
Neutrophils and Neutrophil Extracellular Traps in Cardiovascular Disease: An Overview and Potential Therapeutic Approaches

Department of Cardiology, Keio University School of Medicine, Shinjuku-ku, Tokyo 1608582, Japan
*
Author to whom correspondence should be addressed.
Biomedicines 2022, 10(8), 1850; https://doi.org/10.3390/biomedicines10081850
Submission received: 21 May 2022
/
Revised: 18 July 2022
/
Accepted: 23 July 2022
/
Published: 1 August 2022
(This article belongs to the Collection Advances in Leukocyte Biology)
Abstract
:Recent advances in pharmacotherapy have markedly improved the prognosis of cardiovascular disease (CVD) but have not completely conquered it. Therapies targeting the NOD-like receptor family pyrin domain containing 3 inflammasome and its downstream cytokines have proven effective in the secondary prevention of cardiovascular events, suggesting that inflammation is a target for treating residual risk in CVD. Neutrophil-induced inflammation has long been recognized as important in the pathogenesis of CVD. Circadian rhythm-related and disease-specific microenvironment changes give rise to neutrophil diversity. Neutrophils are primed by various stimuli, such as chemokines, cytokines, and damage-related molecular patterns, and the activated neutrophils contribute to the inflammatory response in CVD through degranulation, phagocytosis, reactive oxygen species generation, and the release of neutrophil extracellular traps (NETs). In particular, NETs promote immunothrombosis through the interaction with vascular endothelial cells and platelets and are implicated in the development of various types of CVD, such as acute coronary syndrome, deep vein thrombosis, and heart failure. NETs are promising candidates for anti-inflammatory therapy in CVD, and their efficacy has already been demonstrated in various animal models of the disease; however, they have yet to be clinically applied in humans. This narrative review discusses the diversity and complexity of neutrophils in the trajectory of CVD, the therapeutic potential of targeting NETs, and the related clinical issues.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
In recent years, inflammation has gained attention as a residual risk in cardiovascular disease (CVD), and it is being investigated in proof-of-concept clinical trials. In clinical practice, the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome-interleukin (IL)-1β-IL-6 high-sensitivity C-reactive protein (hsCRP) pathway has been a therapeutic target against inflammation [1]. Starting with the JUPITER (Justification for the Use of Statin in Prevention: An Intervention Trial Evaluating Rosuvastatin) trial, which showed that—in addition to low-density lipoprotein cholesterol (LDL-C) levels—hsCRP levels can also be a marker for predicting the reduction of cardiovascular event risk with statin therapy [2], the CANTOS (Canakinumab Antiinflammatory Thrombosis Outcome Study) showed that canakinumab, a human anti-IL-1β monoclonal antibody, is effective in the secondary prevention of myocardial infarction (MI) [3]. Subsequently, in COLCOT (Colchicine Cardiovascular Outcomes Trial) and the LoDoCo2 (Low-dose Colchicine 2) trial, colchicine, an inhibitor of the NLRP3 inflammasome, was reported to be effective in the secondary prevention in patients with coronary artery disease (CAD) [4,5]. The ZEUS (Ziltivekimab Cardiovascular Outcomes Study) is currently underway to determine whether ziltivekimab, a human monoclonal antibody that is directed against the IL-6 ligand, is effective in preventing major adverse cardiovascular events (defined as a composite of nonfatal stroke, nonfatal MI, and cardiovascular death) in patients at a high cardiovascular risk who have chronic kidney disease and elevated hsCRP [6].
Neutrophils are immune cells that form the first line of defense against viral, bacterial, fungal, and parasitic infections. They do so through degranulation, phagocytosis, the production of reactive oxygen species (ROS), and the construction of neutrophil extracellular traps (NETs), i.e., web-like structures that are composed of neutrophil DNA with antimicrobial proteins, including neutrophil elastase (NE), myeloperoxidase (MPO), and cathepsin G, histones, and granules [7,8]. Neutrophils are not only essential for defense against infection but also are extensively involved in the pathogenesis of diseases, from inflammation induction to tissue repair [9]. Although neutrophils had been recognized as a homogeneous population, accumulated evidence clearly demonstrates that they acquire diversity in a disease-specific manner and that not only quantitative but also qualitative changes in neutrophils influence cardiovascular outcomes.
In this narrative review, we summarize some recent basic and clinical findings on how the multifaceted trait alterations in neutrophils and NET formation are involved in the pathogenesis of inflammation in CVD. We also highlight the feasibility and challenges of developing anti-inflammatory therapies for CVD that target NETs.
2. Life Cycle of Neutrophils
2.1. Neutrophil Release from the Bone Marrow
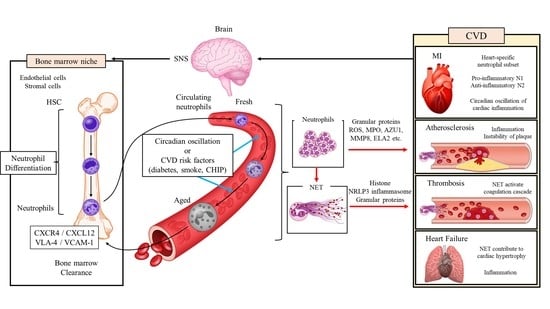
Neutrophils account for approximately 70% of circulating leucocytes and, in humans, more than 1 × 1011 to 2 × 1011 neutrophils are produced daily in the bone marrow (BM) [10]. Neutrophils are mobilized from the BM physiologically or in response to stress conditions, and this mobilization involves the antagonistic action of CXC chemokine receptor 2 (CXCR2) and CXCR4 on the neutrophil surface [11].
In the hematopoietic stem cell niches of BM, neutrophil adhesion to endothelial and stromal cells is mediated via very late antigen-4 (VLA-4)/vascular cell adhesion molecule 1 (VCAM-1) signaling, and this adhesion is enhanced by the CXCR4/CXC ligand 12 (CXCL12) pathway [11,12,13,14,15]. CXCL12 is mainly expressed in osteoblasts [16,17]. During the differentiation process, neutrophils change into CXCR4+CXCR2 pre-proliferating neutrophils, CXCR4-CXCR2low non-proliferating immature neutrophils, and finally, CXCR2high neutrophils. The decreased expression of CXCR4 allows the release of neutrophils from the BM into the circulatory system [12]. The CXCR2 ligands, CXCL1 and CXCL2, are expressed on osteoblasts and endothelial cells in BM, and CXCR2/CXCR2 ligand signaling stimulates neutrophil egress from the BM [11,14]. Of note, the hematopoietic stem cell niche is regulated by the sympathetic nervous system: Noradrenaline that is released by sympathetic nerves acts on the β3-adrenergic receptors of stromal cells to inhibit CXCL12 expression, thereby promoting neutrophil outflow into the circulation [18,19].
2.2. Aging of Neutrophils in Circulating Blood
Neutrophils newly that are released from the BM disappear from the circulating blood in as little as 6 to 12 h, during which time they undergo intrinsic changes, termed neutrophil senescence, that follow circadian patterns [20,21,22,23]. In mice, fresh neutrophils that are produced in the BM are released into the circulation during the night when the mice are active, whereas the aged neutrophils are removed from the peripheral circulation during the daytime, when the mice are inactive [24]. Diurnal changes in phenotypes include the loss of CD62L (also known as L-selectin) and an increase in CXCR4 expression [24].
In mice, the diurnal rhythm of neutrophils is regulated by the clock gene Arntl, which encodes Bmal1; Bmal1 triggers CXCR2-dependent transcriptional changes by regulating the circadian expression of CXCL2 and driving cell-autonomous neutrophil phenotypic and functional changes, namely the aging program [22]. In contrast, CXCR4 antagonizes CXCR2-mediated signaling and thereby blocks the aging program [22].
2.3. Homeostatic Function of Neutrophils
Recent evidence suggests that after their lifespan in the circulating blood, neutrophils also enter healthy tissues other than the BM and help to maintain tissue homeostasis [22]. Most infiltrate the spleen and lungs, but some infiltrate other organs, such as the intestines, heart, adipose tissue, and kidneys [24,25].
In general, neutrophils eliminate invading pathogens through a variety of inflammatory responses, including degranulation, phagocytosis, ROS production, and the release of NETs [8]. The contents of neutrophil granules are cytotoxic and include MPO, cathepsin G, elastase 2, cathelicidin antimicrobial peptide, and matrix metalloproteinase 8 (MMP8), and neutrophils generate a large amount of ROS by activating superoxide-generating enzyme nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, a process that is known as the ″oxidative burst. Neutrophils phagocytose pathogens, then the neutrophil granules fuse with the pathogen-containing phagosomes, and subsequently, ROS and neutrophil proteases work together to kill the ingested pathogens. The NETs that are produced by neutrophils have highly adhesive properties, so they trap pathogens, which are then eliminated by the various types of NET granules [7].
Curiously, despite their abundance of cytotoxic granules and ability to produce ROS, neutrophils rarely cause tissue damage when they are mobilized into normal tissues. Adrover et al. reported that neutrophils that are freshly released from the BM contain abundant granules and are capable of NET formation, but while flowing in the peripheral blood, they degranulate by a mechanism that is regulated by the clock protein Bmal1 and CXCR2 and their NET-forming capacity is also reduced. This cell-intrinsic disarmament program causes aging neutrophils to have blunted inflammatory properties, protecting tissues from excessive inflammation [21]. However, another report showed the opposite, i.e., enhanced ROS production, adhesion, and phagocytosis by aged human neutrophils [30]. Therefore, further studies are needed to determine how the inflammatory properties of neutrophils are altered during neutrophil aging in physiological and pathological settings.
