, X.S.; Rishi, A.K.; Sheikh, M.S.; Han, Q.X.; Ordonez, J.V.; Shroot, B.; Fontana, J.A. p53 independent G0/G1 arrest and apoptosis induced by a novel retinoid in human breast cancer cells. Oncogene 1995, 11, 493–504. [Google Scholar] [PubMed]
- Balmaña, J.; Tung, N.M.; Isakoff, S.J.; Graña, B.; Ryan, P.D.; Saura, C.; Lowe, E.S.; Frewer, P.; Winer, E.; Baselga, J.; et al. Phase I trial of olaparib in combination with cisplatin for the treatment of patients with advanced breast, ovarian and other solid tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2014, 25, 1656–1663. [Google Scholar] [CrossRef] [PubMed]
- Rodler, E.T.; Gralow, J.; Kurland, B.F.; Griffin, M.; Yeh, R.; Thompson, J.A.; Porter, P.; Swisher, E.M.; Gadi, V.K.; Korde, L.A.; et al. Phase I: Veliparib with cisplatin (CP) and vinorelbine (VNR) in advanced triple-negative breast cancer (TNBC) and/or BRCA mutation-associated breast cancer. J. Clin. Oncol. 2014, 32, 2569. [Google Scholar] [CrossRef]
- Somlo, G.; Frankel, P.H.; Luu, T.H.; Ma, C.; Arun, B.; Garcia, A.; Cigler, T.; Fleming, G.F.; Harvey, H.A.; Sparano, J.A.; et al. Efficacy of the combination of ABT-888 (veliparib) and carboplatin in patients with BRCA-associated breast cancer. J. Clin. Oncol. 2013, 31, 1024. [Google Scholar] [CrossRef]
- Pahuja, S.; Beumer, J.H.; Appleman, L.J.; Tawbi, H.A.-H.; Stoller, R.G.; Lee, J.J.; Lin, Y.; Ding, F.; Yu, J.; Belani, C.P.; et al. A phase I study of veliparib (ABT-888) in combination with weekly carboplatin and paclitaxel in advanced solid malignancies and enriched for triple-negative breast cancer (TNBC). J. Clin. Oncol. 2015, 33, 1015. [Google Scholar] [CrossRef]
- Kummar, S.; Chen, A.; Ji, J.; Zhang, Y.; Reid, J.M.; Ames, M.; Jia, L.; Weil, M.; Speranza, G.; Murgo, A.J.; et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011, 71, 5626–5634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.P.; Bradley, J.D.; Hagerty, T.; Wyatt, E.A. Efficacy of DAN-222, a novel investigational polymeric nanoparticle with topoisomerase I inhibitor, as monotherapy in breast cancer models and when combined with PARP inhibitor. J. Clin. Oncol. 2021, 39, 1081. [Google Scholar] [CrossRef]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Jones, M.J.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J., III; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Gatti-Mays, M.E.; Karzai, F.H.; Soltani, S.N.; Zimmer, A.; Green, J.E.; Lee, M.J.; Trepel, J.B.; Yuno, A.; Lipkowitz, S.; Nair, J.; et al. A Phase II Single Arm Pilot Study of the CHK1 Inhibitor Prexasertib (LY2606368) in BRCA Wild-Type, Advanced Triple-Negative Breast Cancer. Oncologist 2020, 25, e1013–e1824. [Google Scholar] [CrossRef] [PubMed]
- Pitts, T.M.; Simmons, D.M.; Bagby, S.M.; Hartman, S.J.; Yacob, B.W.; Gittleman, B.; Tentler, J.J.; Cittelly, D.; Ormond, D.R.; Messersmith, W.A.; et al. Wee1 Inhibition Enhances the Anti-Tumor Effects of Capecitabine in Preclinical Models of Triple-Negative Breast Cancer. Cancers 2020, 12, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sand, A.; Piacsek, M.; Donohoe, D.L.; Duffin, A.T.; Riddell, G.T.; Sun, C.; Tang, M.; Rovin, R.A.; Tjoe, J.A.; Yin, J. WEE1 inhibitor, AZD1775, overcomes trastuzumab resistance by targeting cancer stem-like properties in HER2-positive breast cancer. Cancer Lett. 2020, 472, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Chen, Z.; Medhurst, A.L.; Neal, J.A.; Bao, Z.; Mortusewicz, O.; McGouran, J.; Song, X.; Shen, H.; Hamdy, F.C.; et al. DNA-PKcs and PARP1 Bind to Unresected Stalled DNA Replication Forks Where They Recruit XRCC1 to Mediate Repair. Cancer Res. 2016, 76, 1078–1088. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.F.; Shih, H.Y.; Shang, Z.F.; Kuo, C.T.; Guo, J.; Du, C.; Lee, H.; Chen, B.P.C. PIDD mediates the association of DNA-PKcs and ATR at stalled replication forks to facilitate the ATR signaling pathway. Nucleic Acids Res. 2018, 46, 1847–1859. [Google Scholar] [CrossRef] [Green Version]
