
Defective Proinsulin Handling Modulates the MHC I Bound Peptidome and Activates the Inflammasome in β-Cells

 , , , , , , , , , ,
, , , , , , , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Procurement of Cell Lines and Reagents
2.2. Cell Culture and Pelleting
2.3. Drugs Treatment/Cytokines Exposure
2.4. Flow Cytometry
2.5. Isolation of RT1.A Bound Complexes
2.6. Fractionation by Reverse-Phase High-Performance Liquid Chromatography (RP-HPLC)
2.7. Mass Spectrometry Data Acquisition and Analysis
2.8. Bioinformatics Analyses
2.9. Tryptic Digestion of Heavy Chain
2.10. Western Blotting
2.11. Statistical Analysis
2.12. Data Availability
3. Results
3.1. INS-1E Cells Predominantly Express Class Ia Allele
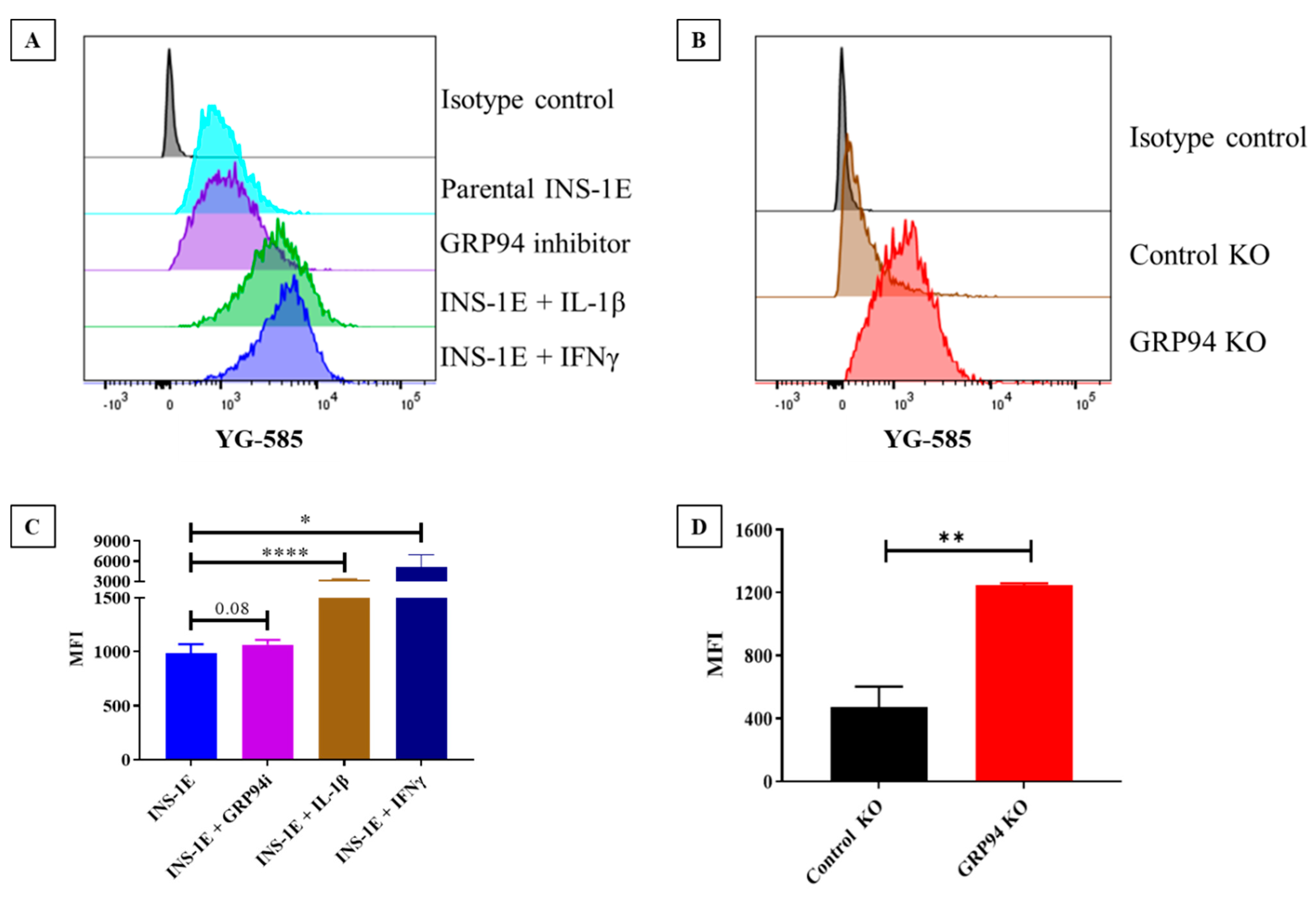
3.2. GRP94 KO/Cytokines Exposure Increase RT1.A Expression
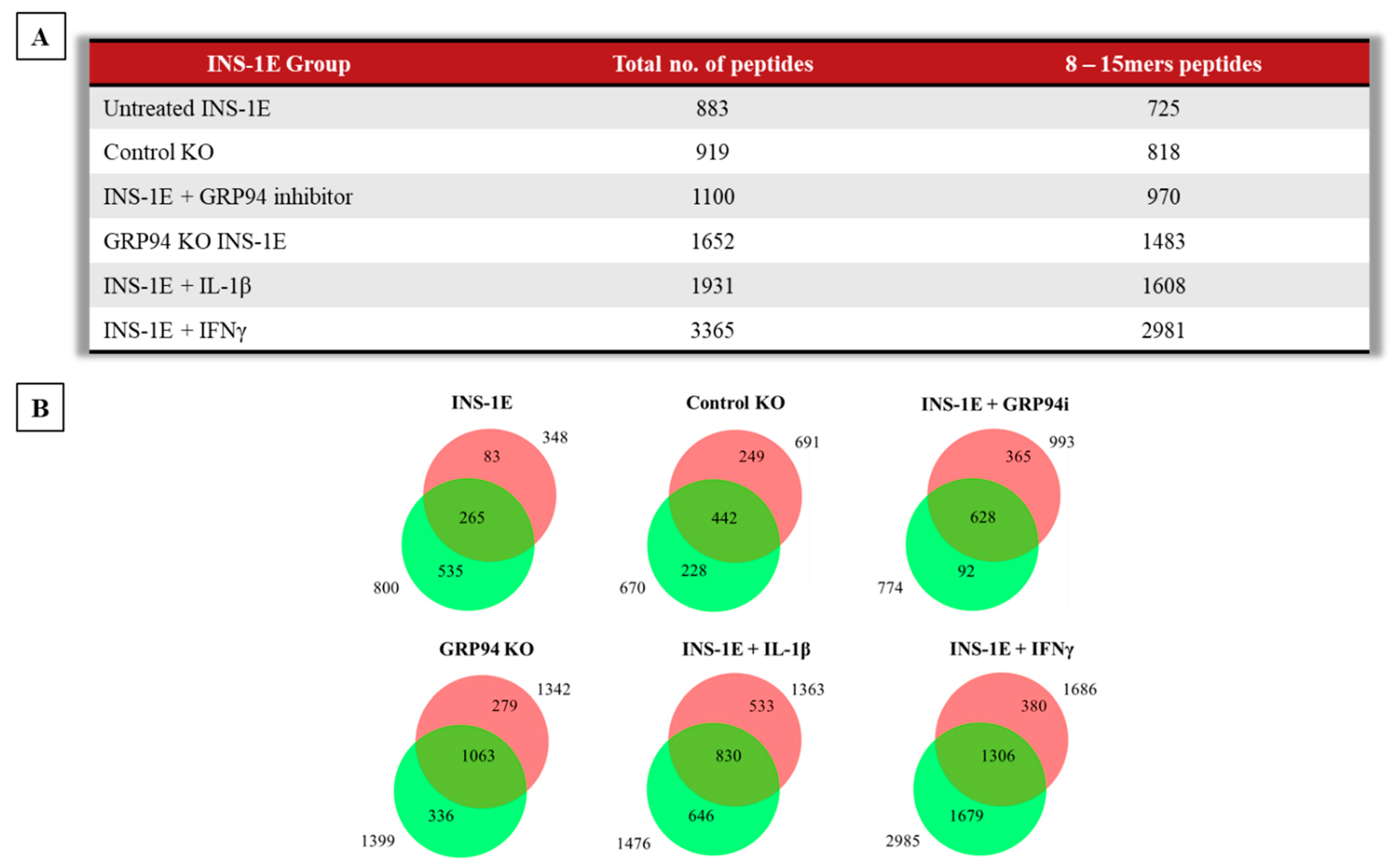
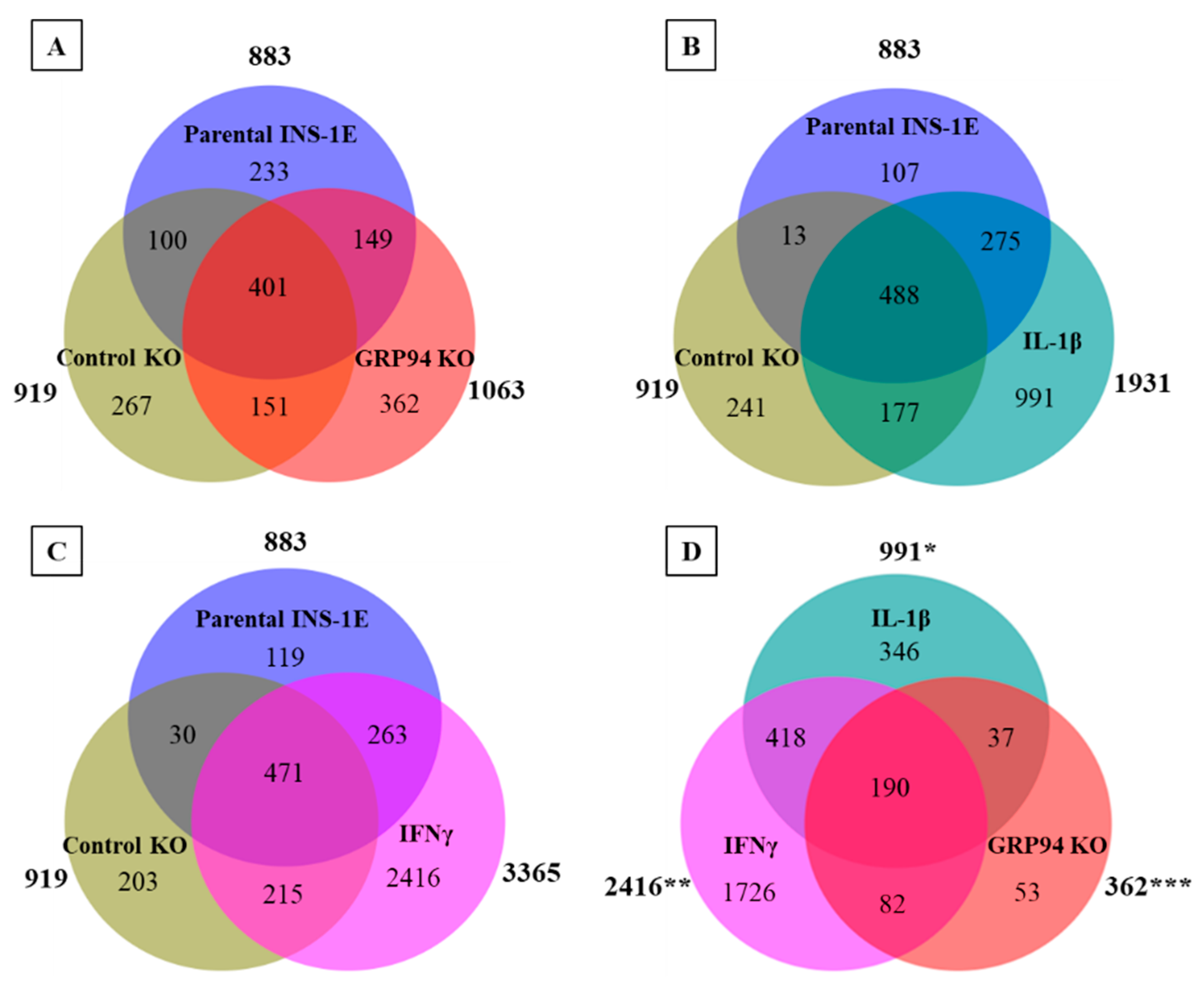
3.3. GRP94 KO Modulates RT1.A-Bound Peptides Yield
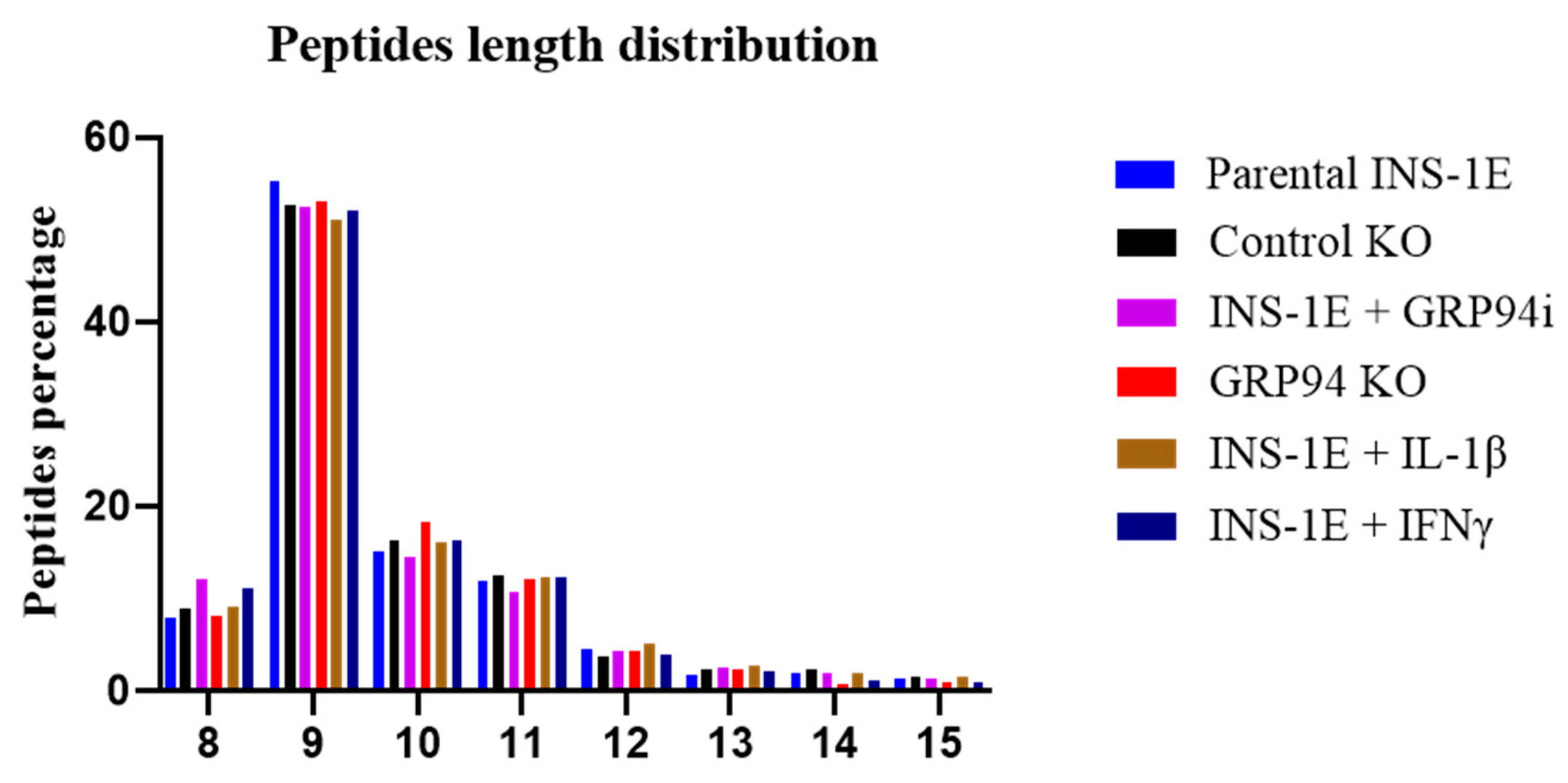
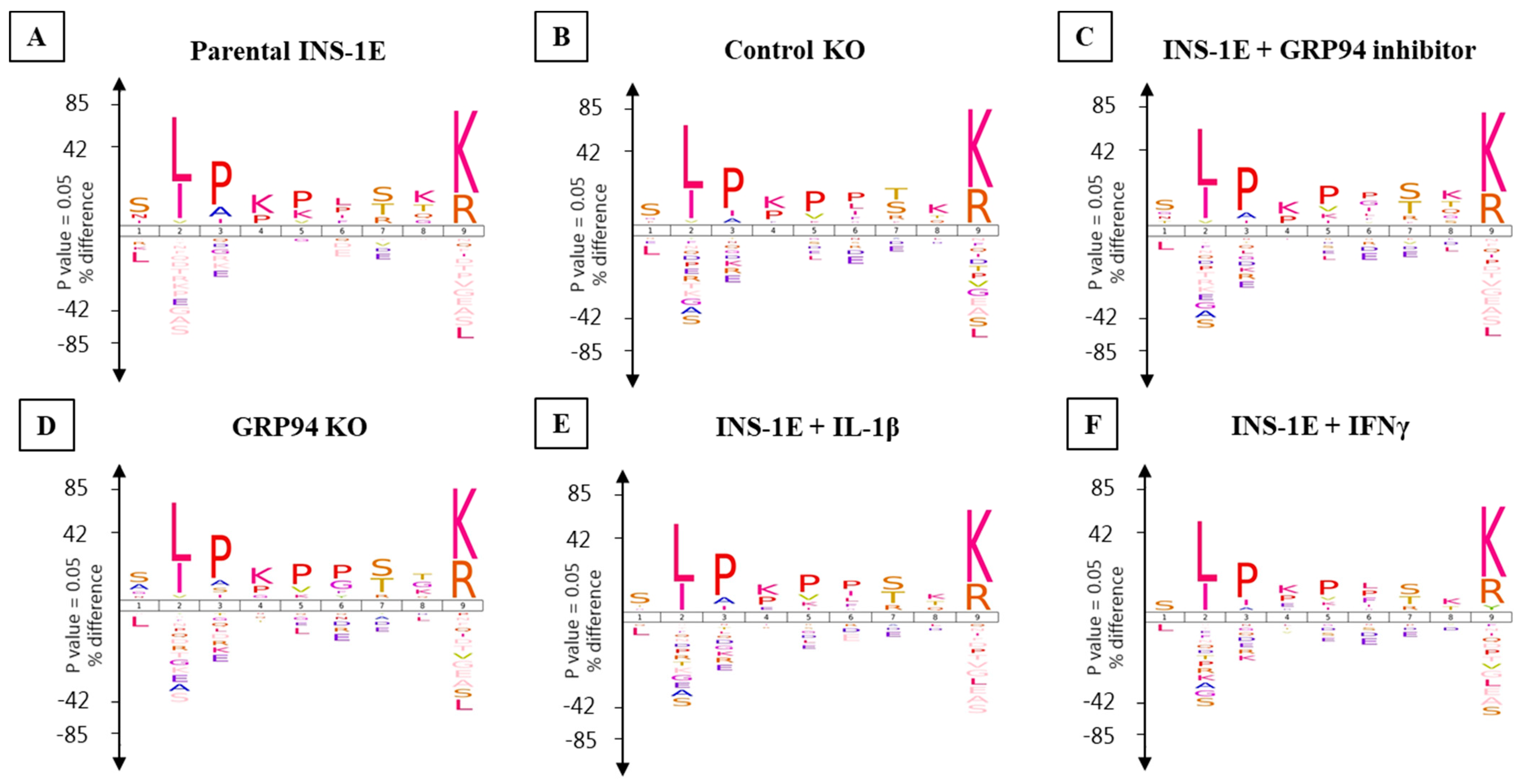
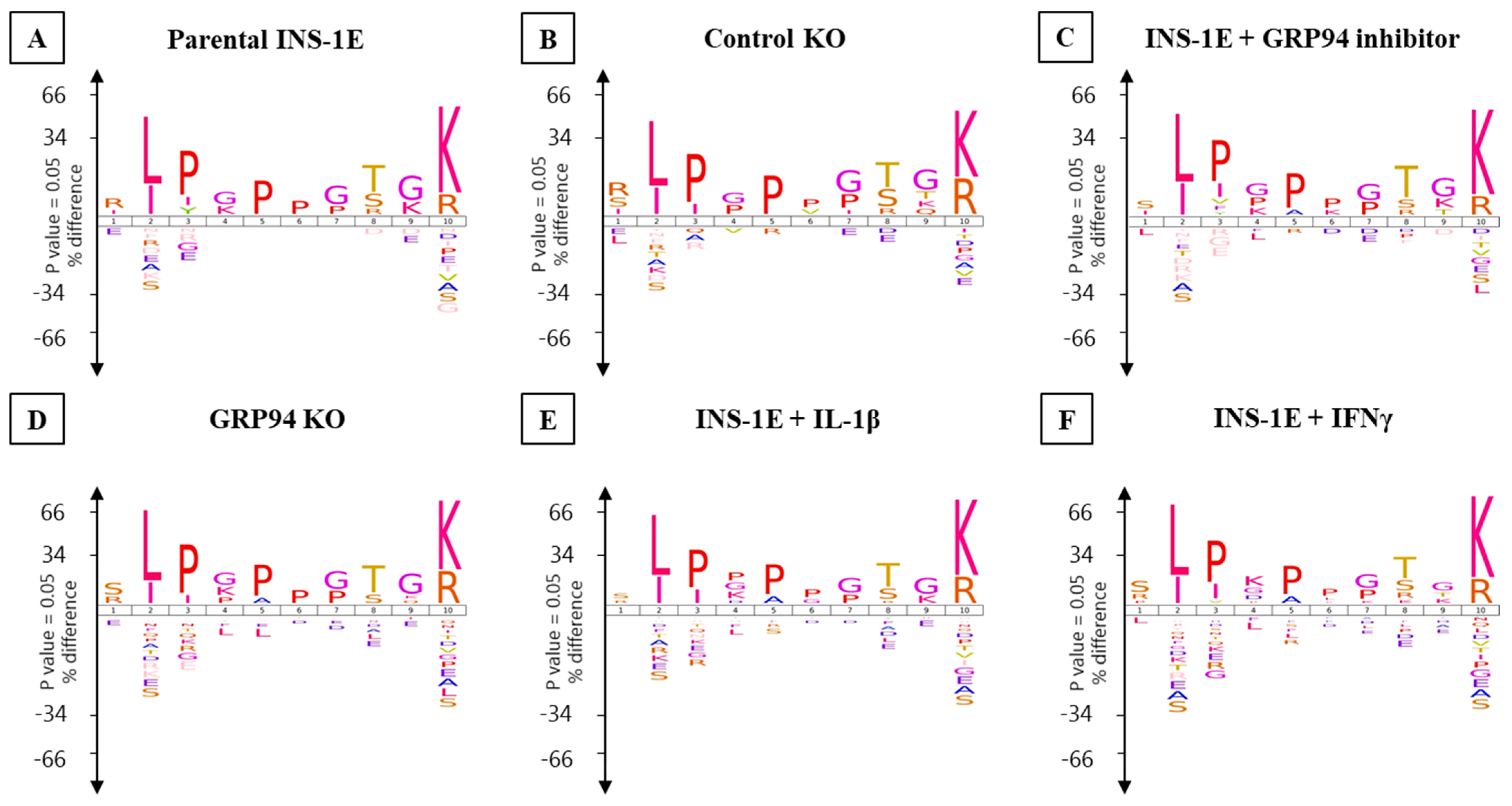
3.4. RT1.Ag Has a Preference for Nonamer Ligands
3.5. GRP94 KO or Cytokine Exposure Modulates Proinsulin Derived RT1.A-Bound Peptides
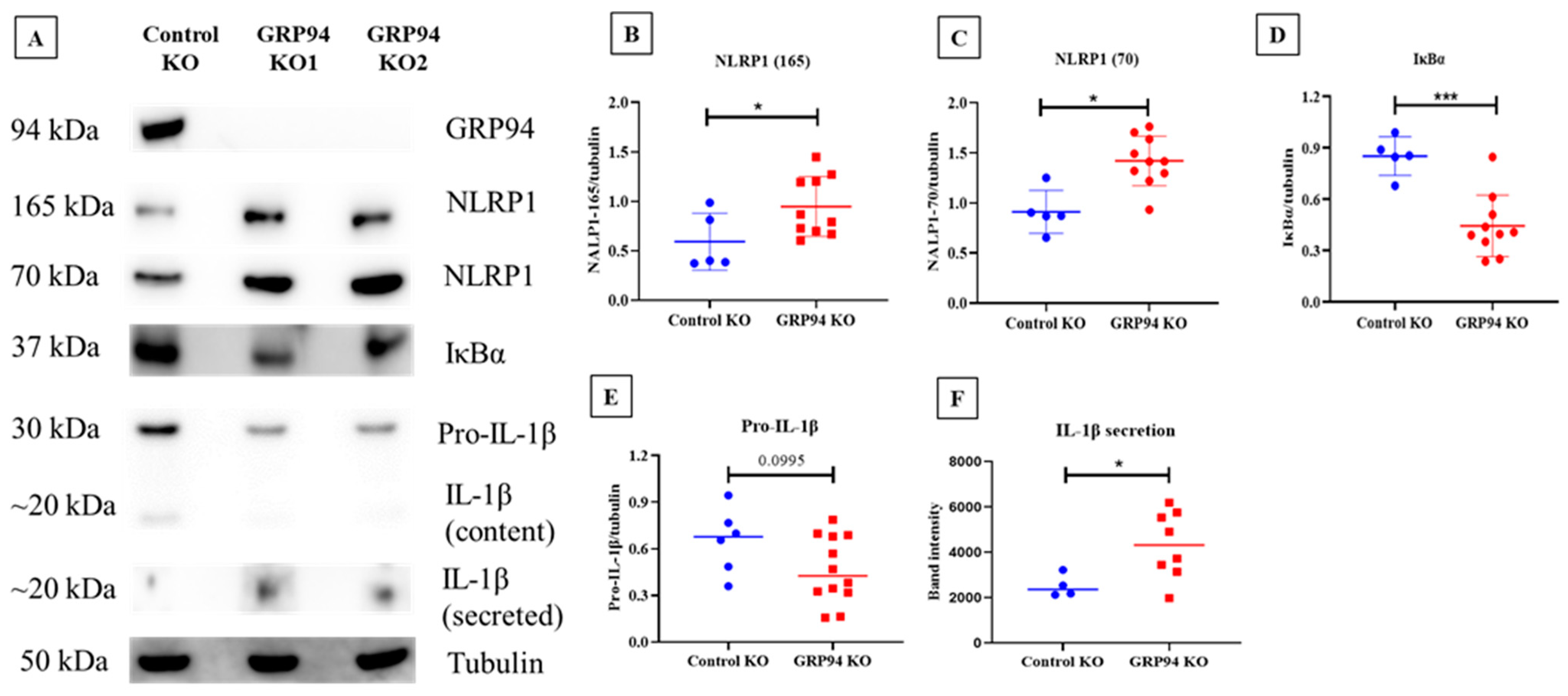
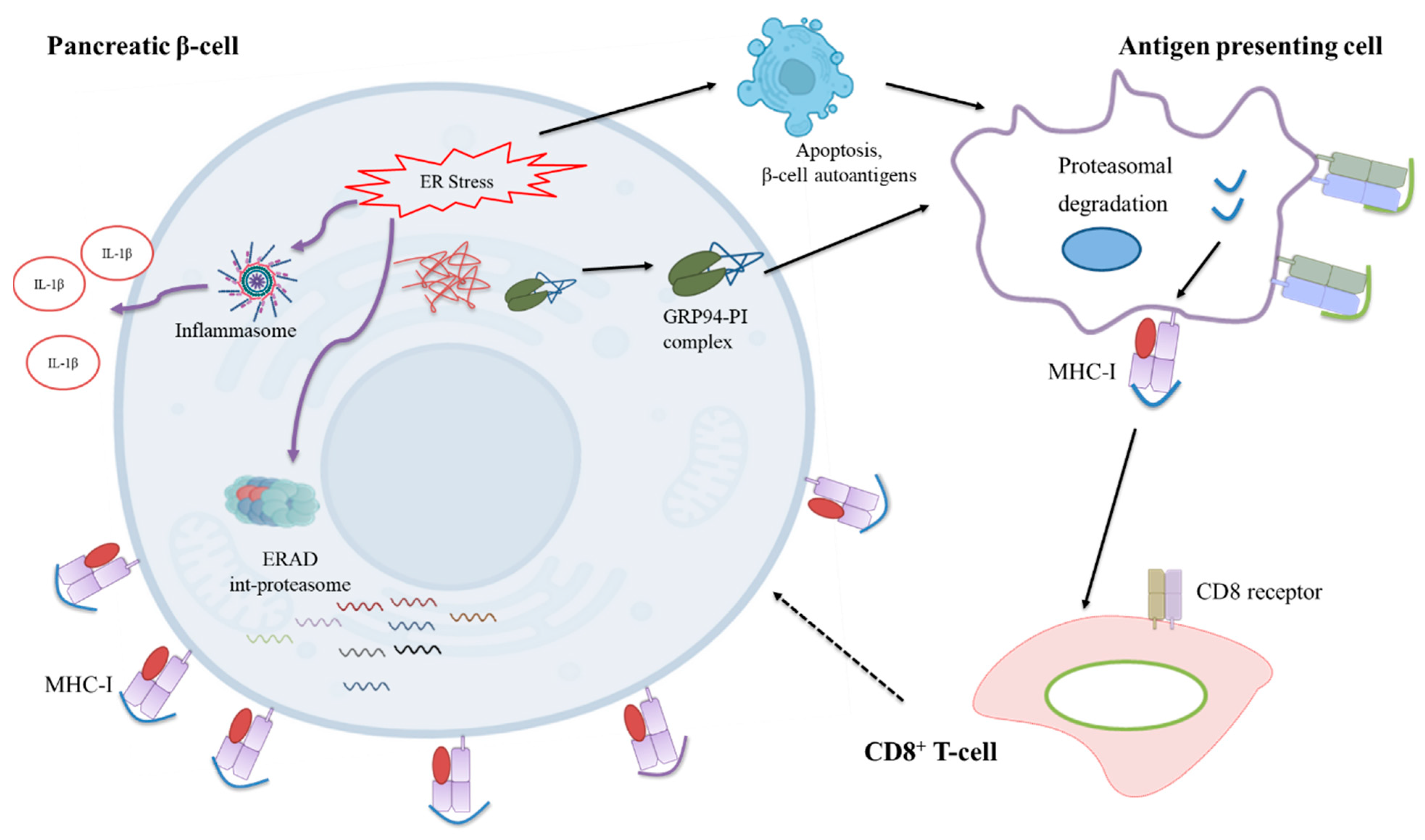
3.6. Limiting β-Cell Folding Capacity Activates Inflammatory Pathways
4. Discussion
5. Limitations of the Study/Further Possibilities
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baker, R.L.; Jamison, B.L.; Wiles, T.A.; Lindsay, R.S.; Barbour, G.; Bradley, B.; Delong, T.; Friedman, R.S.; Nakayama, M.; Haskins, K. CD4 T cells reactive to hybrid insulin peptides are indicators of disease activity in the NOD mouse. Diabetes 2018, 67, 1836–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Duque, S.; Azoury, M.E.; Colli, M.L.; Afonso, G.; Turatsinze, J.-V.; Nigi, L.; Lalanne, A.I.; Sebastiani, G.; Carré, A.; Pinto, S. Conventional and neo-antigenic peptides presented by β cells are targeted by circulating naïve CD8+ T cells in type 1 diabetic and healthy donors. Cell Metab. 2018, 28, 946–960.e946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marré, M.L.; Profozich, J.L.; Coneybeer, J.T.; Geng, X.; Bertera, S.; Ford, M.J.; Trucco, M.; Piganelli, J.D. Inherent ER stress in pancreatic islet β cells causes self-recognition by autoreactive T cells in type 1 diabetes. J. Autoimmun. 