2.4. Leukocyte Adhesion Cascade
Circulating neutrophils are mobilized to inflammatory foci through a process that is called the leukocyte adhesion cascade [31]. The activated vascular endothelial cells adjacent to inflammatory sites express adhesion receptors such as P-selectin and E-selectin, which mediate leukocyte rolling on the activated endothelium. Efficient effector function of neutrophils requires β2 integrins [32]. When integrins bind to ligands such as intercellular adhesion molecule-1 (ICAM-1) and ICAM-2 on endothelial cells, neutrophils can firmly adhere and migrate to peripheral tissues [33]. The expression of CXCL1 and CXCL2 by resident cells is essential for neutrophil recruitment to inflammatory sites [34]. At the inflammatory sites, various stimuli, such as cytokines, chemokines, pathogen-associated molecular patterns, and damage-associated molecular patterns, cause neutrophils to undergo tissue-specific phenotypic changes [20].
2.5. NET Formation
The dysregulation of neutrophil processes, including the overproduction or defective degradation of NETs, can cause excessive inflammation and the development of potentially fatal pathologies, including acute respiratory distress syndrome and immunothrombosis [35,36]. As discussed below, NETs are also deeply involved in CVD.
NET formation can be triggered by a variety of microbial stimuli and inflammatory mediators. In general, it requires the production of ROS, large amounts of which are produced and released by neutrophils during the oxidative burst [36,37]. ROS-dependent activation of protein arginine deiminase 4 (PAD4) promotes citrullination of histones, nuclear membrane loss, and the release of NET components [36]. NET formation is defective in NE knockout mice and MPO-deficient patients, showing that NE and MPO—proteins that are derived from azurophil granules—are essential for NET formation [38,39]. Activation of the NRLP3 inflammasome in macrophages induces NET formation via the production of IL-1β and IL-18 [40]. Conversely, NETs activate the NRLP3 inflammasome in macrophages. Histones that are derived from NETs directly interact with toll-like receptor 2 (TLR2) that is expressed on T-cells, leading to phosphorylation of signal transducer and activator of transcription (STAT) 3 and differentiation of T-cells into T helper 17 (Th17) cells, which are capable of mobilizing high numbers of neutrophils [41,42,43]. In this way, NETs amplify neutrophil-centered inflammation.
3. Neutrophils in CVD
Neutrophils are primed by various lifestyle-related disease risk factor stimuli, such as hyperglycemia, and the activated neutrophils contribute to sterile inflammatory responses such as atherosclerosis and the wound healing processes after MI through degranulation, phagocytosis, generation of ROS, and the release of neutrophil extracellular traps (NETs) [44]. In particular, NETs promote immunothrombosis through their interaction with vascular endothelial cells and platelets and have been implicated in the pathogenesis of various types of CVD, including acute coronary syndrome (ACS), deep vein thrombosis, and heart failure [45].
3.1. Neutrophil Counts as Prognostic Factors in CVD
In a cohort study of 12 types of CVD in a sample of more than 700,000 patients, patients with a neutrophil count in the upper limit of the normal range had higher incidences of unheralded coronary death, nonfatal MI, heart failure, peripheral artery disease, and abdominal aortic aneurysm than those with a neutrophil count at the lower end of the normal range [46]. Another study found that increased neutrophil counts are comparable to hypertension as a risk factor for CVD [47]. Furthermore, neutrophil counts correlate with recurrent ischemic events and mortality in CAD [48,49]. An elevated neutrophil to lymphocyte ratio is associated with CAD mortality, left atrial thrombosis, and impaired myocardial perfusion [47,50,51]. In an analysis of data from the Jackson Heart Study, a study of African American people living in Jackson, Mississippi, USA, a higher neutrophil to lymphocyte ratio was prospectively associated with all-cause mortality, coronary heart disease, and heart failure [52]. However, the causal relationship between the ratio and CVD remains unclear. Further investigations are also required to assess how an increase in the absolute number of neutrophils or qualitative changes in neutrophils affect the pathogenesis of CVD.
3.2. Effects of Cardiovascular Risks on Neutrophil Activation
Hyperglycemia purportedly increases the risk of developing CVD by generating primed inflammatory neutrophils. In a diabetic mouse model, hyperglycemia promoted myelopoiesis and neutrophil activation in the BM [53]. In hyperglycemic conditions, the neutrophil-derived S100 calcium-binding proteins A8/A9 (S100A8/A9) stimulated hepatic Kupffer cells via the receptor for advanced glycation end products, resulting in increased IL-6 production. IL-6 acts on the hepatocytes to stimulate thrombopoietin production, causing inflammatory platelet hyperplasia [54]. Increasing the levels of IL-6 are also associated with cardiovascular events [55]. The activated Kupffer cells are involved in amplifying inflammation and may be a novel therapeutic target in CVD. In mice with high-fat diet-induced obesity, elastase that was released from azurophil granules of neutrophils was found to disrupt insulin receptor substrate 1 in adipocytes and hepatocytes, mediating insulin resistance and lipogenesis [56].
Hyperglycemia promotes NET formation both in vitro and in vivo by activating NADPH oxidase, which increases ROS production [57,58]. NETs are associated with diabetic complications such as diabetic retinopathy and delayed healing [57,59,60]. The neutrophils of patients with Type 1 and 2 diabetes show elevated expression of peptidyl arginine deiminase 4 (PAD4), an important enzyme for histone citrullination and chromatin decondensation [60].
Angiotensin II (Ang II), a vasoconstrictor, plays an important role in hypertension and atherosclerosis. In addition, it induces ROS production in neutrophils, resulting in NET formation, and the Ang II receptor Type I antagonist losartan suppresses NET formation [61].
Hyperglycemia and the activation of the renin-angiotensin system may increase the risk of CVD by causing changes in the neutrophil phenotype. Future studies are needed to investigate whether lifestyle modifications such as weight loss and exercise, as well as the administration of glucose-lowering drugs and renin-angiotensin system inhibitors, can ameliorate changes in the inflammatory phenotype of neutrophils.
3.3. Neutrophils in the Post-MI Heart
Neutrophils are the first responders that amplify the acute inflammatory response after MI. At the onset of MI, the sympathetic nervous system is activated, and signaling at β3 adrenergic receptors in the BM decreases CXCL12 expression in the hematopoietic stem cell niche, which mobilizes neutrophils into the circulating blood [18]. Damage-associated molecular patterns that are released from necrotic cardiomyocytes activate neutrophils, and the activated neutrophils remove the damaged tissue. Neutrophils secrete vascular endothelial growth factor and also play an important role in angiogenesis in injured tissues [62]. However, the persistent infiltration of neutrophils leads to delayed inflammation resolution and tissue repair, resulting in adverse left ventricular remodeling [63]. After fulfilling their role, neutrophils undergo apoptosis and are subsequently ingested by macrophages [64].
Recently, the activation of gasdermin D (GSDMD) in neutrophils at the early stage of MI was found to play an important role in the increased production of neutrophils and their mobilization to the infarcted lesion [65]. GSDMD is a pore-forming protein and is required for the formation and release of NETs [66,67]. During NET formation, GSDMD is proteolytically activated by neutrophil proteases. Cleaved and activated GSDMD forms a pore in the granule membrane, facilitating the release of proteases into the cytoplasm, which leads to further GSDMD activation. These events allow proteases to migrate into the nucleus, where they process histones and enable nuclear expansion. Furthermore, in the final step, the activated GSDMD forms a pore in the plasma membrane, allowing the release of NETs. In mice, genetic deletion of GSDMD reduces neutrophil/monocyte recruitment to the infarcted heart, resulting in reduced infarct size, improved cardiac function, and increased survival after MI [65]. However, the involvement of NETs in this process is unknown.
Neutrophil polarity at the site of MI changes over time [68]. In the early phase (day 1 after MI), the inflammatory neutrophils (known as N1) infiltrate the heart, and on days 5 and 7, the anti-inflammatory neutrophils (known as N2) promote healing by polarizing macrophages to a repair phenotype [9,69]. Recent single-cell analyses have revealed that neutrophils follow different aging trajectories in the circulating blood and in ischemic myocardium [70]. Neutrophils infiltrating the heart immediately after the onset of MI, on day 1, had the same gene expression profile as BM-derived juvenile neutrophils. However, from day 3 onward, a subset of SiglecFhi neutrophils emerged that were not present in the circulating blood and whose gene expression profile suggested that they had increased effector function (phagocytosis, ROS production). This subset of SiglecFhi neutrophils was considered to be a locally acquired phenotype within the microenvironment of the MI lesion.
Thus, fresh neutrophils migrating from the BM undergo an aging process that is specific to the post-MI heart and distinct from the physiological aging process that is regulated by the circadian rhythm (Figure 1). The elimination of inflammatory neutrophils after MI or the promotion of differentiation of anti-inflammatory tissue-repairing neutrophils may be considered as a therapeutic strategy to preserve cardiac function after MI. The detailed mechanisms of the disease- and organ-specific phenotypic changes in neutrophils remain unclear, but their elucidation may help prevent adverse cardiac remodeling post-MI.
3.4. Neutrophil Aging Process and MI Outcome
In a mouse model of acute MI, due to left anterior descending artery ischemia-reperfusion, the extent of myocardial damage showed diurnal variation, with a larger infarct size at zeitgeber time 5 (5 h after the lights were turned on, i.e., in the phase when mice are inactive), when the circulating aged neutrophils are present. Consistent with this, the infarct size was larger in the Cxcr4ΔN mutant mice, which had more aged neutrophils in the peripheral blood, and smaller in the ArntlΔN mutant mice, in which the circulating neutrophils remain fresh and were not aged [22]. The authors concluded that the aged neutrophils increase the infarct size by promoting the formation of immunothrombi in the blood vessels. Neutrophils that are circulating in the peripheral blood degranulate through a proteome release process that is mediated by CXCR2-CXCL2 signaling [21]. As CXCR4 acts antagonistically with CXCR2, one would expect degranulation to be enhanced in Cxcr4ΔN mutants. Thus, at first glance it appears to be contradictory that during ischemia-reperfusion injury, the disarmed aging neutrophils, which should have low NET-forming capacity, promote intravascular thrombus formation. However, it is quite possible that the local functional and morphological changes of neutrophils during aseptic tissue injury are different from the processes that follow circadian rhythms in normal conditions. Indeed, in ex vivo systems, we found that DNA damage by phorbol-12-myristate-13 acetate (PMA) promotes NET induction, resulting in increased MPO granules within neutrophils via the increased expression of CXCR4 on the cell surface. In support of this finding, we observed that more PMA-stimulated dsDNA is released in the granule-rich early morning neutrophils than in the granule-poor daytime neutrophils [71]. CXCR4 expression on neutrophils in the inflammatory setting may inhibit the physiological disarming process by CXCR2-CXCL2 signaling and contribute to increased NET formation.
