
Effect of Prenatal Opioid Exposure on the Human Placental Methylome

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Setting
2.2. Subjects
2.3. Phenotype Data Collection
2.4. Experimental Methods
2.4.1. Placental Collection
2.4.2. DNA Extraction
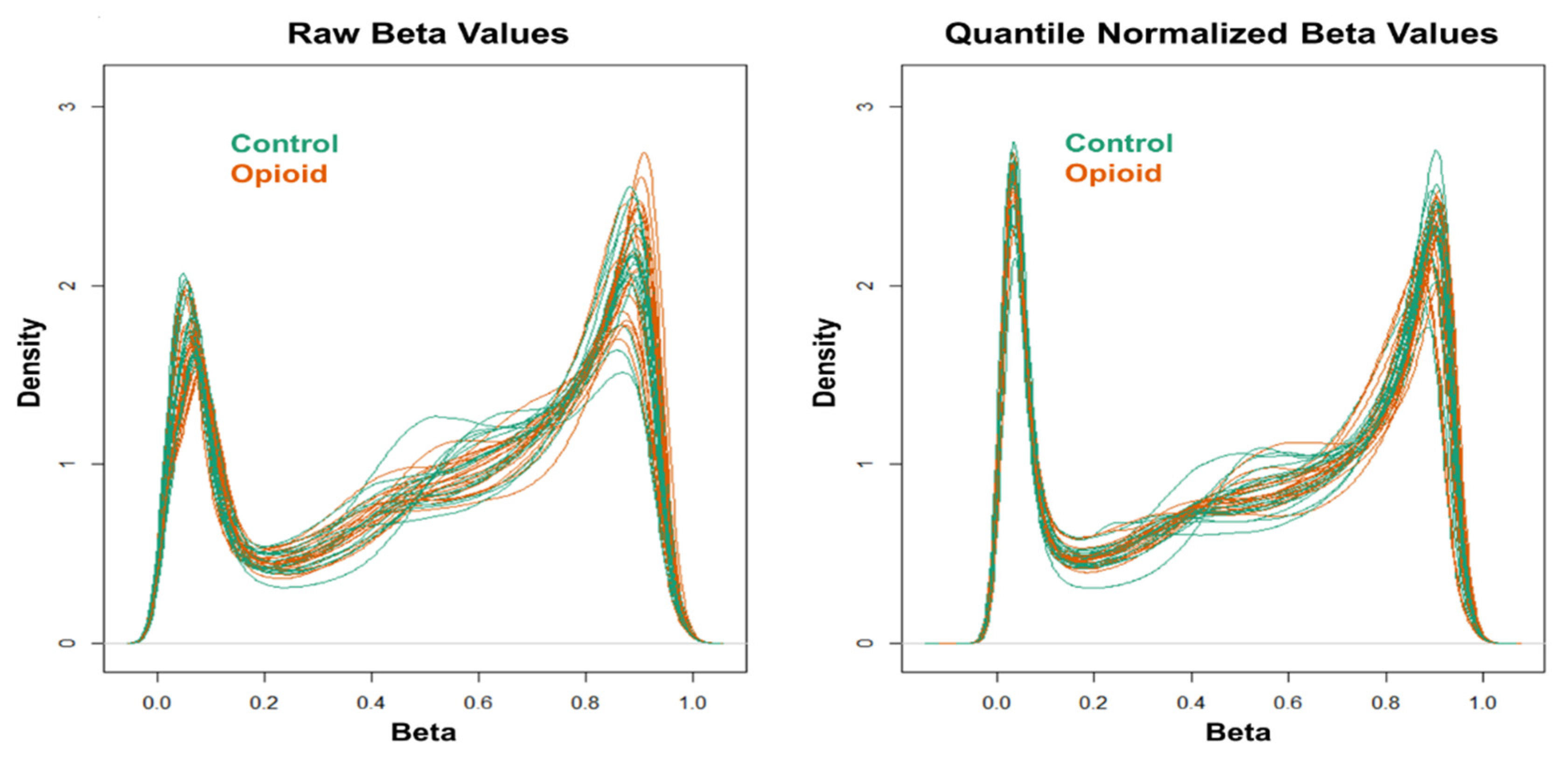
2.4.3. Illumina EPIC DNA Methylation Array Assay and Raw Data Processing
2.4.4. Statistical Analysis
2.4.5. Bioinformatics Analysis
3. Results
3.1. Subjects and Demographic Data
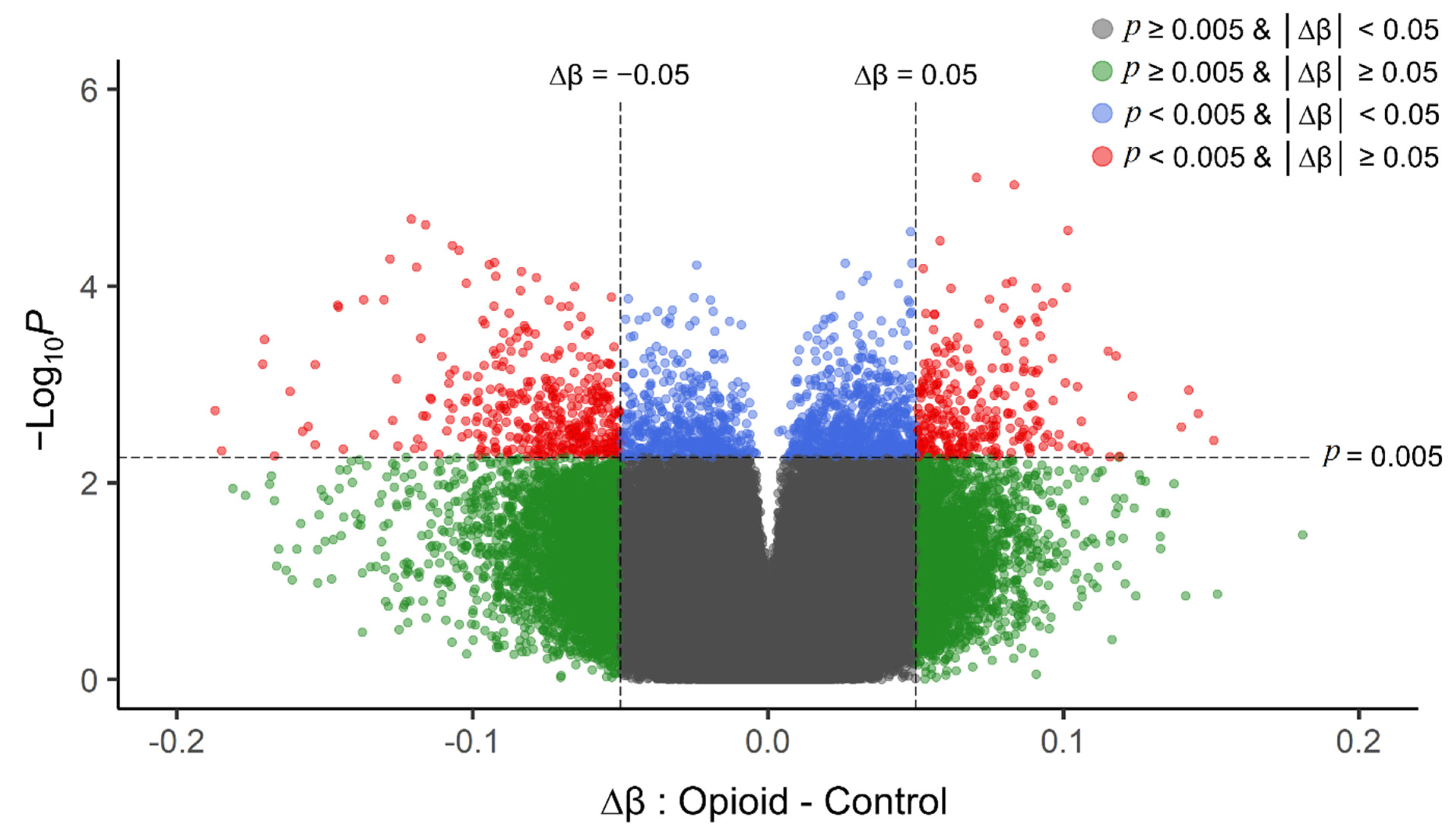
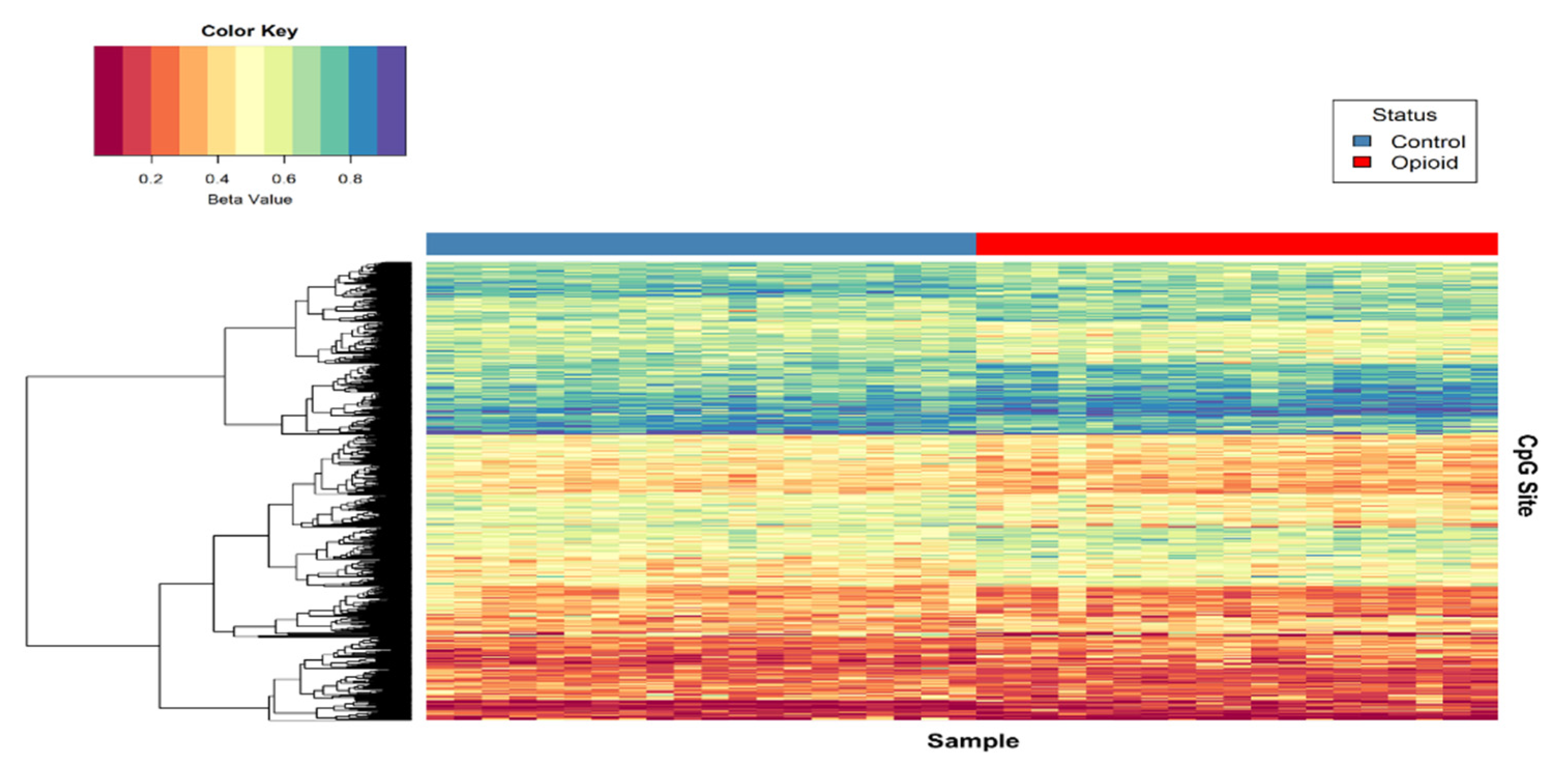
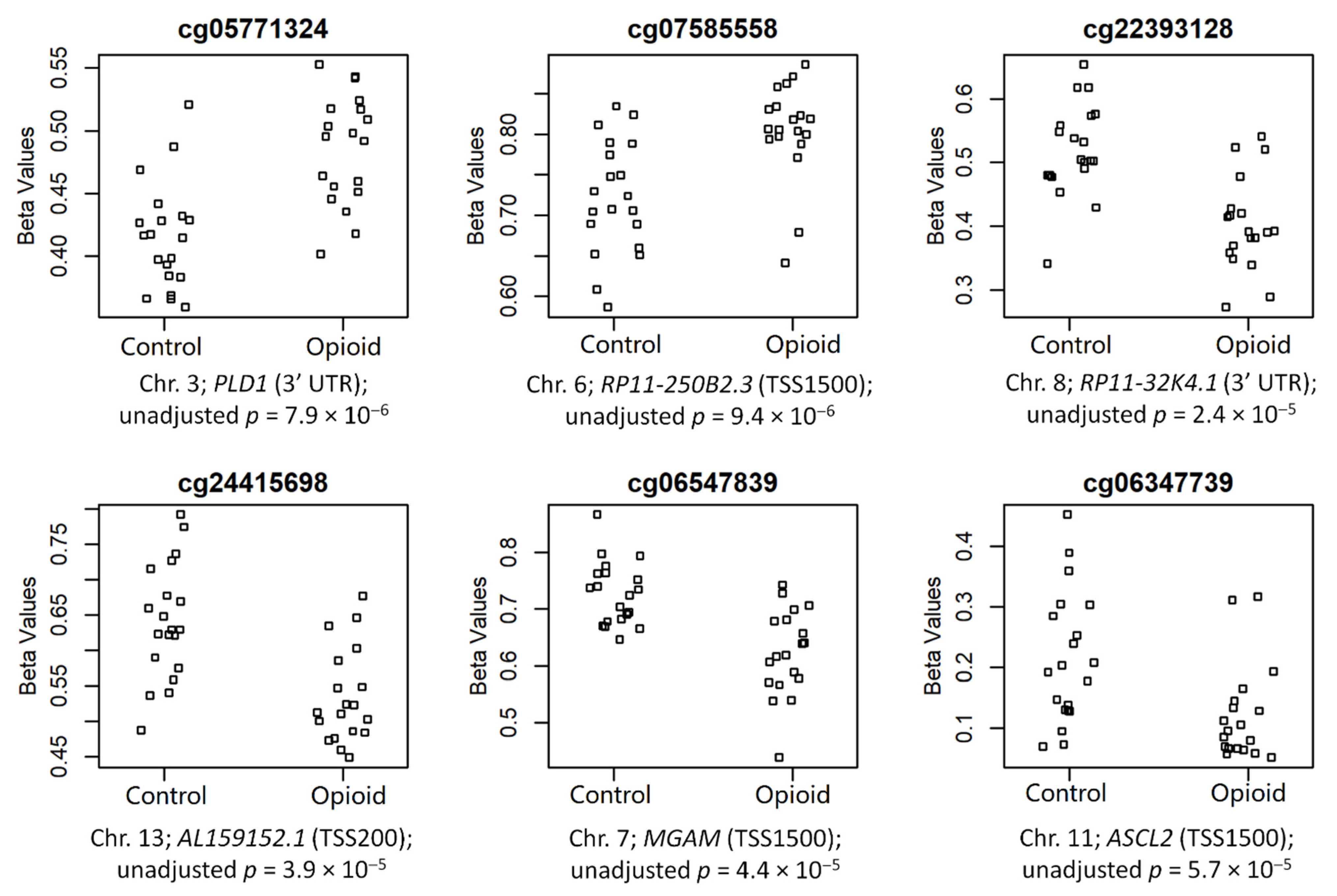
3.2. Differential Methylation in Opioid-Exposed Placentas
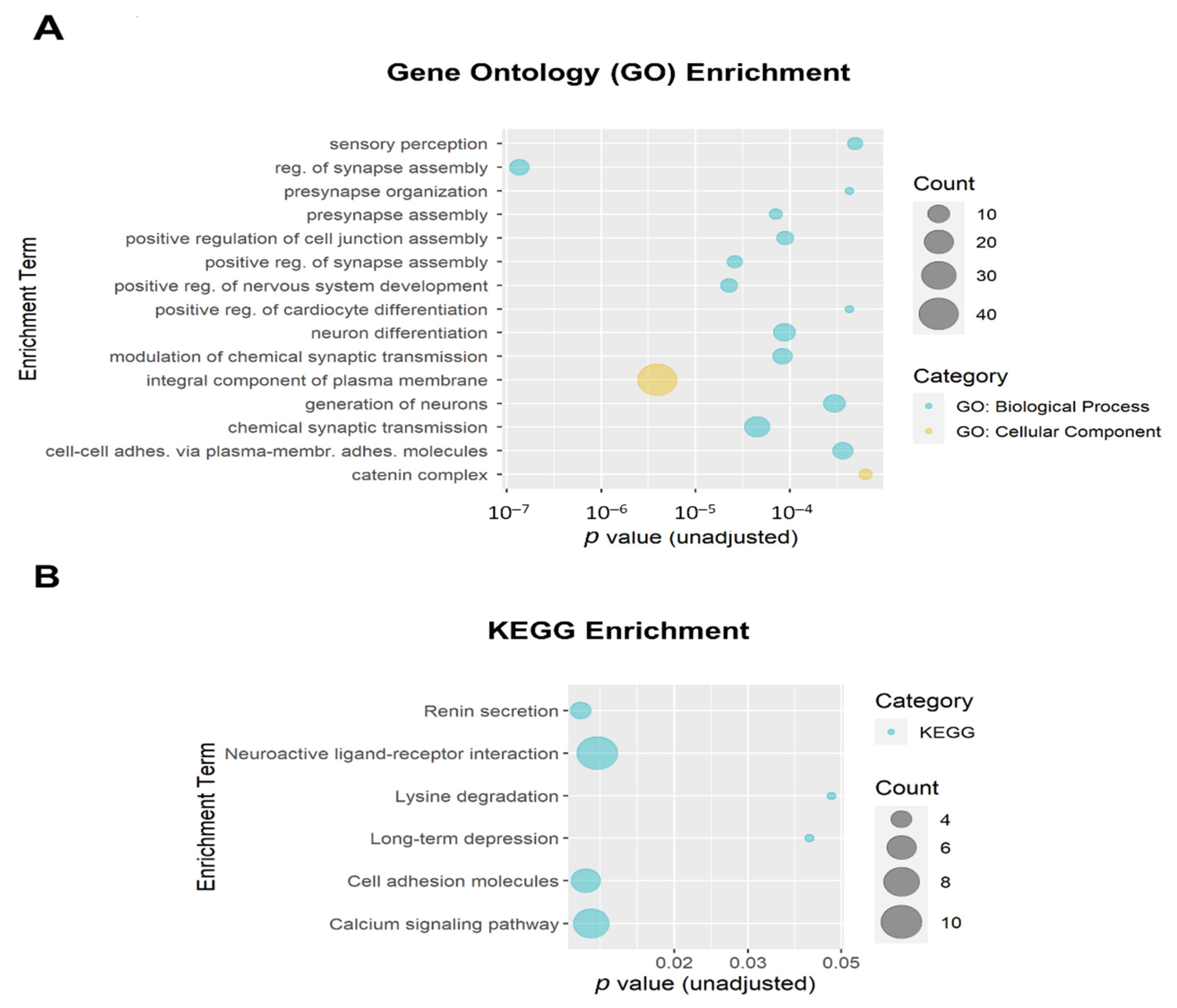
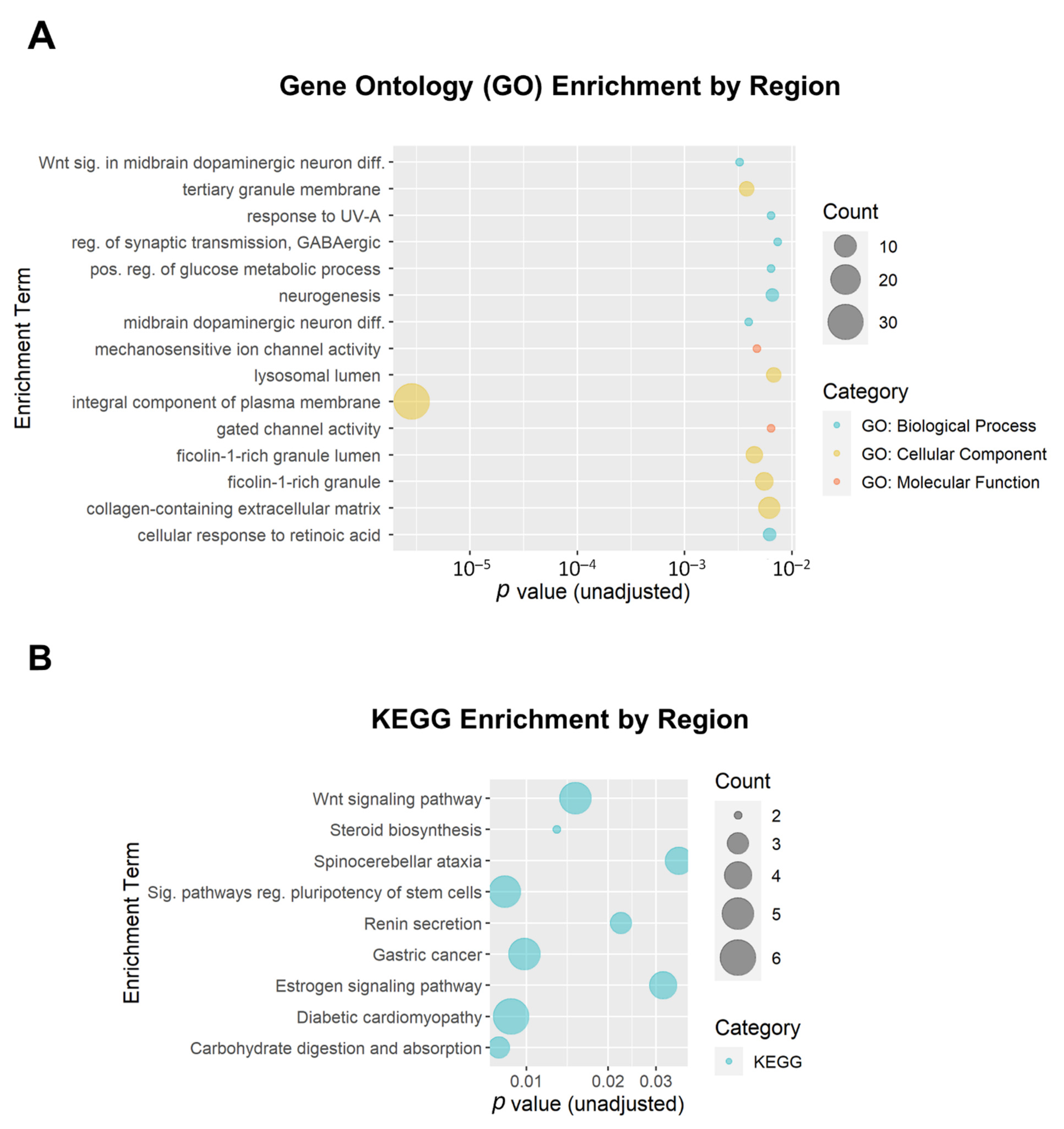
3.3. Functional Enrichment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tolia, V.N.; Patrick, S.W.; Bennett, M.M.; Murthy, K.; Sousa, J.; Smith, P.B.; Clark, R.H.; Spitzer, A.R. Increasing incidence of the neonatal abstinence syndrome in u.S. Neonatal icus. N. Engl. J. Med. 2015, 372, 2118–2126. [Google Scholar] [CrossRef] [PubMed]
- Winkelman, T.N.A.; Villapiano, N.; Kozhimannil, K.B.; Davis, M.M.; Patrick, S.W. Incidence and costs of neonatal abstinence syndrome among infants with medicaid: 2004–2014. Pediatrics 2018, 141, e20173520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrick, S.W.; Davis, M.M.; Lehmann, C.U.; Cooper, W.O. Increasing incidence and geographic distribution of neonatal abstinence syndrome: United states 2009 to 2012. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2015, 35, 650–655. [Google Scholar]