2016, 72, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, M.A.; Bowman, M.A.; Campbell, L.; Darrow, B.L.; Kaufman, D.L.; Maclaren, N.K. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J. Clin. Investig. 1994, 94, 2125–2129. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, S.; Sundaresh, B.; Huang, Q.; Brady, C.; Yoo, T.; Cronin, C.; Rudnicki, C.; Flood, M.; Momeni, B.; Ludvigsson, J. The role of gut microbiota and environmental factors in type 1 diabetes pathogenesis. Front. Endocrinol. 2020, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, H.; Wahlberg, J.; Vaarala, O.; Ludvigsson, J.; Group, A.S. Short duration of breast-feeding as a risk-factor for β-cell autoantibodies in 5-year-old children from the general population. Br. J. Nutr. 2007, 97, 111–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karjalainen, J.; Martin, J.M.; Knip, M.; Ilonen, J.; Robinson, B.H.; Savilahti, E.; Akerblom, H.K.; Dosch, H.-M. A bovine albumin peptide as a possible trigger of insulin-dependent diabetes mellitus. N. Engl. J. Med. 1992, 327, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M. Insulin as a key autoantigen in the development of type 1 diabetes. Diabetes/Metab. Res. Rev. 2011, 27, 773–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Nakayama, M.; Eisenbarth, G.S. Insulin as an autoantigen in NOD/human diabetes. Curr. Opin. Immunol. 2008, 20, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, R.L.; Rihanek, M.; Hohenstein, A.C.; Nakayama, M.; Michels, A.; Gottlieb, P.A.; Haskins, K.; Delong, T. Hybrid insulin peptides are autoantigens in type 1 diabetes. Diabetes 2019, 68, 1830–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delong, T.; Wiles, T.A.; Baker, R.L.; Bradley, B.; Barbour, G.; Reisdorph, R.; Armstrong, M.; Powell, R.L.; Reisdorph, N.; Kumar, N. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 2016, 351, 711–714. [Google Scholar] [CrossRef] [Green Version]
- Sidney, J.; Vela, J.L.; Friedrich, D.; Kolla, R.; von Herrath, M.; Wesley, J.D.; Sette, A. Low HLA binding of diabetes-associated CD8+ T-cell epitopes is increased by post translational modifications. BMC Immunol. 2018, 19, 12. [Google Scholar] [CrossRef] [Green Version]