3.5. Mechanistic Insight into Immunothrombosis Induced by NETs
Neutrophils and neutrophil-derived inflammatory mediators have been implicated in immunothrombosis (Figure 2). Studies of laser-induced endothelial injury in mice showed that neutrophils infiltrate injured endothelial cells before platelets do, promoting platelet activation, the coagulation cascade, and fibrin generation [72]. The neutrophils activate platelets through the production of ROS [73], and the activated platelets present high mobility group box 1 (HMGB1) to the neutrophils, inducing NET formation [74]. The NETs bind to active coagulation factors, such as factor XII and tissue factor, causing the activation of the coagulation cascade and the generation of thrombin and fibrin [75,76]. Neutrophil granules, such as cathepsin G and elastase 2, degrade tissue factor pathway inhibitor, thereby promoting coagulation [75]. The extracellular histone proteins lead to thrombin production and platelet activation via TLR2 and TLR4, enhancing immunothrombosis [77]. Furthermore, NETs bind fibrinogen and promote fibrin deposition and fibrin network formation [78]. DNA that is released from NETs also serves as a scaffold for binding neutrophil granule proteins, von Willebrand factor (vWF), and fibronectin, a process that promotes platelet binding [79]. Neutrophils that are isolated from MI foci are highly activated and form aggregates with platelets [80]. Both granular proteins and ROS that are released by neutrophils activate endothelial cells, and these activated cells further induce neutrophils in the injured tissue [81]. Hence, neutrophils are increasingly recognized as being important in thrombosis formation and as a risk factor for CVD; however, therapies that target neutrophils to suppress immunothrombosis have not been implemented.
3.6. NETs in Coronary Artery Disease
After MI, large numbers of neutrophils accumulate in coronary arteries and contribute to coronary thrombosis by forming NETs, the primary scaffold for platelets, red blood cells, and fibrin [80]. In patients with ST-segment elevation MI (STEMI), the levels of NETs-related factors, such as dsDNA and MPO/DNA complexes, are significantly higher in the coronary blood than in the peripheral blood. Elevated coronary dsDNA levels may predict in-hospital major adverse cardiovascular events more sensitively than creatine kinase muscle and brain and troponin T [82]. Furthermore, in patients with STEMI, the extent of NET formation in coronary thrombosis correlated positively with the area under the curve of creatine kinase muscle and brain and infarct size that was measured by cardiac magnetic resonance and negatively with ST segment resolution on electrocardiogram [80]. In a cohort of 30 patients with STEMI and stable angina who underwent successful percutaneous coronary intervention, a gradual decrease in dsDNA was reported in all the patients 14 days after the intervention, although during the observation period the dsDNA levels remained higher in patients with STEMI than in those with stable angina [83]. NETs have also been implicated in the pathophysiology of stent thrombosis [84]. MPO levels correlate with the prevalence and severity of CVD [85,86,87]. A clinical study examining whether double-stranded DNA and MPO DNA in the circulating blood are associated with clinical outcomes and increased coagulability in patients with stable CAD found that only the double-stranded DNA levels were significantly associated with adverse clinical outcomes at 2 years (unstable angina, nonbleeding stroke, MI, and death) [88]. Men, smokers, patients with metabolic syndrome, and patients with previous MI had significantly higher double-stranded DNA levels; however, the association between the double-stranded DNA levels and hypercoagulability was weak. Double-stranded DNA is not necessarily derived solely from NETs. Further studies with more specific NETs markers (e.g., histone citrullination of neutrophils) are warranted.
3.7. NETs in Atherosclerotic Plaques
In animal models, several neutrophil-derived inflammatory factors, such as IL-37, heparin-binding protein (HBP/CAP37/azurocidin), MMP8, and ROS, are implicated in the development and instability of atherosclerotic plaques. IL-37 and heparin-binding protein (HBP/CAP37/azurocidin) specifically stimulate the mobilization of inflammatory monocytes via the activation of formyl peptide receptors [89]. During the development of atherosclerosis, infiltrated monocytes differentiate into macrophages, which contribute to plaque formation as lipid-rich foam cells [90]. MMP8 degrades collagen and induces plaque instability [91]. ROS activates endothelial cells, leading to neutrophil mobilization and activation, and oxidizes LDL, which may contribute to plaque vulnerability [92].
NETs have been identified in human atherosclerotic foci, and the presence of NETs indicates a more severe atherothrombotic state [93,94]. In human atherosclerotic plaques, neutrophils and NETs are seen near clusters of apoptotic endothelial and smooth muscle cells that contribute to plaque disruption [95,96]. The rupture of the fibrous capsule of atherosclerotic plaques has been considered as the primary cause of ACS, and there is evidence for the involvement of NETs in plaque rupture [74]. However, advances in imaging techniques, such as optical coherence tomography, now indicate that erosion of the plaque while it retains its fibrous capsule is responsible for about one-third of cases of ACS [97]. NETs seem to be involved in the process of plaque erosion. The process begins with a change in the endothelial shear stress gradient, which activates TLR2 on the endothelial cells, leading to a loss of basement membrane integrity and shedding of endothelial cells, followed by the formation of NETs and thrombosis [95]. Cholesterol crystals activate IL-1β transcription in response to macrophages and simultaneously activate inflammasomes and stimulate IL-1β secretion. NETs act synergistically with cholesterol crystals in the transcriptional activation of IL-1β in macrophages. The secreted IL-1β upregulates the T-cell-derived cytokine IL-17 and drives CXCL1 and CXCL2 to amplify neutrophil recruitment to atherosclerotic plaques [98]. NETs are also triggered by P-selectin, which is expressed by the activated platelets or the activated endothelium [99]. NETs components, such as cathelicidin antimicrobial peptide and cathepsin G, can induce the migration of monocytes and macrophages into a plaque [100,101]. After plaque rupture, the interaction between neutrophils and thrombin-activated platelets at the injured lesion further promotes NET formation [76].
As mentioned above, evidence is accumulating that NETs are involved in both plaque rupture and erosion, but the effect of NETs on atherosclerotic plaque formation itself is still unclear [96,102]. In an atherosclerosis model in which low-density lipoprotein receptor-deficient mice were fed a high-fat diet, the loss of Pad4 in hematopoietic cells by BM transplantation to suppress NET formation had no effect on fatty streak and plaque formation [96].
In summary, although the contribution of NETs to the formation of atherosclerotic plaques is debatable, they appear to play an important role in the development of ACSs that are associated with plaque rupture and erosion. In addition to anticoagulation and antiplatelet therapies, clinical trials assessing whether neutrophil-targeted anti-immunothrombotic therapies are effective in preventing or treating ACS may be worthwhile.
3.8. NETs in Venous Thrombosis
Venous thrombosis is usually initiated by endothelial dysfunction [103]. Endothelial cells contain Weibel-Palade bodies (WPBs), granules that store vWF, P-selectin, and other vasoregulatory factors; in response to vascular injury, the endothelial cells secrete WPBs, which release vWF. Secreted as ultra-high molecular weight multimers, vWF adheres to both platelets and collagen in subendothelial tissue that is exposed at the site of vascular injury. P-selectin is translocated from within the WPBs to the extracellular surface and is released by exocytosis, and externalized P-selectin that is anchored to the plasma membrane interacts with a leukocyte ligand, P-selectin glycoprotein ligand-1. Some platelets that are bound to vWF multimers are activated, express P-selectin, and adhere to neutrophils [99], and the platelet-neutrophil interaction promotes the formation of NETs [104]. Histones in NETs stimulate the release of WPBs from vascular endothelial cells [105], forming a feedforward loop.
In a mouse model of deep venous thrombosis that was induced by flow restriction of the inferior vena cava, venous thrombi contained large amounts of NET components, such as the extracellular citrullinated histone H3, in the vicinity of vWF [106]. Genetic depletion of PAD4 and intravenous administration of deoxyribonuclease 1 ameliorated thrombus formation [106,107]. However, in humans, thromboses that were recovered from patients with venous thrombosis contained significantly fewer NETs than coronary thromboses from patients with MI [80]. Further studies are needed on the involvement of NETs in venous thrombosis and their potential as therapeutic targets in humans.
3.9. NETs in Heart Failure
Although the pathogenesis of heart failure is heterogeneous and involves many different types of inflammation, recent evidence suggests that NETs are involved in chronic cardiac dysfunction. In mice, Ang II-induced cardiac hypertrophy and dysfunction were alleviated by neutrophil depletion with anti-Ly6G antibodies, whereas they were exacerbated by acute neutrophilia that was achieved by neutrophils transfusion [108]. Mechanistically, Ang II induced neutrophil activation, resulting in adhesion to the vessel wall and NET formation. NETs triggered microthrombosis and the impairment of myocardial microcirculation. Chronic microthrombosis leads to capillary rarefaction, further exacerbating myocardial hypoxia and impairing cardiac function. Leukocytes from human and murine models of heart failure were characterized by markedly decreased expression of Kruppel-like factor 2 (Klf2), a suppressor of myeloid inflammatory activation. The downregulation of Klf2 enhances the inflammatory phenotype of neutrophils via the activation of hypoxia-inducible factor 1 and other factors, activating the NET-thrombosis pathway [108]. Currently, the main approach to treating heart failure is to reduce the hemodynamic burden on the heart by inhibiting the excessive activation of neurohumoral factors. NET-mediated immunothrombotic mechanisms are promising new therapeutic targets for heart failure.