- Milliren, C.E.; Gupta, M.; Graham, D.A.; Melvin, P.; Jorina, M.; Ozonoff, A. Hospital variation in neonatal abstinence syndrome incidence, treatment modalities, resource use, and costs across pediatric hospitals in the United States, 2013 to 2016. Hosp. Pediatr. 2018, 8, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Corsi, D.J.; Hsu, H.; Fell, D.B.; Wen, S.W.; Walker, M. Association of maternal opioid use in pregnancy with adverse perinatal outcomes in Ontario, Canada, from 2012 to 2018. JAMA Netw. Open 2020, 3, e208256. [Google Scholar] [CrossRef]
- Patrick, S.W.; Barfield, W.D.; Poindexter, B.B.; Committee on Fetus and Newborn, Committee on Substance Use and Prevention. Neonatal opioid withdrawal syndrome. Pediatrics 2020, 146, e2020029074. [Google Scholar] [CrossRef]
- Wouldes, T.A.; Woodward, L.J. Neurobehavior of newborn infants exposed prenatally to methadone and identification of a neurobehavioral profile linked to poorer neurodevelopmental outcomes at age 24 months. PLoS ONE 2020, 15, e0240905. [Google Scholar]
- Benninger, K.L.; Borghese, T.; Kovalcik, J.B.; Moore-Clingenpeel, M.; Isler, C.; Bonachea, E.M.; Stark, A.R.; Patrick, S.W.; Maitre, N.L. Prenatal exposures are associated with worse neurodevelopmental outcomes in infants with neonatal opioid withdrawal syndrome. Front. Pediatr 2020, 8, 462. [Google Scholar] [CrossRef]
- Czynski, A.J.; Davis, J.M.; Dansereau, L.M.; Engelhardt, B.; Marro, P.; Bogen, D.L.; Hudak, M.L.; Shenberger, J.; Wachman, E.M.; Oliveira, E.L.; et al. Neurodevelopmental outcomes of neonates randomized to morphine or methadone for treatment of neonatal abstinence syndrome. J. Pediatr. 2020, 219, 146–151. [Google Scholar] [CrossRef]