- Ghiasi, S.M.; Dahlby, T.; Andersen, C.H.; Haataja, L.; Petersen, S.; Omar-Hmeadi, M.; Yang, M.; Pihl, C.; Bresson, S.E.; Khilji, M.S. Endoplasmic reticulum chaperone glucose-regulated protein 94 is essential for proinsulin handling. Diabetes 2019, 68, 747–760. [Google Scholar] [CrossRef]
- Liu, M.; Haataja, L.; Wright, J.; Wickramasinghe, N.P.; Hua, Q.-X.; Phillips, N.F.; Barbetti, F.; Weiss, M.A.; Arvan, P. Mutant INS-gene induced diabetes of youth: Proinsulin cysteine residues impose dominant-negative inhibition on wild-type proinsulin transport. PLoS ONE 2010, 5, e13333. [Google Scholar] [CrossRef]
- Liu, M.; Weiss, M.A.; Arunagiri, A.; Yong, J.; Rege, N.; Sun, J.; Haataja, L.; Kaufman, R.J.; Arvan, P. Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes. Metab. 2018, 20, 28–50. [Google Scholar] [CrossRef]
- Scheuner, D.; Vander Mierde, D.; Song, B.; Flamez, D.; Creemers, J.W.; Tsukamoto, K.; Ribick, M.; Schuit, F.C.; Kaufman, R.J. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat. Med. 2005, 11, 757–764. [Google Scholar] [CrossRef]
- Khilji, M.S.; Bresson, S.E.; Verstappen, D.; Pihl, C.; Andersen, P.A.K.; Agergaard, J.B.; Dahlby, T.; Bryde, T.H.; Klindt, K.; Nielsen, C.K. The inducible β5i proteasome subunit contributes to proinsulin degradation in GRP94-deficient β-cells and is overexpressed in type 2 diabetes pancreatic islets. Am. J. Physiol.-Endocrinol. Metab. 2020, 318, E892–E900. [Google Scholar] [CrossRef]
- Tersey, S.A.; Nishiki, Y.; Templin, A.T.; Cabrera, S.M.; Stull, N.D.; Colvin, S.C.; Evans-Molina, C.; Rickus, J.L.; Maier, B.; Mirmira, R.G. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 2012, 61, 818–827. [Google Scholar] [CrossRef] [Green Version]
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.; Atkinson, M.A.; Roep, B.O.; von Herrath, M.G. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 2012, 209, 51–60. [Google Scholar] [CrossRef]