- Olcina, M.M.; Foskolou, I.P.; Anbalagan, S.; Senra, J.M.; Pires, I.M.; Jiang, Y.; Ryan, A.J.; Hammond, E.M. Replication stress and chromatin context link ATM activation to a role in DNA replication. Mol. Cell 2013, 52, 758–766. [Google Scholar] [CrossRef] [Green Version]
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine ± cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 510–519. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Lücking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bömer, U.; Moosmayer, D.; Eberspächer, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage–Inducing or Repair–Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef] [Green Version]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef] [Green Version]
- Keenan, T.E.; Li, T.; Vallius, T.; Guerriero, J.L.; Tayob, N.; Kochupurakkal, B.; Davis, J.; Pastorello, R.; Tahara, R.K.; Anderson, L.; et al. Clinical Efficacy and Molecular Response Correlates of the WEE1 Inhibitor Adavosertib Combined with Cisplatin in Patients with Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 983–991. [Google Scholar] [CrossRef]
- Jin, J.; Fang, H.; Yang, F.; Ji, W.; Guan, N.; Sun, Z.; Shi, Y.; Zhou, G.; Guan, X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia 2018, 20, 478–488. [Google Scholar] [CrossRef]
- Nanda, R.; Chow, L.Q.M.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Braiteh, F.S.; Cassier, P.; Delord, J.-P.; Eder, J.P.; Fasso, M.; Xiao, Y.; Wang, Y.; Molinero, L.; Chen, D.S.; et al. Abstract 2859: Inhibition of PD-L1 by MPDL3280A leads to clinical activity in patients with metastatic triple-negative breast cancer (TNBC). Cancer Res. 2015, 75, 2859. [Google Scholar] [CrossRef]
- Lin, Y.-L.; Pasero, P. Replication stress: From chromatin to immunity and beyond. Curr. Opin. Genet. Dev. 2021, 71, 136–142. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.-P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from Irradiated Cancer Cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrail, D.J.; Pilié, P.G.; Dai, H.; Lam, T.N.A.; Liang, Y.; Voorwerk, L.; Kok, M.; Zhang, X.H.-F.; Rosen, J.M.; Heimberger, A.B.; et al. Replication stress response defects are associated with response to immune checkpoint blockade in nonhypermutated cancers. Sci. Transl. Med. 2021, 13, eabe6201. [Google Scholar] [CrossRef]
- Sun, L.L.; Yang, R.Y.; Li, C.W.; Chen, M.K.; Shao, B.; Hsu, J.M.; Chan, L.C.; Yang, Y.; Hsu, J.L.; Lai, Y.J.; et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am. J. Cancer Res. 2018, 8, 1307–1316. [Google Scholar]
- Asleh, K.; Riaz, N.; Cheng, A.S.; Gao, D.; Leung, S.C.Y.; Anurag, M.; Nielsen, T.O. Proteomics-derived basal biomarker DNA-PKcs is associated with intrinsic subtype and long-term clinical outcomes in breast cancer. NPJ Breast Cancer 2021, 7, 114. [Google Scholar] [CrossRef]
- Proctor, M.; Gonzalez Cruz, J.L.; Daignault-Mill, S.M.; Veitch, M.; Zeng, B.; Ehmann, A.; Sabdia, M.; Snell, C.; Keane, C.; Dolcetti, R.; et al. Targeting Replication Stress Using CHK1 Inhibitor Promotes Innate and NKT Cell Immune Responses and Tumour Regression. Cancers 2021, 13, 3733. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.E.; Vidal, G.A.; Mita, M.M.; Fleming, G.F.; Holloway, R.W.; Le, L.V.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. J. Clin. Oncol. 2018, 36, 106. [Google Scholar] [CrossRef]