- Serra, A.E.; Lemon, L.S.; Mokhtari, N.B.; Parks, W.T.; Catov, J.M.; Venkataramanan, R.; Caritis, S.N. Delayed villous maturation in term placentas exposed to opioid maintenance therapy: A retrospective cohort study. Am. J. Obstet. Gynecol. 2017, 216, 418.e1–418.e5. [Google Scholar] [CrossRef]
- Miller, C.B.; Wright, T. Investigating mechanisms of stillbirth in the setting of prenatal substance use. Acad. Forensic Pathol. 2018, 8, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Ortigosa, S.; Friguls, B.; Joya, X.; Martinez, S.; Marinoso, M.L.; Alameda, F.; Vall, O.; Garcia-Algar, O. Feto-placental morphological effects of prenatal exposure to drugs of abuse. Reprod. Toxicol. 2012, 34, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, A.; Lutz, S.M.; Everson, T.M.; Perron, P.; Bouchard, L.; Hivert, M.F. Mediation by placental DNA methylation of the association of prenatal maternal smoking and birth weight. Am. J. Epidemiol. 2019, 188, 1878–1886. [Google Scholar] [CrossRef]
- Novakovic, B.; Saffery, R. The ever growing complexity of placental epigenetics—Role in adverse pregnancy outcomes and fetal programming. Placenta 2012, 33, 959–970. [Google Scholar] [CrossRef]
- Vlahos, A.; Mansell, T.; Saffery, R.; Novakovic, B. Human placental methylome in the interplay of adverse placental health, environmental exposure, and pregnancy outcome. PLoS Genet. 2019, 15, e1008236. [Google Scholar] [CrossRef] [Green Version]