- Patterson, C.C.; Harjutsalo, V.; Rosenbauer, J.; Neu, A.; Cinek, O.; Skrivarhaug, T.; Rami-Merhar, B.; Soltesz, G.; Svensson, J.; Parslow, R.C. Trends and cyclical variation in the incidence of childhood type 1 diabetes in 26 European centres in the 25 year period 1989–2013: A multicentre prospective registration study. Diabetologia 2019, 62, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Lamb, M.M.; Yin, X.; Barriga, K.; Hoffman, M.R.; Baron, A.E.; Eisenbarth, G.S.; Rewers, M.; Norris, J.M. Dietary glycemic index, development of islet autoimmunity, and subsequent progression to type 1 diabetes in young children. J. Clin. Endocrinol. Metab. 2008, 93, 3936–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludvigsson, J. Why diabetes incidence increases-a unifying theory. Ann. N. Y. Acad. Sci. 2006, 1079, 374. [Google Scholar] [CrossRef] [PubMed]
- Marré, M.L.; James, E.A.; Piganelli, J.D. β cell ER stress and the implications for immunogenicity in type 1 diabetes. Front. Cell Dev. Biol. 2015, 3, 67. [Google Scholar] [CrossRef] [Green Version]
- Marre, M.L.; McGinty, J.W.; Chow, I.-T.; DeNicola, M.E.; Beck, N.W.; Kent, S.C.; Powers, A.C.; Bottino, R.; Harlan, D.M.; Greenbaum, C.J. Modifying enzymes are elicited by ER stress, generating epitopes that are selectively recognized by CD4+ T cells in patients with type 1 diabetes. Diabetes 2018, 67, 1356–1368. [Google Scholar] [CrossRef] [Green Version]
- Rewers, M.; Ludvigsson, J. Environmental risk factors for type 1 diabetes. Lancet 2016, 387, 2340–2348. [Google Scholar] [CrossRef] [Green Version]
- Sims, E.K.; Syed, F.; Nyalwidhe, J.; Bahnson, H.T.; Haataja, L.; Speake, C.; Morris, M.A.; Balamurugan, A.N.; Mirmira, R.G.; Nadler, J. Abnormalities in proinsulin processing in islets from individuals with longstanding T1D. Transl. Res. 2019, 213, 90–99. [Google Scholar] [CrossRef]
- Sun, J.; Cui, J.; He, Q.; Chen, Z.; Arvan, P.; Liu, M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol. Asp. Med. 2015, 42, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Heath, W.R.; Carbone, F.R. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 2009, 10, 1237–1244. [Google Scholar] [CrossRef]
- Steinman, R.M.; Gutchinov, B.; Witmer, M.D.; Nussenzweig, M.C. Dendritic cells are the principal stimulators of the primary mixed leukocyte reaction in mice. J. Exp. Med. 1983, 157, 613–627. [Google Scholar] [CrossRef] [Green Version]
- Norbury, C.C.; Basta, S.; Donohue, K.B.; Tscharke, D.C.; Princiotta, M.F.; Berglund, P.; Gibbs, J.; Bennink, J.R.; Yewdell, J.W. CD8+ T cell cross-priming via transfer of proteasome substrates. Science 2004, 304, 1318–1321. [Google Scholar] [CrossRef] [Green Version]