- Domchek, S.; Postel-Vinay, S.; Bang, Y.-J.; Park, Y.; Alexandre, J.; Delord, J.-P.; Italiano, A.; You, B.; Bastian, S.; Krebs, M.; et al. Abstract PD6-11: An open-label, multitumor, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in germline BRCA-mutated (gBRCAm) HER2-negative metastatic breast cancer (MBC). Cancer Res. 2018, 78, PD6-11. [Google Scholar] [CrossRef]
- Bendell, J.C.; Shafique, M.R.; Perez, B.; Chennoufi, S.; Beier, F.; Trang, K.; Antonia, S.J. Phase 1, open-label, dose-escalation study of M3814 + avelumab ± radiotherapy (RT) in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2019, 37, TPS3169. [Google Scholar] [CrossRef]
- Patel, M.R.; Falchook, G.S.; Wang, J.S.-Z.; Imedio, E.R.; Kumar, S.; Motlagh, P.; Miah, K.; Mugundu, G.M.; Jones, S.F.; Spigel, D.R.; et al. Open-label, multicenter, phase I study to assess safety and tolerability of adavosertib plus durvalumab in patients with advanced solid tumors. J. Clin. Oncol. 2019, 37, 2562. [Google Scholar] [CrossRef]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burleigh, K.; Maltbaek, J.H.; Cambier, S.; Green, R.; Gale, M., Jr.; James, R.C.; Stetson, D.B. Human DNA-PK activates a STING-independent DNA sensing pathway. Sci. Immunol. 2020, 5, eaba4219. [Google Scholar] [CrossRef] [Green Version]
- Birkbak, N.J.; Li, Y.; Pathania, S.; Greene-Colozzi, A.; Dreze, M.; Bowman-Colin, C.; Sztupinszki, Z.; Krzystanek, M.; Diossy, M.; Tung, N.; et al. Overexpression of BLM promotes DNA damage and increased sensitivity to platinum salts in triple-negative breast and serous ovarian cancers. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 903–909. [Google Scholar] [CrossRef]
- Puppe, J.; Opdam, M.; Schouten, P.C.; Jóźwiak, K.; Lips, E.; Severson, T.; van de Ven, M.; Brambillasca, C.; Bouwman, P.; van Tellingen, O.; et al. EZH2 Is Overexpressed in BRCA1-like Breast Tumors and Predictive for Sensitivity to High-Dose Platinum-Based Chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4351–4362. [Google Scholar] [CrossRef] [Green Version]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target. Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef] [Green Version]
- Krajewska, M.; Fehrmann, R.S.N.; Schoonen, P.M.; Labib, S.; de Vries, E.G.E.; Franke, L.; van Vugt, M.A.T.M. ATR inhibition preferentially targets homologous recombination-deficient tumor cells. Oncogene 2015, 34, 3474–3481. [Google Scholar] [CrossRef]
- Aarts, M.; Bajrami, I.; Herrera-Abreu, M.T.; Elliott, R.; Brough, R.; Ashworth, A.; Lord, C.J.; Turner, N.C. Functional Genetic Screen Identifies Increased Sensitivity to WEE1 Inhibition in Cells with Defects in Fanconi Anemia and HR Pathways. Mol. Cancer Ther. 2015, 14, 865–876. [Google Scholar] [CrossRef] [Green Version]
- Dean, E.; Krebs, M.G.; Im, S.-A.; Campone, M.; Postel-Vinay, S.; Arkenau, T.; Lopez, J.; Abida, W.; Jodrell, D.; Lee, K.-W.; et al. Abstract PS11-18: Ceralasertib (cer) in combination with olaparib (ola) in patients (pts) with advanced breast cancer (BC): Results of phase I expansion cohorts. Cancer Res. 2021, 81, PS11-18. [Google Scholar] [CrossRef]
- Baillie, K.E.; Stirling, P.C. Beyond Kinases: Targeting Replication Stress Proteins in Cancer Therapy. Trends Cancer 2021, 7, 430–446. [Google Scholar] [CrossRef]
- Wang, S.; Konorev, E.A.; Kotamraju, S.; Joseph, J.; Kalivendi, S.; Kalyanaraman, B. Doxorubicin Induces Apoptosis in Normal and Tumor Cells via Distinctly Different Mechanisms: INTERMEDIACY OF H2O2-AND p53-DEPENDENT PATHWAYS. J. Biol. Chem 2004, 279, 25535–25543. [Google Scholar] [CrossRef] [Green Version]
- Tamaki, T.; Naomoto, Y.; Kimura, S.; Kawashima, R.; Shirakawa, Y.; Shigemitsu, K.; Yamatsuji, T.; Haisa, M.; Gunduz, M.; Tanaka, N. Apoptosis in normal tissues induced by anti-cancer drugs. J. Int. Med. Res. 2003, 31, 6–16. [Google Scholar] [CrossRef]
Figure 1.