- Conradt, E.; Fei, M.; LaGasse, L.; Tronick, E.; Guerin, D.; Gorman, D.; Marsit, C.J.; Lester, B.M. Prenatal predictors of infant self-regulation: The contributions of placental DNA methylation of nr3c1 and neuroendocrine activity. Front. Behav. Neurosci. 2015, 9, 130. [Google Scholar] [CrossRef] [Green Version]
- Paquette, A.G.; Lester, B.M.; Lesseur, C.; Armstrong, D.A.; Guerin, D.J.; Appleton, A.A.; Marsit, C.J. Placental epigenetic patterning of glucocorticoid response genes is associated with infant neurodevelopment. Epigenomics 2015, 7, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Palma-Gudiel, H.; Cordova-Palomera, A.; Leza, J.C.; Fananas, L. Glucocorticoid receptor gene (nr3c1) methylation processes as mediators of early adversity in stress-related disorders causality: A critical review. Neurosci. Biobehav. Rev. 2015, 55, 520–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Otterdijk, S.D.; Binder, A.M.; Michels, K.B. Locus-specific DNA methylation in the placenta is associated with levels of pro-inflammatory proteins in cord blood and they are both independently affected by maternal smoking during pregnancy. Epigenetics 2017, 12, 875–885. [Google Scholar] [CrossRef] [Green Version]
- Barua, S.; Junaid, M.A. Lifestyle, pregnancy and epigenetic effects. Epigenomics 2015, 7, 85–102. [Google Scholar] [CrossRef]
- Nielsen, D.A.; Utrankar, A.; Reyes, J.A.; Simons, D.D.; Kosten, T.R. Epigenetics of drug abuse: Predisposition or response. Pharmacogenomics 2012, 13, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Wachman, E.M.; Farrer, L.A. The genetics and epigenetics of neonatal abstinence syndrome. Semin. Fetal Neonatal Med. 2019, 24, 105–110. [Google Scholar] [CrossRef]