- Pagetta, A.; Folda, A.; Brunati, A.; Finotti, P. Identification and purification from the plasma of Type 1 diabetic subjects of a proteolytically active Grp94. Diabetologia 2003, 46, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Murshid, A.; Gong, J.; Calderwood, S.K. Heat shock protein 90 mediates efficient antigen cross presentation through the scavenger receptor expressed by endothelial cells-I. J. Immunol. 2010, 185, 2903–2917. [Google Scholar] [CrossRef] [Green Version]
- Purcell, A.W.; Ramarathinam, S.H.; Ternette, N. Mass spectrometry–based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019, 14, 1687–1707. [Google Scholar] [CrossRef]
- Faridi, P.; Woods, K.; Ostrouska, S.; Deceneux, C.; Aranha, R.; Duscharla, D.; Wong, S.Q.; Chen, W.; Ramarathinam, S.; Sian, T.C.L.K. Spliced peptides and cytokine driven changes in the immunopeptidome of melanoma. Cancer Immunol. Res. 2020, 8, 1322–1334. [Google Scholar] [CrossRef]
- Maddelein, D.; Colaert, N.; Buchanan, I.; Hulstaert, N.; Gevaert, K.; Martens, L. The iceLogo web server and SOAP service for determining protein consensus sequences. Nucleic Acids Res. 2015, 43, W543–W546. [Google Scholar] [CrossRef]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn–a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [Green Version]
- Andreatta, M.; Alvarez, B.; Nielsen, M. GibbsCluster: Unsupervised clustering and alignment of peptide sequences. Nucleic Acids Res. 2017, 45, W458–W463. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Andreatta, M. NNAlign: A platform to construct and evaluate artificial neural network models of receptor–ligand interactions. Nucleic Acids Res. 2017, 45, W344–W349. [Google Scholar] [CrossRef]
- Khilji, M.S.; Verstappen, D.; Dahlby, T.; Burstein Prause, M.C.; Pihl, C.; Bresson, S.E.; Bryde, T.H.; Keller Andersen, P.A.; Klindt, K.; Zivkovic, D. The intermediate proteasome is constitutively expressed in pancreatic beta cells and upregulated by stimulatory, low concentrations of interleukin 1 β. PLoS ONE 2020, 15, e0222432. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Jameson, S.C.; Tope, W.D.; Tredgett, E.M.; Windle, J.M.; Diamond, A.G.; Howard, J.C. Cloning and expression of class I major histocompatibility complex genes of the rat. J. Exp. Med. 1992, 175, 1749–1757. [Google Scholar] [CrossRef] [Green Version]