Cellular responses to DNA replication stress. Depending on the types of RS, cells activate numerous responses to ensure accurate completion of genome duplication, including regulation of origin firing, replication fork remodeling, activation of RSR signaling, and involvement of DNA damage response pathways.
Figure 1.
Cellular responses to DNA replication stress. Depending on the types of RS, cells activate numerous responses to ensure accurate completion of genome duplication, including regulation of origin firing, replication fork remodeling, activation of RSR signaling, and involvement of DNA damage response pathways.

Figure 2.
Targeting DNA replication stress in breast cancer. Breast cancer cells harboring RS can be targeted through three major mechanisms. First, RS can be harnessed in breast cancer cells with high intrinsic RS to induce replication catastrophe. Second, breast cancer cells can be targeted by abrogating their G2–M cell-cycle checkpoint to induce mitotic catastrophe. Third, breast cancer cells with high intrinsic RS exhibit high levels of cytosolic DNA leaking from nucleus, which can trigger interferon signaling to prime tumor for immune therapies.
Figure 2.
Targeting DNA replication stress in breast cancer. Breast cancer cells harboring RS can be targeted through three major mechanisms. First, RS can be harnessed in breast cancer cells with high intrinsic RS to induce replication catastrophe. Second, breast cancer cells can be targeted by abrogating their G2–M cell-cycle checkpoint to induce mitotic catastrophe. Third, breast cancer cells with high intrinsic RS exhibit high levels of cytosolic DNA leaking from nucleus, which can trigger interferon signaling to prime tumor for immune therapies.


{kind=link}
{kind=link}
Table 1.
Therapeutic agents that enhance replication stress in breast cancer.
| Class of Agents or Target | Mechanism of Action | Compounds | Clinical Stage | Limitations | Refs. |
|---|---|---|---|---|---|
| Nucleoside analogues | Incorporation into DNA to inhibit replication | 5-Fluorouracil | Approved | [58] | |
| Gemcitabine | Approved | [21] | |||
| Azacitidine | Phase 2 | [59] | |||
| Alkylating agents and platinum compounds | Modification of DNA | Carboplatin | Phase 2 and 3 | [60] | |
| Cisplatin | Phase 2 | [61] | |||
| Cyclophosphamide | Approved | [21] | |||
| Methotrexate | Approved | [21] | |||
| Temozolomide | Phase 2 | Lack of systemic activity for temozolomide in a heavily pretreated, heterogeneous group of breast cancer patients | [62,63] | ||
| Topoisomerase I and II | Relaxation of DNA supercoiling | Doxorubicin | Approved | [21] | |
| Epirubicin | Approved | ||||
| Etoposide | Phase 2 | Dose-limiting toxicities | [60] | ||
| Mitoxantrone | Phase 2 | [21] | |||
| PARP | Recruitment of MRE11 to stalled reverse forks, regulation of fork restart, ssDNA break repair, and regulation of fork elongation | Olaparib | Approved | [64,65,66] | |
| Niraparib | Approved | ||||
| Rucaparib | Approved | ||||
| Talazoparib | Approved | ||||
| Veliparib | Phase 3 | Veliparib plus carboplatin-containing chemotherapy did not impact long-term outcomes in TNBC | |||
| RP12146 | Phase 1/1b | ||||
| DNA polymerases | Regulation of DNA replication | CD437 | Preclinical | [67] | |
| Zelpolib | Preclinical | [68] | |||