4. Neutrophils in CVD Complicated by COVID-19
CVD is the most common comorbidity in patients with COVID-19 and has been associated with adverse outcomes [109]. Cardiovascular complications due to SARS-CoV-2 infection include ACS, venous thrombosis and pulmonary embolism (associated with abnormal coagulation), and myocarditis [110]. Neutrophil-induced inflammation and NET-induced immunological thromboembolism may contribute to the pathophysiology of COVID-19–related CVD. Indeed, NET formation by circulating and infiltrating neutrophils is increased in patients with COVID-19 [111] and correlates with the clinical severity and prognosis in these patients [112]. The oxidative burst that is associated with SARS-CoV-2 infection stimulates NET formation [113], and the spike proteins and viral RNA of SARS-CoV-2, together with the proinflammatory cytokines TNFα and IL-8, can activate neutrophils. When SARS-CoV-2 or its components adhere to platelets, they promote platelet activation and aggregation and induce NET formation [114,115]. The cytotoxicity of histones that are released from NETs promotes endothelial dysfunction and death [116]. The experience of SARS-CoV-2 infections during the COVID-19 pandemic is reaffirming the universal importance of neutrophils and NETs in the pathophysiology of CVD.
4.1. ACS
Excess NET formation that is induced by SARS-CoV-2 infection is important for the etiology of ACS that are associated with COVID-19 [117]. In fact, in a case series of five patients with STEMI that was associated with COVID-19, co-expression of MPO-DNA complexes and citrullinated histone H3, both markers of NETs, was found in the thrombi aspirated from coronary arteries in all the patients [117]. The coronary thrombogenic mechanism involving NETs is a distinct feature of ACS in general. Although coronary thrombosis may be induced by multiple mechanisms, such as platelet activation by SARS-CoV-2 and endothelial dysfunction, NETs formation is undoubtedly the main cause of coronary thrombosis and may be a therapeutic target.
4.2. Venous Thrombosis and Pulmonary Embolism
The incidence of venous thromboembolism in patients with COVID-19 is estimated to be around 7.4% to 16.5% [118,119]; this incidence appears to be higher than in other viral infections, suggesting that a thrombogenic mechanism that is specific to COVID-19 is involved. SARS-CoV-2 infects cells via angiotensin-converting enzyme 2 (ACE2), which is expressed on the vascular endothelial cells [120]. ACE2 plays an important role in vascular endothelial cell homeostasis by converting Ang II to Ang 1–7 and antagonizing Ang II-induced ROS production and consequent vascular endothelial injury [121]. SARS-CoV-2 impairs ACE2 function, leading to an increase in oxidative stress in the vascular endothelial cells and progressive endothelial cell injury [122]. This process causes vascular endothelial cells to release vWF, which binds to platelets; vWF stabilizes blood coagulation factor VIII and activates the coagulation cascade, resulting in thrombosis formation [123]. In addition, NET induction via ACE2 receptors on neutrophils and the subsequent activation of the coagulation cascade may also be involved in the development of venous thrombi.
4.3. Myocarditis
Acute myocarditis has been reported as a complication of COVID-19. In a study of 1597 U.S. athletes that were infected with SARS-CoV-2 (mean age, 19 years), cardiac magnetic resonance imaging scans revealed that 37 patients (2.3%) had asymptomatic or mild myocarditis [124]. In addition, among 148 patients with COVID-19 with elevated cardiac troponin, 26% of patients had a myocarditis-like pattern on cardiovascular magnetic resonance examination imaging [125]. In 22 patients with COVID-19 who showed elevated troponin, cardiac pathological findings showed no cases of the interstitial lymphocytic infiltrates or extensive myocardial necrosis that are characteristic of common viral myocarditis [126]. NETs have been detected in the endocardium of patients with myocarditis [127]. In addition, in an experimental myocarditis model of mice with coxsackie virus B3 infection, NET production peaked one week after infection, and neutrophil-specific PAD4 knockout reduced cardiac necrosis and inflammation. These findings suggest that NET production that is induced by viral infection is important for the pathogenesis of myocarditis [128]. Although no studies have clarified the extent to which NETs are involved in myocarditis in patients with COVID-19, various cytokines and chemokines that are released during SARS-CoV-2 infection may induce NET formation and lead to the onset of myocarditis.
The cytokine midkine, which is important for neutrophil adhesion during acute inflammation and subsequent extravasation, promotes neutrophil recruitment and NET formation in the heart and contributes significantly to the pathology of myocarditis [127]. In patients with COVID-19, the serum midkine levels are elevated about three-fold compared with healthy controls [129]. It has been suggested that midkine may increase the affinity of SARS-CoV-2 for ACE2 receptors and promote viral invasion and accumulation [130]. These results suggest that midkine and other inflammatory cascades in the pathogenesis of COVID-19 may contribute to the development of myocarditis by promoting NET formation.
4.4. Antithrombotic Therapy in Patients with COVID-19
Anticoagulation with low molecular weight heparin for the prevention of thrombosis has been shown to improve survival in patients with COVID-19 without increasing the risk of serious bleeding, so prophylactic anticoagulation with low molecular weight heparin is recommended in all hospitalized patients with COVID-19 who do not have any contraindications [131,132]. Recently, thrombosis prophylaxis with rivaroxaban was reported to have a beneficial effect on clinical outcomes in patients with COVID-19 who have a high thrombotic risk [133]. However, despite prophylactic anticoagulation, the incidence of venous thrombosis remains high in COVID-19 [134], suggesting that thrombogenic mechanisms are involved that cannot be prevented by existing strategies. Although the risk of exacerbation of infection due to suppression of NET production must be considered, the suppression of neutrophil activation and NET production may help to improve vascular endothelial damage and immunothrombosis and may reduce the prevalence of CVD that is associated with COVID-19.
5. Potential Therapeutics Targeting NETs
Given that NET formation that is associated with neutrophil activation is deeply involved in the pathogenesis of CVD, NET formation and NETs themselves may be promising therapeutic targets for the prevention and treatment of various types of CVD [82]. Some potential therapeutics are discussed below.
5.1. PAD4 Blockade and DNase I
Both PAD4 blockade and Dnase I-driven removal have been evaluated as promising NET-targeting therapeutic strategies. In apolipoprotein E-deficient mice that were fed a high-fat diet, the PAD inhibitor Cl-amidine blocked NET formation, decreased the size of the atherosclerotic lesion, and delayed carotid thrombosis onset time upon photochemical injury [135]. In acute carotid injury experiments, hematopoietic PAD4 deficiency or DNase I treatment protected mice from plaque erosion [96]. In a rat model of myocardial ischemia-reperfusion injury, a combination of DNase I and recombinant tissue-type plasminogen activator reduced NET formation and improved coronary microvasculature flow, leading to a reduction in infarct size and left ventricular remodeling [136].
However, in sterile inflammation the suppression of NETs has not only advantages but also disadvantages. DNase therapy may release highly active enzymes and toxic molecules such as histones that can damage healthy cells [137]. Furthermore, NETs exhibit anti-inflammatory effects such as degradation of cytokines and chemokines [138]. NETs also promote macrophage polarization toward a reparative phenotype. In fact, the wound healing process after MI is impaired in NET-deficient mice [139]. In addition, as stated above, NETs are important as a defense mechanism against infection. In mouse models of acute lung injury that was induced by bacteria such as methicillin-resistant Staphylococcus aureus and Pseudomonas aeruginosa, the partial deletion of PAD4 (heterozygous PAD4-deficient mice) and DNase I treatment had a beneficial effect on attenuating the lung injury without negatively affecting the bacterial burden and also improved survival; in contrast, the complete loss of PAD4 (homozygous PAD4-deficient mice) slightly reduced the lung injury and reduced bacterial clearance but ultimately did not improve survival [140]. Even in sterile inflammation, the possibility of an increased risk of infection should always be considered when targeting NETs.
5.2. Th2 Cytokines
So-called Type 2 immune responses, in which Th2 cytokines such as IL-4 and IL-13 play a central role, are involved in the pathogenesis of atopic and allergic diseases. Interestingly, patients with allergic diseases such as atopic dermatitis have fewer skin neutrophils than people with healthy skin and are more susceptible to bacterial skin infections [141], and the MPO levels and NET formation capacity are known to be reduced in neutrophils from patients with allergic diseases compared with those from healthy individuals [142]. Experimental evidence indicates that when the Type 2 cytokines IL-4 and IL-13 predominate, signaling via the Type 2 IL-4 receptors on neutrophils suppresses neutrophil effector functions such as mobilization, chemotaxis, and NET formation. Type 2 IL-4 receptor-mediated signaling suppresses neutrophil recruitment from the BM by decreasing CXCR2 expression and increasing CXCR4 expression via the activation of signal transducer and activator of transcription 6 and p38 mitogen-activated protein kinases. It also inhibits neutrophil chemotaxis by activating p38 mitogen-activated protein kinases and thus directly inhibiting the CXCR2-phosphoinositide 3-kinase pathway. Further studies are needed to determine how Th2 cytokines affect the aging process that is associated with the circadian rhythm of neutrophils in the peripheral blood and the qualitative changes of neutrophils in the inflammatory setting.
5.3. Molecular Hydrogen
Recently, we demonstrated the therapeutic potential of molecular hydrogen (H2) to inhibit neutrophil activation and NET formation [71]. In various pathological conditions, H2 has been experimentally and clinically proven to have therapeutic effects by scavenging hydroxyl radicals and improving inflammation [143,144,145,146,147,148,149,150]. In our previous study, we reported that inhalation of H2 during a percutaneous coronary intervention in patients with STEMI reduces left ventricular remodeling at 6 months [150]. We also confirmed the positive effects of H2 inhalation on brain damage after cardiopulmonary resuscitation in rats [151]. A multicenter randomized clinical trial on the clinical application of H2 inhalation in post-cardiac arrest syndrome is ongoing in Japan [152].