- Montalvo-Ortiz, J.L.; Cheng, Z.; Kranzler, H.R.; Zhang, H.; Gelernter, J. Genomewide study of epigenetic biomarkers of opioid dependence in european- american women. Sci. Rep. 2019, 9, 4660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, D.A.; Yuferov, V.; Hamon, S.; Jackson, C.; Ho, A.; Ott, J.; Kreek, M.J. Increased oprm1 DNA methylation in lymphocytes of methadone-maintained former heroin addicts. Neuropsychopharmacology 2009, 34, 867–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wachman, E.M.; Hayes, M.J.; Shrestha, H.; Nikita, F.N.U.; Nolin, A.; Hoyo, L.; Daigle, K.; Jones, H.E.; Nielsen, D.A. Epigenetic variation in oprm1 gene in opioid-exposed mother-infant dyads. Genes Brain Behav. 2018, 17, e12476. [Google Scholar] [CrossRef] [PubMed]
- Wachman, E.M.; Hayes, M.J.; Lester, B.M.; Terrin, N.; Brown, M.S.; Nielsen, D.A.; Davis, J.M. Epigenetic variation in the mu-opioid receptor gene in infants with neonatal abstinence syndrome. J. Pediatr. 2014, 165, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Wachman, E.M.; Wang, A.; Isley, B.C.; Boateng, J.; Beierle, J.A.; Hansbury, A.; Shrestha, H.; Bryant, C.; Zhang, H. Placental oprm1 DNA methylation and associations with neonatal opioid withdrawal syndrome, a pilot study. Explor. Med. 2020, 1, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the illumina methylationepic beadchip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Blighe, K.; Rana, S.; Lewis, M. Enhancedvolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2021. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 23 July 2019).