- Reizis, B.; Schild, H.; Stefanović, S.; Mor, F.; Rammensee, H.-G.; Cohen, I.R. Peptide binding motifs of the MHC class I molecules (RT1. A l) of the Lewis rat. Immunogenetics 1997, 45, 278–279. [Google Scholar] [CrossRef]
- Stevens, J.; Wiesmüller, K.H.; Walden, P.; Joly, E. Peptide length preferences for rat and mouse MHC class I molecules using random peptide libraries. Eur. J. Immunol. 1998, 28, 1272–1279. [Google Scholar] [CrossRef]
- Le Rolle, A.-F.; Hutchings, A.; Butcher, G.W.; Joly, E. Cloning of three different species of MHC class I cDNAs of the RT1 g haplotype from the NEDH rat. Immunogenetics 2000, 51, 503–507. [Google Scholar] [PubMed]
- Hafner-Bratkovič, I.; Sušjan, P.; Lainšček, D.; Tapia-Abellán, A.; Cerović, K.; Kadunc, L.; Angosto-Bazarra, D.; Pelegrin, P.; Jerala, R. NLRP3 lacking the leucine-rich repeat domain can be fully activated via the canonical inflammasome pathway. Nat. Commun. 2018, 9, 5182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostankovitch, M.; Robila, V.; Engelhard, V.H. Regulated folding of tyrosinase in the endoplasmic reticulum demonstrates that misfolded full-length proteins are efficient substrates for class I processing and presentation. J. Immunol. 2005, 174, 2544–2551. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.J.; Rodriguez-Calvo, T.; Gerling, I.C.; Mathews, C.E.; Kaddis, J.S.; Russell, M.A.; Zeissler, M.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K. Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 2016, 59, 2448–2458. [Google Scholar] [CrossRef] [Green Version]
- Pugliese, A. Autoreactive T cells in type 1 diabetes. J. Clin. Investig. 2017, 127, 2881–2891. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Battaglia-Hsu, S.-F.; Arnold, C. Endoplasmic reticulum stress in metabolic disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- Salvado, L.; Palomer, X.; Barroso, E.; Vázquez-Carrera, M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol. Metab. 2015, 26, 438–448. [Google Scholar] [CrossRef]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 2004, 24, 10161–10168. [Google Scholar] [CrossRef] [Green Version]