A published multicenter randomized clinical trial demonstrated that H2 inhalation improves symptoms and prognosis in patients with moderate COVID-19 pneumonia [153]. As NETs have been implicated in both the severity of COVID-19 pneumonia and in left ventricular remodeling after MI, we sought to test the hypothesis that H2 could inhibit excessive neutrophil activation and NET formation. In vitro experiments on human neutrophils revealed that H2 can suppress both PMA-induced (ROS-dependent) and Ca2+ ionophore-induced (ROS-independent) neutrophil aggregation and subsequent NET formation [71]. We found that histone H2AX, a DNA damage marker, is present in hyperactivated neutrophils and induces CXCR4 expression as part of the DNA damage response. The upregulation of CXCR4 inhibits homeostatic degranulation by CXCR2, leading to the accumulation of neutrophil granules and creating a situation that facilitates NET formation. Consistent with this finding, NET-forming neutrophils were reported to highly express CXCR4 on their surface [154]. The effects of H2 on DNA damage in stimulated neutrophils included the suppression of CXCR4 expression, maintenance of constant degranulation, and the lowering of the NET formation threshold. In vivo, H2 inhalation also suppressed NETs formed in the pulmonary arteries of lipopolysaccharide-induced sepsis models of aged micro-miniature pigs [154]. Thus, H2 may exert anti-NET effects by various mechanisms, such as the elimination of ROS, the suppression of Ca2+-dependent PAD4 activation, the inhibition of neutrophil aggregation, and the suppression of pathogenic cellular senescence of neutrophils.
Neutrophil activation also has beneficial effects in sterile inflammation. In experimental acute sterile lung injury in mice, alveolar neutrophils acquire the ability to take up and degrade extracellular DNA fragments and allow optimal organ repair only when they are exposed to an inflammatory environment [155]. Furthermore, neutrophils are said to be able to transport matrix from normal to damaged organs to promote wound healing [156]. Consequently, the development of treatments targeting neutrophils in aseptic inflammation must also consider their anti-inflammatory and tissue repair functions. The same is true for treatments targeting NETs. Furthermore, treatments that use DNase I to lysate NETs that have already formed and been released may instead risk causing systemic inflammation by spreading NET contents, such as histones and MPO, throughout the body. H2 inhibits neutrophil aggregation and NET formation by PMA-stimulated human neutrophils without affecting gene expression profiles or phagocytosis and chemotaxis. Consequently, H2 therapy is expected to be effective in the prevention and treatment of NET-related CVD because it can potently inhibit the formation only of excessive NETs on activated neutrophils without affecting the essential tissue homeostatic function of neutrophils (Figure 3).
6. Conclusions
Although neutrophils have a very short lifespan, their functional dynamism and localization are tightly regulated by circadian rhythms, which explains the clinical observation that disease severity varies with the time of day. In addition, in inflammatory milieu neutrophils undergo disease- and organ-specific phenotypic changes, which have a profound impact on the outcome of reversible and sometimes irreversible organ damage. Understanding the life cycle of neutrophils and the dynamic changes in their function in the local inflammatory setting may lead to the discovery of new targets for the prevention and treatment of CVD.
Neutrophil-mediated inflammation and thrombosis are among the common mechanisms underlying CVD, and the moderate suppression of neutrophil overactivation and NET formation is expected to improve the pathogenesis of various types of CVD, including ACS, thromboembolism, atherosclerosis, heart failure, and myocarditis. In particular, inhalation of H2 can efficiently prevent excessive NET formation without suppressing the basic functions of neutrophils. Consequently, H2 can reduce chronic inflammation and irreversible organ damage and is expected to have a therapeutic effect not only in infectious diseases but also in various types of CVD. One important strategy to overcome inflammation as a residual risk in CVD will be to develop neutrophil-targeted, NET-suppressive therapies that can be implemented clinically.
Author Contributions
Both K.S. and M.S. contributed to conceptualization, writing (original draft preparation, review, and editing), and funding acquisition; both authors have read and agreed to the published version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Japan Society for the Promotion of Science, JSPS Grant-in-Aid for Young Scientists JP21K16096 (2021–2022), Grant for The Japan Research Foundation for Healthy Aging, Grant for Japan Foundation for Applied Enzymology, Grant for The Japanese Heart Failure Society, Grant for Takeda Science Foundation and The Vehicle Racing Commemorative Foundation. The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ridker, P.M. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream to Identify Novel Targets for Atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rane, M. Interleukin-6 Signaling and Anti-Interleukin-6 Therapeutics in Cardiovascular Disease. Circ. Res. 2021, 128, 1728–1746. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.W. Current Understanding in Neutrophil Differentiation and Heterogeneity. Immune Netw. 2017, 17, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Görgens, A.; Radtke, S.; Horn, P.A.; Giebel, B. New relationships of human hematopoietic lineages facilitate detection of multipotent hematopoietic stem and progenitor cells. Cell Cycle 2013, 12, 3478–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- De Filippo, K.; Rankin, S.M. CXCR4, the master regulator of neutrophil trafficking in homeostasis and disease. Eur. J. Clin. Investig. 2018, 48 (Suppl. S2), e12949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eash, K.J.; Means, J.M.; White, D.W.; Link, D.C. CXCR4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions. Blood 2009, 113, 4711–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadik, C.D.; Kim, N.D.; Luster, A.D. Neutrophils cascading their way to inflammation. Trends Immunol. 2011, 32, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Petty, J.M.; Lenox, C.C.; Weiss, D.J.; Poynter, M.E.; Suratt, B.T. Crosstalk between CXCR4/stromal derived factor-1 and VLA-4/VCAM-1 pathways regulates neutrophil retention in the bone marrow. J. Immunol. 2009, 182, 604–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Semerad, C.L.; Christopher, M.J.; Liu, F.; Short, B.; Simmons, P.J.; Winkler, I.; Levesque, J.P.; Chappel, J.; Ross, F.P.; Link, D.C. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood 2005, 106, 3020–3027. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 2008, 452, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Ferrer, S.; Battista, M.; Frenette, P.S. Cooperation of beta(2)- and beta(3)-adrenergic receptors in hematopoietic progenitor cell mobilization. Ann. N. Y. Acad. Sci. 2010, 1192, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Adrover, J.M.; Aroca-Crevillén, A.; Crainiciuc, G.; Ostos, F.; Rojas-Vega, Y.; Rubio-Ponce, A.; Cilloniz, C.; Bonzón-Kulichenko, E.; Calvo, E.; Rico, D.; et al. Programmed ’disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat. Immunol. 2020, 21, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Adrover, J.M.; Del Fresno, C.; Crainiciuc, G.; Cuartero, M.I.; Casanova-Acebes, M.; Weiss, L.A.; Huerga-Encabo, H.; Silvestre-Roig, C.; Rossaint, J.; Cossío, I.; et al. A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity 2019, 50, 390–402.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aroca-Crevillén, A.; Adrover, J.M.; Hidalgo, A. Circadian Features of Neutrophil Biology. Front. Immunol. 2020, 11, 576. [Google Scholar] [CrossRef]
- Casanova-Acebes, M.; Pitaval, C.; Weiss, L.A.; Nombela-Arrieta, C.; Chèvre, R.; González, N.A.; Kunisaki, Y.; Zhang, D.; van Rooijen, N.; Silberstein, L.E.; et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 2013, 153, 1025–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furze, R.C.; Rankin, S.M. Neutrophil mobilization and clearance in the bone marrow. Immunology 2008, 125, 281–288. [Google Scholar] [CrossRef]
- Martin, C.; Burdon, P.C.; Bridger, G.; Gutierrez-Ramos, J.C.; Williams, T.J.; Rankin, S.M. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 2003, 19, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Rankin, S.M. The bone marrow: A site of neutrophil clearance. J. Leukoc. Biol. 2010, 88, 241–251. [Google Scholar] [CrossRef]
- von Vietinghoff, S.; Ley, K. Homeostatic regulation of blood neutrophil counts. J. Immunol. 2008, 181, 5183–5188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bratton, D.L.; Henson, P.M. Neutrophil clearance: When the party is over, clean-up begins. Trends Immunol. 2011, 32, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Uhl, B.; Vadlau, Y.; Zuchtriegel, G.; Nekolla, K.; Sharaf, K.; Gaertner, F.; Massberg, S.; Krombach, F.; Reichel, C.A. Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood 2016, 128, 2327–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Bouti, P.; Webbers, S.D.S.; Fagerholm, S.C.; Alon, R.; Moser, M.; Matlung, H.L.; Kuijpers, T.W. β2 Integrin Signaling Cascade in Neutrophils: More Than a Single Function. Front. Immunol. 2020, 11, 619925. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Chavakis, T. Endogenous modulators of inflammatory cell recruitment. Trends Immunol. 2013, 34, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.S.; Randall, K.L.; Pettitt, J.A.; Ellyard, J.I.; Blumenthal, A.; Enders, A.; Quah, B.J.; Bopp, T.; Parish, C.R.; Brüstle, A. Neutrophil extracellular traps and their histones promote Th17 cell differentiation directly via TLR2. Nat. Commun. 2022, 13, 528. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Costantini, C.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010, 115, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Zenobia, C.; Hajishengallis, G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol. 2000 2015, 69, 142–159. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef]