- Gu, Z.; Eils, R.; Schlesner, M.; Ishaque, N. Enrichedheatmap: An r/bioconductor package for comprehensive visualization of genomic signal associations. BMC Genom. 2018, 19, 234. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Reginald, V.L.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative html5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Brown, H.A.; Thomas, P.G.; Lindsley, C.W. Targeting phospholipase d in cancer, infection and neurodegenerative disorders. Nat. Rev. Drug Discov. 2017, 16, 351–367. [Google Scholar] [CrossRef]
- Lebenthal, E.; Khin Maung, U.; Zheng, B.Y.; Lu, R.B.; Lerner, A. Small intestinal glucoamylase deficiency and starch malabsorption: A newly recognized alpha-glucosidase deficiency in children. J. Pediatr. 1994, 124, 541–546. [Google Scholar] [CrossRef]
- Jubb, A.M.; Chalasani, S.; Frantz, G.D.; Smits, R.; Grabsch, H.I.; Kavi, V.; Maughan, N.J.; Hillan, K.J.; Quirke, P.; Koeppen, H. Achaete-scute like 2 (ascl2) is a target of wnt signalling and is upregulated in intestinal neoplasia. Oncogene 2006, 25, 3445–3457. [Google Scholar] [CrossRef] [Green Version]
- Kee, K.; Angeles, V.T.; Flores, M.; Nguyen, H.N.; Reijo Pera, R.A. Human dazl, daz and boule genes modulate primordial germ-cell and haploid gamete formation. Nature 2009, 462, 222–225. [Google Scholar] [CrossRef]
- Han, S.; Yang, B.Z.; Kranzler, H.R.; Liu, X.; Zhao, H.; Farrer, L.A.; Boerwinkle, E.; Potash, J.B.; Gelernter, J. Integrating gwass and human protein interaction networks identifies a gene subnetwork underlying alcohol dependence. Am. J. Hum. Genet. 2013, 93, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, M.T.; Jayanthi, S.; Cadet, J.L. Potassium channels and their potential roles in substance use disorders. Int. J. Mol. Sci. 2021, 22, 1249. [Google Scholar] [CrossRef]
- Shen, Y.; Xu, L.; Foster, D.A. Role for phospholipase d in receptor-mediated endocytosis. Mol. Cell. Biol. 2001, 21, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Park, J.B.; Kim, J.H.; Kim, Y.; Kim, J.H.; Shin, K.J.; Lee, J.S.; Ha, S.H.; Suh, P.G.; Ryu, S.H. Actin directly interacts with phospholipase d, inhibiting its activity. J. Biol. Chem. 2001, 276, 28252–28260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, T.; Brandenburg, L.O.; Schulz, S.; Liang, Y.; Klein, J.; Hollt, V. Adp-ribosylation factor-dependent phospholipase d2 activation is required for agonist-induced mu-opioid receptor endocytosis. J. Biol. Chem. 2003, 278, 9979–9985. [Google Scholar] [CrossRef] [Green Version]
- Quezada-Calvillo, R.; Robayo-Torres, C.C.; Ao, Z.; Hamaker, B.R.; Quaroni, A.; Brayer, G.D.; Sterchi, E.E.; Baker, S.S.; Nichols, B.L. Luminal substrate “brake” on mucosal maltase-glucoamylase activity regulates total rate of starch digestion to glucose. J. Pediatr. Gastroenterol. Nutr. 2007, 45, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Hanhineva, K.; Torronen, R.; Bondia-Pons, I.; Pekkinen, J.; Kolehmainen, M.; Mykkanen, H.; Poutanen, K. Impact of dietary polyphenols on carbohydrate metabolism. Int. J. Mol. Sci. 2010, 11, 1365–1402. [Google Scholar] [CrossRef] [PubMed]
- Eaves, E.R.; Barber, J.; Whealy, R.; Clancey, S.A.; Wright, R.; Cocking, J.H.; Spadafino, J.; Hepp, C.M. Characterization of neonatal opioid withdrawal syndrome in arizona from 2010–2017. PLoS ONE 2021, 16, e0248476. [Google Scholar] [CrossRef]