| DNA2 | Regulation of Fork restart and ART activation | C5 | Preclinical | [69] | |
| NSC-105808 | Preclinical | [37] | |||
| RAD52 | Regulator of initiation and resolution of fork reversal | D-G23 | Preclinical | [70,71] | |
| D-I03 | Preclinical | ||||
| F79 | Preclinical | ||||
| A5MP | Preclinical | ||||
| AICAR | Preclinical | ||||
| ATR | Essential kinase in replication stress response | M4344 | Phase 1 | [72,73] | |
| BAY1895344 | Phase 1 | Dose-limiting hematological toxicities | |||
| RP-3500 | Phase 1 | ||||
| AZD6738 | Phase 1 and 2 | Toxicities such as hematological toxicities and immune toxicities were evident in ≥20% of subjects | |||
| M6620 | Phase 1 and 2 | ||||
| CHK1 | Key downstream effector kinase of the ATR response | GDC-0575 | Phase 1 | [74] | |
| Prexasertib | Phase 2 | [7] | |||
| UCN-01 | Phase 2 | The clinical outcome was inconclusive | [75] | ||
| AZD7762 | Phase 1 | The development was stopped due to cardiac toxicity | [76] | ||
| WEE1 | G2/M checkpoint kinase | AZD1775 | Phase 2 | [77] | |
| ZN-c3 | Phase 1 | [78] | |||
| Debio0123 | Phase 1 | [79] | |||
| DNA-PK | Regulation of stalled forks restart and ATR signaling | M3814 | Phase 1 | [80] | |
| AZD7648 | Phase 1 | [81] | |||
| ATM | Preserving replication fork integrity and maintaining DNA replication | AZD0156 | Phase 1 | Dose-limiting hematological toxicities | [82] |
Table 2.
Combination strategies of ICB with agents targeting RS in breast cancer.
| Class of Agents or Target | Compounds | Trials | Clinical Stage | Biomarker |
|---|---|---|---|---|
| Alkylating agents and platinum compounds | Carboplatin + pembrolizumab | NCT03121352 | Phase 2 | N/A |
| NCT03639948 | Phase 2 | N/A | ||
| Cyclophosphamide + pembrolizumab | NCT03139851 | Phase 2 | N/A | |
| NCT02768701 | Phase 2 | N/A | ||
| NCT03971045 | Phase 2 | N/A | ||
| PARP | Niraparib + pembrolizumab | NCT02657889 | Phase ½ | BRCA1/2 mutant |
| Niraparib + dostarlimab | NCT04673448 | Phase 1 | BRCA mutant | |
| NCT04837209 | Phase 2 | N/A | ||
| Olaparib + pembrolizumab | NCT03025035 | Phase 2 | BRCA mutant or HDR-defect | |
| Olaparib + atezolizumab | NCT02849496 | Phase 2 | BRCA mutant HER2-negative | |
| Talazoparib + avelumb | NCT03330405 | Phase ½ | N/A | |
| NCT03964532 | Phase ½ | N/A | ||
| Olaparib + durvalumab | NCT03544125 | Phase 1 | N/A | |
| NCT02484404 | Phase ½ | N/A | ||
| NCT03801369 | Phase 2 | N/A | ||
| NCT05498155 | Phase 2 | BRCA mutant HER2-negative | ||
| NCT03167619 | Phase 2 | N/A | ||
| ATR | AZD6738 + durvalumab | NCT03740893 | Phase 2 | N/A |
| WEE1 | ZN-c3 + pembrolizumab | NCT05431582 | Phase 1 | N/A |
| DNA-PK | M3814 + avelumab | NCT03724890 | Phase 1 | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, J.; Chan, D.W.; Lin, S.-Y. Exploiting DNA Replication Stress as a Therapeutic Strategy for Breast Cancer. Biomedicines 2022, 10, 2775. https://doi.org/10.3390/biomedicines10112775
AMA Style
Zhang J, Chan DW, Lin S-Y. Exploiting DNA Replication Stress as a Therapeutic Strategy for Breast Cancer. Biomedicines. 2022; 10(11):2775. https://doi.org/10.3390/biomedicines10112775
Chicago/Turabian StyleZhang, Jing, Doug W. Chan, and Shiaw-Yih Lin. 2022. "Exploiting DNA Replication Stress as a Therapeutic Strategy for Breast Cancer" Biomedicines 10, no. 11: 2775. https://doi.org/10.3390/biomedicines10112775
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.