- Klopf, J.; Brostjan, C.; Eilenberg, W.; Neumayer, C. Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. Int. J. Mol. Sci. 2021, 22, 559. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yu, J.; Zeng, R.; Zhao, L.; Wan, Z.; Zeng, Z.; Cao, Y. Neutrophil Count Is Associated With Risks of Cardiovascular Diseases. J. Am. Coll. Cardiol. 2017, 70, 911–912. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.D.; Denaxas, S.; Nicholas, O.; Hingorani, A.D.; Hemingway, H. Neutrophil Counts and Initial Presentation of 12 Cardiovascular Diseases: A CALIBER Cohort Study. J. Am. Coll. Cardiol. 2017, 69, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Avanzas, P.; Arroyo-Espliguero, R.; Cosín-Sales, J.; Quiles, J.; Zouridakis, E.; Kaski, J.C. Multiple complex stenoses, high neutrophil count and C-reactive protein levels in patients with chronic stable angina. Atherosclerosis 2004, 175, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Dinerman, J.L.; Mehta, J.L.; Saldeen, T.G.; Emerson, S.; Wallin, R.; Davda, R.; Davidson, A. Increased neutrophil elastase release in unstable angina pectoris and acute myocardial infarction. J. Am. Coll. Cardiol. 1990, 15, 1559–1563. [Google Scholar] [CrossRef] [Green Version]
- Koza, Y. What is the clinical benefit of neutrophil-lymphocyte ratio in cardiovascular patients? J. Cardiovasc. Thorac. Res. 2014, 6, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, M.; Aparci, M.; Uz, O.; Isilak, Z.; Balta, S.; Dogan, M.; Kardesoglu, E.; Uzun, M. Neutrophil-lymphocyte ratio may predict left atrial thrombus in patients with nonvalvular atrial fibrillation. Clin. Appl. Thromb. Hemost. 2015, 21, 166–171. [Google Scholar] [CrossRef]
- Kim, S.; Eliot, M.; Koestler, D.C.; Wu, W.C.; Kelsey, K.T. Association of Neutrophil-to-Lymphocyte Ratio With Mortality and Cardiovascular Disease in the Jackson Heart Study and Modification by the Duffy Antigen Variant. JAMA Cardiol. 2018, 3, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.S.; et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraakman, M.J.; Lee, M.K.; Al-Sharea, A.; Dragoljevic, D.; Barrett, T.J.; Montenont, E.; Basu, D.; Heywood, S.; Kammoun, H.L.; Flynn, M.; et al. Neutrophil-derived S100 calcium-binding proteins A8/A9 promote reticulated thrombocytosis and atherogenesis in diabetes. J. Clin. Investig. 2017, 127, 2133–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Rifai, N.; Stampfer, M.J.; Hennekens, C.H. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation 2000, 101, 1767–1772. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol. 2015, 52, 497–503. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2018, 9, 3076. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Menegazzo, L.; Rigato, M.; Scattolini, V.; Poncina, N.; Bruttocao, A.; Ciciliot, S.; Mammano, F.; Ciubotaru, C.D.; Brocco, E.; et al. NETosis Delays Diabetic Wound Healing in Mice and Humans. Diabetes 2016, 65, 1061–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrysanthopoulou, A.; Gkaliagkousi, E.; Lazaridis, A.; Arelaki, S.; Pateinakis, P.; Ntinopoulou, M.; Mitsios, A.; Antoniadou, C.; Argyriou, C.; Georgiadis, G.S.; et al. Angiotensin II triggers release of neutrophil extracellular traps, linking thromboinflammation with essential hypertension. JCI Insight. 2021, 6, e148668. [Google Scholar] [CrossRef]
- Gong, Y.; Koh, D.R. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 2010, 339, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Tu, Z.; Chen, K.; Xu, Y.; Chen, F.; Xu, S.; Shi, T.; Qian, J.; Shen, L.; Hwa, J.; et al. Gasdermin D inhibition confers antineutrophil-mediated cardioprotection in acute myocardial infarction. J. Clin. Investig. 2022, 132, e151268. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, aar6676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döring, Y.; Libby, P.; Soehnlein, O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Vafadarnejad, E.; Rizzo, G.; Krampert, L.; Arampatzi, P.; Arias-Loza, A.P.; Nazzal, Y.; Rizakou, A.; Knochenhauer, T.; Bandi, S.R.; Nugroho, V.A.; et al. Dynamics of Cardiac Neutrophil Diversity in Murine Myocardial Infarction. Circ. Res. 2020, 127, e232–e249. [Google Scholar] [CrossRef]
- Shirakawa, K.; Kobayashi, E.; Ichihara, G.; Kitakata, H.; Katsumata, Y.; Sugai, K.; Hakamata, Y.; Sano, M. H(2) Inhibits the Formation of Neutrophil Extracellular Traps. JACC Basic Transl. Sci. 2022, 7, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Darbousset, R.; Thomas, G.M.; Mezouar, S.; Frère, C.; Bonier, R.; Mackman, N.; Renné, T.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood 2012, 120, 2133–2143. [Google Scholar] [CrossRef] [Green Version]
- Vanichakarn, P.; Blair, P.; Wu, C.; Freedman, J.E.; Chakrabarti, S. Neutrophil CD40 enhances platelet-mediated inflammation. Thromb Res. 2008, 122, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, M.; Jakowitsch, J.; Panzenböck, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Boer, O.J.; Li, X.; Teeling, P.; Mackaay, C.; Ploegmakers, H.J.; van der Loos, C.M.; Daemen, M.J.; de Winter, R.J.; van der Wal, A.C. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb. Haemost. 2013, 109, 290–297. [Google Scholar] [CrossRef]
- Liu, J.; Yang, D.; Wang, X.; Zhu, Z.; Wang, T.; Ma, A.; Liu, P. Neutrophil extracellular traps and dsDNA predict outcomes among patients with ST-elevation myocardial infarction. Sci. Rep. 2019, 9, 11599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helseth, R.; Solheim, S.; Arnesen, H.; Seljeflot, I.; Opstad, T.B. The Time Course of Markers of Neutrophil Extracellular Traps in Patients Undergoing Revascularisation for Acute Myocardial Infarction or Stable Angina Pectoris. Mediat. Inflamm. 2016, 2016, 2182358. [Google Scholar] [CrossRef] [PubMed]
- Riegger, J.; Byrne, R.A.; Joner, M.; Chandraratne, S.; Gershlick, A.H.; Ten Berg, J.M.; Adriaenssens, T.; Guagliumi, G.; Godschalk, T.C.; Neumann, F.J.; et al. Histopathological evaluation of thrombus in patients presenting with stent thrombosis. A multicenter European study: A report of the prevention of late stent thrombosis by an interdisciplinary global European effort consortium. Eur. Heart J. 2016, 37, 1538–1549. [Google Scholar] [CrossRef] [Green Version]
- Heslop, C.L.; Frohlich, J.J.; Hill, J.S. Myeloperoxidase and C-reactive protein have combined utility for long-term prediction of cardiovascular mortality after coronary angiography. J. Am. Coll. Cardiol. 2010, 55, 1102–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salonen, I.; Huttunen, K.; Hirvonen, M.R.; Dufva, J.; Groundstroem, K.; Dufva, H.; Pekkanen, J.; Salonen, R.O. Serum myeloperoxidase is independent of the risk factors of atherosclerosis. Coron. Artery Dis. 2012, 23, 251–258. [Google Scholar] [CrossRef]
- Zhang, R.; Brennan, M.L.; Fu, X.; Aviles, R.J.; Pearce, G.L.; Penn, M.S.; Topol, E.J.; Sprecher, D.L.; Hazen, S.L. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA 2001, 286, 2136–2142. [Google Scholar] [CrossRef]
- Langseth, M.S.; Opstad, T.B.; Bratseth, V.; Solheim, S.; Arnesen, H.; Pettersen, A.; Seljeflot, I.; Helseth, R. Markers of neutrophil extracellular traps are associated with adverse clinical outcome in stable coronary artery disease. Eur. J. Prev. Cardiol. 2018, 25, 762–769. [Google Scholar] [CrossRef]
- Soehnlein, O.; Zernecke, A.; Eriksson, E.E.; Rothfuchs, A.G.; Pham, C.T.; Herwald, H.; Bidzhekov, K.; Rottenberg, M.E.; Weber, C.; Lindbom, L. Neutrophil secretion products pave the way for inflammatory monocytes. Blood 2008, 112, 1461–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: A double-edged sword. Nat. Rev. Immunol. 2006, 6, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.P.; Sukhova, G.K.; Libby, P.; Gerdes, N.; Tang, N.; Horton, D.B.; Kilbride, M.; Breitbart, R.E.; Chun, M.; Schönbeck, U. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: A novel collagenolytic pathway suggested by transcriptional profiling. Circulation 2001, 104, 1899–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, T.; Kumon, Y.; Kobayashi, T.; Enzan, H.; Nishioka, Y.; Yuri, K.; Wakiguchi, H.; Sugiura, T. Neutrophil infiltration and oxidant-production in human atherosclerotic carotid plaques. Histol. Histopathol. 2011, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Megens, R.T.; Vijayan, S.; Lievens, D.; Döring, Y.; van Zandvoort, M.A.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Gallant, M.; Martinod, K.; Ten Cate, H.; Hofstra, L.; et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arter. Thromb. Vasc. Biol. 2013, 33, 2032–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quillard, T.; Araújo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. Eur. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef] [Green Version]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Fahed, A.C.; Jang, I.K. Plaque erosion and acute coronary syndromes: Phenotype, molecular characteristics and future directions. Nat. Rev. Cardiol. 2021, 18, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Döring, Y.; Drechsler, M.; Wantha, S.; Kemmerich, K.; Lievens, D.; Vijayan, S.; Gallo, R.L.; Weber, C.; Soehnlein, O. Lack of neutrophil-derived CRAMP reduces atherosclerosis in mice. Circ. Res. 2012, 110, 1052–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sjöberg, S.; Tang, T.T.; Oörni, K.; Wu, W.; Liu, C.; Secco, B.; Tia, V.; Sukhova, G.K.; Fernandes, C.; et al. Cathepsin G activity lowers plasma LDL and reduces atherosclerosis. Biochim. Biophys. Acta 2014, 1842, 2174–2183. [Google Scholar] [CrossRef] [Green Version]