- Hayashi, Y.; Morinaga, S.; Zhang, J.; Satoh, Y.; Meredith, A.L.; Nakata, T.; Wu, Z.; Kohsaka, S.; Inoue, K.; Nakanishi, H. Bk channels in microglia are required for morphine-induced hyperalgesia. Nat. Commun. 2016, 7, 11697. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.R.; Cai, Y.Q.; Pan, H.L. Plasticity and emerging role of bkca channels in nociceptive control in neuropathic pain. J. Neurochem. 2009, 110, 352–362. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.M.; Chen, M.; Wong, T.M. The k (ca) channel as a trigger for the cardioprotection induced by kappa-opioid receptor stimulation—Its relationship with protein kinase c. Br. J. Pharmacol. 2005, 145, 984–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez-Marrero, C.; Burgos, A.; Garcia, J.O.; Palacio, S.; Marrero, H.G.; Bernardo, A.; Perez-Laspiur, J.; Rivera-Oliver, M.; Seale, G.; Treistman, S.N. Alcohol regulates bk surface expression via wnt/beta-catenin signaling. J. Neurosci. 2016, 36, 10625–10639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, M.T.; Noronha, A.; Goldman, D.; Koob, G.F. Genetic studies of alcohol dependence in the context of the addiction cycle. Neuropharmacology 2017, 122, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Jayanthi, S.; Torres, O.V.; Ladenheim, B.; Cadet, J.L. A single prior injection of methamphetamine enhances methamphetamine self-administration (sa) and blocks sa-induced changes in DNA methylation and mrna expression of potassium channels in the rat nucleus accumbens. Mol. Neurobiol. 2020, 57, 1459–1472. [Google Scholar] [CrossRef] [Green Version]
- Pavlicev, M.; Wagner, G.P.; Chavan, A.R.; Owens, K.; Maziarz, J.; Dunn-Fletcher, C.; Kallapur, S.G.; Muglia, L.; Jones, H. Single-cell transcriptomics of the human placenta: Inferring the cell communication network of the maternal-fetal interface. Genome Res. 2017, 27, 349–361. [Google Scholar] [CrossRef]
- Goldfarb, S.S.; Stanwood, G.D.; Flynn, H.A.; Graham, D.L. Developmental opioid exposures: Neurobiological underpinnings, behavioral impacts, and policy implications. Exp. Biol. Med. 2020, 245, 131–137. [Google Scholar] [CrossRef]
- Matus-Leibovitch, N.; Ezra-Macabee, V.; Saya, D.; Attali, B.; Avidor-Reiss, T.; Barg, J.; Vogel, Z. Increased expression of synapsin i mrna in defined areas of the rat central nervous system following chronic morphine treatment. Mol. Brain Res. 1995, 34, 221–230. [Google Scholar] [CrossRef]
- Yang, S.N.; Liu, C.A.; Chung, M.Y.; Huang, H.C.; Yeh, G.C.; Wong, C.S.; Lin, W.W.; Yang, C.H.; Tao, P.L. Alterations of postsynaptic density proteins in the hippocampus of rat offspring from the morphine-addicted mother: Beneficial effect of dextromethorphan. Hippocampus 2006, 16, 521–530. [Google Scholar] [CrossRef]
- Walton, E.; Hass, J.; Liu, J.; Roffman, J.L.; Bernardoni, F.; Roessner, V.; Kirsch, M.; Schackert, G.; Calhoun, V.; Ehrlich, S. Correspondence of DNA methylation between blood and brain tissue and its application to schizophrenia research. Schizophr. Bull. 2016, 42, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Gourlay, G.K.; Cherry, D.A.; Cousins, M.J. A comparative study of the efficacy and pharmacokinetics of oral methadone and morphine in the treatment of severe pain in patients with cancer. Pain 1986, 25, 297–312. [Google Scholar] [CrossRef]
- Welsh, C.; Valadez-Meltzer, A. Buprenorphine: A (relatively) new treatment for opioid dependence. Psychiatry 2005, 2, 29–39. [Google Scholar] [PubMed]
- Fairbairn, N.; Coffin, P.O.; Walley, A.Y. Naloxone for heroin, prescription opioid, and illicitly made fentanyl overdoses: Challenges and innovations responding to a dynamic epidemic. Int. J. Drug Policy 2017, 46, 172–179. [Google Scholar] [CrossRef]
- Xu, Z.; Taylor, J.A. Genome-wide age-related DNA methylation changes in blood and other tissues relate to histone modification, expression and cancer. Carcinogenesis 2014, 35, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.; Ma, J.; Harris, A.; Patterson, L.; Brown, K.A.; Shope, C.; Showalter, L.; Abramovici, A.; Aagaard-Tillery, K.M. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics 2011, 6, 1284–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Zhang, C.; Cheng, L.; Reilly, J.L.; Bishop, J.R.; Sweeney, J.A.; Chen, H.Y.; Gershon, E.S.; Liu, C. Correlation between DNA methylation and gene expression in the brains of patients with bipolar disorder and schizophrenia. Bipolar Disord. 2014, 16, 790–799. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographics | Opioid (n = 19) | Control (n = 20) | p-Value |
|---|---|---|---|
| Maternal age in years, mean (SD) | 28.8 (5.1) | 32.7 (3.4) | 0.010 |
| Missing, n | 3 | 2 | |
| Maternal Ethnicity/Race, n (%) | Non-Hispanic | Non-Hispanic | |
| White | 18 (94.7%) | 19 (95%) | 0.970 |
| Black | 1 (5.3%) | 1 (5%) | |
| Maternal Opioid, n (%) | N/A | N/A | |
| Methadone | 10 (52.6%) | ||
| Buprenorphine | 3 (15.8%) | ||
| Other prescription opioid | 1 (5.3%) | ||
| Unprescribed fentanyl | 5 (26.3%) | ||
| Gestational age at delivery (weeks), median (IQR) | 39.5 (36.6–40.1) | 39.6 (38.5–40.5) | 0.220 |
| Missing, n (%) | 3 (15.8%) | 2 (10%) | |
| Cesarean section delivery, n (%) | 6 (37.5%) | 6 (33.3%) | 1.000 |
| Missing, n (%) | 3 (15.8%) | 2 (10%) | |
| Infant sex, n (%) | |||
| Female | 10 (52.6%) | 8 (40%) | 0.44 |
| Male | 9 (47.4%) | 12 (60%) | |
| Birthweight (g), median (IQR) | 3061 (2825–3462) | 3401 (3065–3795) | 0.05 |
| Chr. # | Width (bp) | # CpGs | Min Smoothed FDR | Mean Diff. (β) | Overlapping Genes |
|---|---|---|---|---|---|
| chr2 | 1477 | 12 | 2.4 × 10−14 | −0.085 | ANKRD53, AC007040.11 |
| chr2 | 1308 | 12 | 5.0 × 10−11 | 0.059 | C2orf70 |
| chr15 | 548 | 7 | 1.6 × 10−10 | 0.065 | TSPAN3 |
| chr11 | 1136 | 14 | 1.5 × 10−9 | 0.098 | MSANTD4 |
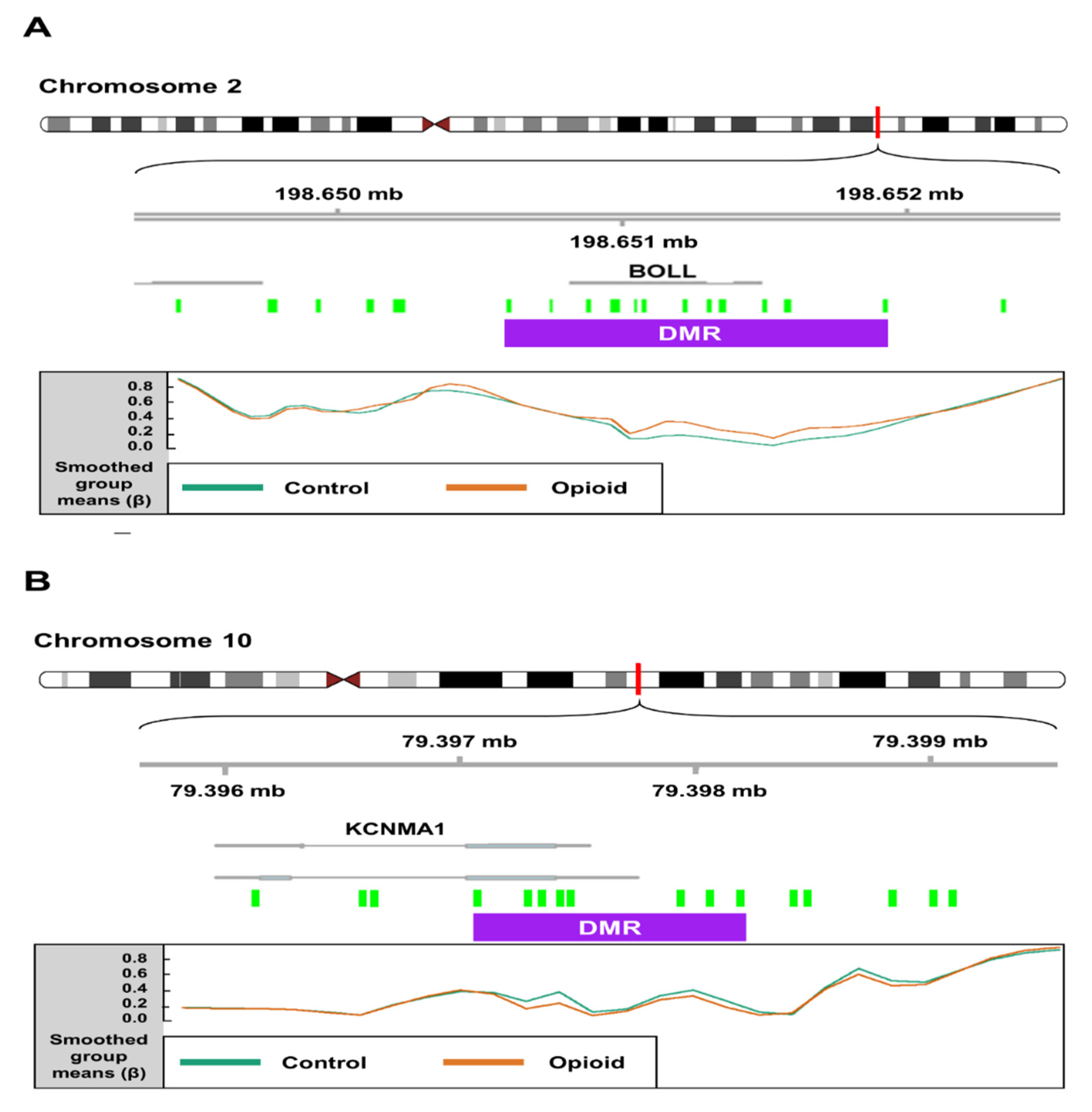
| chr2 | 1322 | 16 | 8.5 × 10−9 | 0.09 | BOLL |
| chr8 | 530 | 9 | 4.2 × 10−6 | −0.061 | FDFT1 |
| chr8 | 1164 | 10 | 1.2 × 10−5 | −0.109 | ZNF572 |
| chr16 | 1144 | 16 | 2.1 × 10−5 | 0.056 | CYBA |
| chr10 | 1120 | 10 | 2.6 × 10−5 | −0.118 | KCNMA1 |
| chr16 | 1396 | 8 | 2.6 × 10−5 | 0.058 | IRX3 |
| chr3 | 822 | 5 | 3.2 × 10−5 | 0.054 | MIR4792 |
| chr3 | 999 | 11 | 3.3 × 10−5 | −0.056 | HLTF-AS1, HLTF |
| chr19 | 978 | 7 | 4.8 × 10−5 | −0.122 | B3GNT3 |
| chr7 | 1063 | 8 | 8.0 × 10−5 | −0.079 | WNT2 |
| chr12 | 469 | 2 | 1.2 × 10−4 | −0.081 | MGAM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borrelli, K.N.; Wachman, E.M.; Beierle, J.A.; Taglauer, E.S.; Jain, M.; Bryant, C.D.; Zhang, H. Effect of Prenatal Opioid Exposure on the Human Placental Methylome. Biomedicines 2022, 10, 1150. https://doi.org/10.3390/biomedicines10051150
Borrelli KN, Wachman EM, Beierle JA, Taglauer ES, Jain M, Bryant CD, Zhang H. Effect of Prenatal Opioid Exposure on the Human Placental Methylome. Biomedicines. 2022; 10(5):1150. https://doi.org/10.3390/biomedicines10051150
Chicago/Turabian StyleBorrelli, Kristyn N., Elisha M. Wachman, Jacob A. Beierle, Elizabeth S. Taglauer, Mayuri Jain, Camron D. Bryant, and Huiping Zhang. 2022. "Effect of Prenatal Opioid Exposure on the Human Placental Methylome" Biomedicines 10, no. 5: 1150. https://doi.org/10.3390/biomedicines10051150