- Soehnlein, O.; Bazioti, V.; Westerterp, M. A Pad 4 Plaque Erosion. Circ. Res. 2018, 123, 6–8. [Google Scholar] [CrossRef]
- Di Nisio, M.; van Es, N.; Büller, H.R. Deep vein thrombosis and pulmonary embolism. Lancet 2016, 388, 3060–3073. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.; Albánez, S.; Mewburn, J.; Nesbitt, K.; Gould, T.J.; Liaw, P.C.; James, P.D.; Swystun, L.L.; Lillicrap, D. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. J. Thromb. Haemost. 2016, 14, 2274–2286. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Fuchs, T.A.; Chauhan, A.K.; Yang, J.J.; De Meyer, S.F.; Köllnberger, M.; Wakefield, T.W.; Lämmle, B.; Massberg, S.; Wagner, D.D. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood 2011, 117, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Wang, P.; Zhang, R.; Watanabe, I.; Chang, E.; Vinayachandran, V.; Nayak, L.; Lapping, S.; Liao, S.; Madera, A.; et al. KLF2 regulates neutrophil activation and thrombosis in cardiac hypertrophy and heart failure progression. J. Clin. Investig. 2022, 132, e147191. [Google Scholar] [CrossRef]
- Xu, J.; Xiao, W.; Liang, X.; Shi, L.; Zhang, P.; Wang, Y.; Wang, Y.; Yang, H. A meta-analysis on the risk factors adjusted association between cardiovascular disease and COVID-19 severity. BMC Public Health 2021, 21, 1533. [Google Scholar] [CrossRef]
- Kwenandar, F.; Japar, K.V.; Damay, V.; Hariyanto, T.I.; Tanaka, M.; Lugito, N.P.H.; Kurniawan, A. Coronavirus disease 2019 and cardiovascular system: A narrative review. Int. J. Cardiol. Heart Vasc. 2020, 29, 100557. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, X.; Liu, X. NETosis and Neutrophil Extracellular Traps in COVID-19: Immunothrombosis and Beyond. Front. Immunol. 2022, 13, 838011. [Google Scholar] [CrossRef] [PubMed]
- Huckriede, J.; Anderberg, S.B.; Morales, A.; de Vries, F.; Hultström, M.; Bergqvist, A.; Ortiz-Pérez, J.T.; Sels, J.W.; Wichapong, K.; Lipcsey, M.; et al. Evolution of NETosis markers and DAMPs have prognostic value in critically ill COVID-19 patients. Sci. Rep. 2021, 11, 15701. [Google Scholar] [CrossRef]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaad, C.; Nuss, P.; Benoliel, J.J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets Can Associate with SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Dupont, A.; Rauch, A.; Staessens, S.; Moussa, M.; Rosa, M.; Corseaux, D.; Jeanpierre, E.; Goutay, J.; Caplan, M.; Varlet, P.; et al. Vascular Endothelial Damage in the Pathogenesis of Organ Injury in Severe COVID-19. Arter. Thromb. Vasc. Biol. 2021, 41, 1760–1773. [Google Scholar] [CrossRef]
- Blasco, A.; Coronado, M.J.; Hernández-Terciado, F.; Martín, P.; Royuela, A.; Ramil, E.; García, D.; Goicolea, J.; Del Trigo, M.; Ortega, J.; et al. Assessment of Neutrophil Extracellular Traps in Coronary Thrombus of a Case Series of Patients With COVID-19 and Myocardial Infarction. JAMA Cardiol. 2020, 6, 1–6. [Google Scholar] [CrossRef]
- Pellicori, P.; Doolub, G.; Wong, C.M.; Lee, K.S.; Mangion, K.; Ahmad, M.; Berry, C.; Squire, I.; Lambiase, P.D.; Lyon, A.; et al. COVID-19 and its cardiovascular effects: A systematic review of prevalence studies. Cochrane Database Syst. Rev. 2021, 3, Cd013879. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.J.; Hong, H.; Ohana, M.; Bompard, F.; Revel, M.P.; Valle, C.; Gervaise, A.; Poissy, J.; Susen, S.; Hékimian, G.; et al. Pulmonary Embolism and Deep Vein Thrombosis in COVID-19: A Systematic Review and Meta-Analysis. Radiology 2021, 298, E70–E80. [Google Scholar] [CrossRef] [PubMed]
- Teuwen, L.A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef]
- Koster, T.; Blann, A.D.; Briët, E.; Vandenbroucke, J.P.; Rosendaal, F.R. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995, 345, 152–155. [Google Scholar] [CrossRef]
- Daniels, C.J.; Rajpal, S.; Greenshields, J.T.; Rosenthal, G.L.; Chung, E.H.; Terrin, M.; Jeudy, J.; Mattson, S.E.; Law, I.H.; Borchers, J.; et al. Prevalence of Clinical and Subclinical Myocarditis in Competitive Athletes With Recent SARS-CoV-2 Infection: Results From the Big Ten COVID-19 Cardiac Registry. JAMA Cardiol. 2021, 6, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, T.; Knight, D.S.; Razvi, Y.; Kumar, K.; Vimalesvaran, K.; Thornton, G.; Patel, R.; Chacko, L.; Brown, J.T.; Coyle, C.; et al. Patterns of myocardial injury in recovered troponin-positive COVID-19 patients assessed by cardiovascular magnetic resonance. Eur. Heart J. 2021, 42, 1866–1878. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.E.; Li, G.; Akmatbekov, A.; Harbert, J.L.; Lameira, F.S.; Brown, J.Q.; Vander Heide, R.S. Unexpected Features of Cardiac Pathology in COVID-19 Infection. Circulation 2020, 142, 1123–1125. [Google Scholar] [CrossRef]
- Weckbach, L.T.; Grabmaier, U.; Uhl, A.; Gess, S.; Boehm, F.; Zehrer, A.; Pick, R.; Salvermoser, M.; Czermak, T.; Pircher, J.; et al. Midkine drives cardiac inflammation by promoting neutrophil trafficking and NETosis in myocarditis. J. Exp. Med. 2019, 216, 350–368. [Google Scholar] [CrossRef]
- Carai, P.; Florit González, L.; Van Bruggen, S.; Spalart, V.; De Giorgio, D.; Geuens, N.; Martinod, K.; Jones, E.A.V.; Heymans, S. Neutrophil inhibition improves acute inflammation in a murine model of viral myocarditis. Cardiovasc. Res. 2022, 00, 1–15. [Google Scholar] [CrossRef]
- Ketenci, S.; Uygar Kalaycı, M.; Dündar, B.; Duranay, R.; Şükrü Aynacıoğlu, A. Elevated serum midkine levels in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infected patients. Int. Immunopharmacol. 2022, 110, 108939. [Google Scholar] [CrossRef]
- Sanino, G.; Bosco, M.; Terrazzano, G. Physiology of Midkine and Its Potential Pathophysiological Role in COVID-19. Front. Physiol. 2020, 11, 616552. [Google Scholar] [CrossRef] [PubMed]
- Moores, L.K.; Tritschler, T.; Brosnahan, S.; Carrier, M.; Collen, J.F.; Doerschug, K.; Holley, A.B.; Jimenez, D.; Le Gal, G.; Rali, P.; et al. Prevention, Diagnosis, and Treatment of VTE in Patients with Coronavirus Disease 2019: CHEST Guideline and Expert Panel Report. Chest 2020, 158, 1143–1163. [Google Scholar] [CrossRef]
- Thålin, C.; Hisada, Y.; Lundström, S.; Mackman, N.; Wallén, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arter. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef]
- Ramacciotti, E.; Barile Agati, L.; Calderaro, D.; Aguiar, V.C.R.; Spyropoulos, A.C.; de Oliveira, C.C.C.; Lins Dos Santos, J.; Volpiani, G.G.; Sobreira, M.L.; Joviliano, E.E.; et al. Rivaroxaban versus no anticoagulation for post-discharge thromboprophylaxis after hospitalisation for COVID-19 (MICHELLE): An open-label, multicentre, randomised, controlled trial. Lancet 2022, 399, 50–59. [Google Scholar] [CrossRef]
- Llitjos, J.F.; Leclerc, M.; Chochois, C.; Monsallier, J.M.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef]
- Knight, J.S.; Luo, W.; O’ Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Ge, L.; Zhou, X.; Ji, W.J.; Lu, R.Y.; Zhang, Y.; Zhang, Y.D.; Ma, Y.Q.; Zhao, J.H.; Li, Y.M. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: Therapeutic potential of DNase-based reperfusion strategy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H500-9. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V. Neutrophil Extracellular Traps in the Second Decade. J. Innate Immun. 2018, 10, 414–421. [Google Scholar] [CrossRef]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhöfer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Eghbalzadeh, K.; Georgi, L.; Louis, T.; Zhao, H.; Keser, U.; Weber, C.; Mollenhauer, M.; Conforti, A.; Wahlers, T.; Paunel-Görgülü, A. Compromised Anti-inflammatory Action of Neutrophil Extracellular Traps in PAD4-Deficient Mice Contributes to Aggravated Acute Inflammation After Myocardial Infarction. Front. Immunol. 2019, 10, 2313. [Google Scholar] [CrossRef]
- Lefrançais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight. 2018, 3, e98178. [Google Scholar] [CrossRef] [Green Version]
- Egholm, C.; Heeb, L.E.M.; Impellizzieri, D.; Boyman, O. The Regulatory Effects of Interleukin-4 Receptor Signaling on Neutrophils in Type 2 Immune Responses. Front. Immunol. 2019, 10, 2507. [Google Scholar] [CrossRef]
- Impellizzieri, D.; Ridder, F.; Raeber, M.E.; Egholm, C.; Woytschak, J.; Kolios, A.G.A.; Legler, D.F.; Boyman, O. IL-4 receptor engagement in human neutrophils impairs their migration and extracellular trap formation. J. Allergy Clin. Immunol. 2019, 144, 267–279.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, T.; Suzuki, M.; Hayashida, K.; Kobayashi, Y.; Yoshizawa, J.; Shibusawa, T.; Sano, M.; Hori, S.; Sasaki, J. Hydrogen gas inhalation alleviates oxidative stress in patients with post-cardiac arrest syndrome. J. Clin. Biochem. Nutr. 2020, 67, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, T.; Sano, M.; Matsuoka, T.; Yoshizawa, J.; Yamamoto, R.; Katsumata, Y.; Endo, J.; Homma, K.; Kajimura, M.; Suzuki, M.; et al. Hydrogen Gas Inhalation Attenuates Endothelial Glycocalyx Damage and Stabilizes Hemodynamics in a Rat Hemorrhagic Shock Model. Shock 2020, 54, 377–385. [Google Scholar] [CrossRef]
- Sugai, K.; Tamura, T.; Sano, M.; Uemura, S.; Fujisawa, M.; Katsumata, Y.; Endo, J.; Yoshizawa, J.; Homma, K.; Suzuki, M.; et al. Daily inhalation of hydrogen gas has a blood pressure-lowering effect in a rat model of hypertension. Sci. Rep. 2020, 10, 20173. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Tamura, T. Hydrogen Gas Therapy: From Preclinical Studies to Clinical Trials. Curr. Pharm. Des. 2021, 27, 650–658. [Google Scholar] [CrossRef]
- Sano, M.; Suzuki, M.; Homma, K.; Hayashida, K.; Tamura, T.; Matsuoka, T.; Katsumata, Y.; Onuki, S.; Sasaki, J. Promising novel therapy with hydrogen gas for emergency and critical care medicine. Acute Med. Surg. 2018, 5, 113–118. [Google Scholar] [CrossRef]
- Nishi, K.; Iwai, S.; Tajima, K.; Okano, S.; Sano, M.; Kobayashi, E. Prevention of Chronic Rejection of Marginal Kidney Graft by Using a Hydrogen Gas-Containing Preservation Solution and Adequate Immunosuppression in a Miniature Pig Model. Front. Immunol. 2020, 11, 626295. [Google Scholar] [CrossRef]
- Kobayashi, E.; Sano, M. Organ preservation solution containing dissolved hydrogen gas from a hydrogen-absorbing alloy canister improves function of transplanted ischemic kidneys in miniature pigs. PLoS ONE 2019, 14, e0222863. [Google Scholar] [CrossRef]
- Katsumata, Y.; Sano, F.; Abe, T.; Tamura, T.; Fujisawa, T.; Shiraishi, Y.; Kohsaka, S.; Ueda, I.; Homma, K.; Suzuki, M.; et al. The Effects of Hydrogen Gas Inhalation on Adverse Left Ventricular Remodeling After Percutaneous Coronary Intervention for ST-Elevated Myocardial Infarction—First Pilot Study in Humans. Circ. J. 2017, 81, 940–947. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, K.; Sano, M.; Kamimura, N.; Yokota, T.; Suzuki, M.; Ohta, S.; Fukuda, K.; Hori, S. Hydrogen inhalation during normoxic resuscitation improves neurological outcome in a rat model of cardiac arrest independently of targeted temperature management. Circulation 2014, 130, 2173–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, T.; Hayashida, K.; Sano, M.; Suzuki, M.; Shibusawa, T.; Yoshizawa, J.; Kobayashi, Y.; Suzuki, T.; Ohta, S.; Morisaki, H.; et al. Feasibility and Safety of Hydrogen Gas Inhalation for Post-Cardiac Arrest Syndrome—First-in-Human Pilot Study. Circ. J. 2016, 80, 1870–1873. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Wei, C.H.; Chen, A.L.; Sun, X.C.; Guo, G.Y.; Zou, X.; Shi, J.D.; Lai, P.Z.; Zheng, Z.G.; Zhong, N.S. Hydrogen/oxygen mixed gas inhalation improves disease severity and dyspnea in patients with Coronavirus disease 2019 in a recent multicenter, open-label clinical trial. J. Thorac. Dis. 2020, 12, 3448–3452. [Google Scholar] [CrossRef]
- Radermecker, C.; Sabatel, C.; Vanwinge, C.; Ruscitti, C.; Maréchal, P.; Perin, F.; Schyns, J.; Rocks, N.; Toussaint, M.; Cataldo, D.; et al. Locally instructed CXCR4(hi) neutrophils trigger environment-driven allergic asthma through the release of neutrophil extracellular traps. Nat. Immunol. 2019, 20, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Oved, J.H.; Paris, A.J.; Gollomp, K.; Dai, N.; Rubey, K.; Wang, P.; Spruce, L.A.; Seeholzer, S.H.; Poncz, M.; Worthen, G.S. Neutrophils promote clearance of nuclear debris following acid-induced lung injury. Blood 2021, 137, 392–397. [Google Scholar] [CrossRef]
- Fischer, A.; Wannemacher, J.; Christ, S.; Koopmans, T.; Kadri, S.; Zhao, J.; Gouda, M.; Ye, H.; Mück-Häusl, M.; Krenn, P.W.; et al. Neutrophils direct preexisting matrix to initiate repair in damaged tissues. Nat. Immunol. 2022, 23, 518–531. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
In response to myocardial infarction (MI), neutrophils migrate into the damaged myocardium. In the post-MI heart, neutrophils amplify cardiac inflammation by releasing cytotoxic granules and stimulating the differentiation of proinflammatory macrophages. During the wound healing phase, the heart-specific microenvironment induces neutrophil aging, which results in the resolution of cardiac inflammation. Neutrophils also contribute to the differentiation of alternatively activated reparative macrophages in the post-MI heart. Neutrophil aging is regulated by the circadian rhythm, but the local microenvironment also has a marked effect on cardiovascular homeostasis. DAMPs, damage-associated molecular patterns; MI, myocardial infarction; ROS, reactive oxygen species.
Figure 1.
In response to myocardial infarction (MI), neutrophils migrate into the damaged myocardium. In the post-MI heart, neutrophils amplify cardiac inflammation by releasing cytotoxic granules and stimulating the differentiation of proinflammatory macrophages. During the wound healing phase, the heart-specific microenvironment induces neutrophil aging, which results in the resolution of cardiac inflammation. Neutrophils also contribute to the differentiation of alternatively activated reparative macrophages in the post-MI heart. Neutrophil aging is regulated by the circadian rhythm, but the local microenvironment also has a marked effect on cardiovascular homeostasis. DAMPs, damage-associated molecular patterns; MI, myocardial infarction; ROS, reactive oxygen species.

Figure 2.
Neutrophils activate platelets through the production of reactive oxygen species (ROS) and the activated platelets present high mobility group box 1 to neutrophils and induce neutrophil extracellular trap (NET) formation. NETs bind to FXIIa and tissue factor (TF), causing the activation of the coagulation cascade. Neutrophil-derived granules degrade the TF pathway inhibitors. Extracellular histone proteins lead to thrombin production and platelet activation via toll-like receptor 2 (TLR2) and TLR4, enhancing immunothrombosis. DNA that is released from NETs also serves as a scaffold for binding neutrophil granule proteins, von Willebrand factor, and fibronectin, a process that promotes platelet binding. HMGB1, high mobility group box 1; ICAM-1, intercellular adhesion molecule 1; ROS, reactive oxygen species; vWF, von Willebrand factor.
Figure 2.
Neutrophils activate platelets through the production of reactive oxygen species (ROS) and the activated platelets present high mobility group box 1 to neutrophils and induce neutrophil extracellular trap (NET) formation. NETs bind to FXIIa and tissue factor (TF), causing the activation of the coagulation cascade. Neutrophil-derived granules degrade the TF pathway inhibitors. Extracellular histone proteins lead to thrombin production and platelet activation via toll-like receptor 2 (TLR2) and TLR4, enhancing immunothrombosis. DNA that is released from NETs also serves as a scaffold for binding neutrophil granule proteins, von Willebrand factor, and fibronectin, a process that promotes platelet binding. HMGB1, high mobility group box 1; ICAM-1, intercellular adhesion molecule 1; ROS, reactive oxygen species; vWF, von Willebrand factor.

Figure 3.
Hydrogen gas causes reduced neutrophil aggregation. Hydrogen gas-mediated neutralization of hypochlorous acid that is produced by the oxidative burst suppresses DNA damage, which is followed by a decrease in CXC chemokine receptor 4 expression on neutrophils. Maintaining the proper disarming process with hydrogen gas leads to reduced citrullination of histones and decreased release of neutrophil extracellular trap (NET) components, making hydrogen gas therapy a potential therapeutic strategy for cardiovascular disease that involves neutrophil inflammation and NET formation. CXCR4, CXC chemokine receptor 4; H2O2, hydrogen peroxide; HOCl, hypochlorous acid; LPS, lipopolysaccharide; MPO, myeloperoxidase; NADPH, nicotinamide adenine dinucleotide phosphate; PMA, phorbol-12-myristate-13acetate.
Figure 3.
Hydrogen gas causes reduced neutrophil aggregation. Hydrogen gas-mediated neutralization of hypochlorous acid that is produced by the oxidative burst suppresses DNA damage, which is followed by a decrease in CXC chemokine receptor 4 expression on neutrophils. Maintaining the proper disarming process with hydrogen gas leads to reduced citrullination of histones and decreased release of neutrophil extracellular trap (NET) components, making hydrogen gas therapy a potential therapeutic strategy for cardiovascular disease that involves neutrophil inflammation and NET formation. CXCR4, CXC chemokine receptor 4; H2O2, hydrogen peroxide; HOCl, hypochlorous acid; LPS, lipopolysaccharide; MPO, myeloperoxidase; NADPH, nicotinamide adenine dinucleotide phosphate; PMA, phorbol-12-myristate-13acetate.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shirakawa, K.; Sano, M. Neutrophils and Neutrophil Extracellular Traps in Cardiovascular Disease: An Overview and Potential Therapeutic Approaches. Biomedicines 2022, 10, 1850. https://doi.org/10.3390/biomedicines10081850
AMA Style
Shirakawa K, Sano M. Neutrophils and Neutrophil Extracellular Traps in Cardiovascular Disease: An Overview and Potential Therapeutic Approaches. Biomedicines. 2022; 10(8):1850. https://doi.org/10.3390/biomedicines10081850
Chicago/Turabian StyleShirakawa, Kohsuke, and Motoaki Sano. 2022. "Neutrophils and Neutrophil Extracellular Traps in Cardiovascular Disease: An Overview and Potential Therapeutic Approaches" Biomedicines 10, no. 8: 1850. https://doi.org/10.3390/biomedicines10081850
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.