- Forloni, M.; Albini, S.; Limongi, M.Z.; Cifaldi, L.; Boldrini, R.; Nicotra, M.R.; Giannini, G.; Natali, P.G.; Giacomini, P.; Fruci, D. NF-κB, and not MYCN, regulates MHC class I and endoplasmic reticulum aminopeptidases in human neuroblastoma cells. Cancer Res. 2010, 70, 916–924. [Google Scholar] [CrossRef] [Green Version]
- Marroqui, L.; Dos Santos, R.S.; de Brachène, A.C.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-α mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 2017, 60, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Culina, S.; Lalanne, A.I.; Afonso, G.; Cerosaletti, K.; Pinto, S.; Sebastiani, G.; Kuranda, K.; Nigi, L.; Eugster, A.; Østerbye, T. Islet-reactive CD8+ T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci. Immunol. 2018, 3, eaa04013. [Google Scholar] [CrossRef] [Green Version]
- James, E.A.; Mallone, R.; Kent, S.C.; DiLorenzo, T.P. T-cell epitopes and neo-epitopes in type 1 diabetes: A comprehensive update and reappraisal. Diabetes 2020, 69, 1311–1335. [Google Scholar] [CrossRef]
- Spinas, G.A.; Mandrup-Poulsen, T.; Mølvig, J.; Bæk, L.; Bendtzen, K.; Dinarello, C.A.; Nerup, J. Low concentrations of interleukin-1 stimulate and high concentrations inhibit insulin release from isolated rat islets of Langerhans. Eur. J. Endocrinol. 1986, 113, 551–558. [Google Scholar] [CrossRef]
- Hasnain, S.Z.; Lourie, R.; Das, I.; Chen, A.C.H.; McGuckin, M.A. The interplay between endoplasmic reticulum stress and inflammation. Immunol. Cell Biol. 2012, 90, 260–270. [Google Scholar] [CrossRef]
- D’Osualdo, A.; Anania, V.G.; Yu, K.; Lill, J.R.; Kaufman, R.J.; Matsuzawa, S.-i.; Reed, J.C. Transcription factor ATF4 induces NLRP1 inflammasome expression during endoplasmic reticulum stress. PLoS ONE 2015, 10, e0130635. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Schroder, K.; Sagulenko, V.; Zamoshnikova, A.; Richards, A.A.; Cridland, J.A.; Irvine, K.M.; Stacey, K.J.; Sweet, M.J. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 2012, 217, 1325–1329. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Shen, X.; Liu, J.; Chen, W.; Wu, F.; Wu, W.; Meng, Z.; Zhu, M.; Miao, C. High glucose mediates NLRP3 inflammasome activation via upregulation of ELF3 expression. Cell Death Dis. 2020, 11, 383. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wali, J.A.; Gurzov, E.N.; Fynch, S.; Elkerbout, L.; Kay, T.W.; Masters, S.L.; Thomas, H.E. Activation of the NLRP3 inflammasome complex is not required for stress-induced death of pancreatic islets. PLoS ONE 2014, 9, e113128. [Google Scholar]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta-Mol. Cell Res. 2012, 1823, 774–787. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-s.; Song, L.; Wang, J.; Wu, H.; Gu, G.; Sugi, Y.; Li, Z.; Wang, H. GRP94 is an essential regulator of pancreatic β-cell development, mass, and function in male mice. Endocrinology 2018, 159, 1062–1073. [Google Scholar] [CrossRef]
- Ghiasi, S.M.; Dahllöf, M.S.; Osmai, Y.; Osmai, M.; Jakobsen, K.K.; Aivazidis, A.; Tyrberg, B.; Perruzza, L.; Prause, M.C.B.; Christensen, D.P. Regulation of the β-cell inflammasome and contribution to stress-induced cellular dysfunction and apoptosis. Mol. Cell. Endocrinol. 2018, 478, 106–114. [Google Scholar] [CrossRef]
- Pagetta, A.; Tramentozzi, E.; Corbetti, L.; Frasson, M.; Brunati, A.M.; Finotti, P. Characterization of immune complexes of idiotypic catalytic and anti-idiotypic inhibitory antibodies in plasma of type 1 diabetic subjects. Mol. Immunol. 2007, 44, 2870–2883. [Google Scholar] [CrossRef]
- Roveri, A.; Zaccarin, M.; Pagetta, A.; Tramentozzi, E.; Finotti, P. Proteomic investigation on Grp94-IgG complexes circulating in plasma of type 1 diabetic subjects. J. Diabetes Res. 2015, 2015, 815839. [Google Scholar] [CrossRef] [Green Version]
- Berwin, B.; Reed, R.C.; Nicchitta, C.V. Virally induced lytic cell death elicits the release of immunogenic GRP94/gp96. J. Biol. Chem. 2001, 276, 21083–21088. [Google Scholar] [CrossRef] [Green Version]
- Calderwood, S.K.; Mambula, S.S.; Gray, P.J., Jr.; Theriault, J.R. Extracellular heat shock proteins in cell signaling. FEBS Lett. 2007, 581, 3689–3694. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Binder, R.J.; Ramalingam, T.; Srivastava, P.K. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 2001, 14, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Berwin, B.; Hart, J.P.; Pizzo, S.V.; Nicchitta, C.V. Cutting edge: CD91-independent cross-presentation of GRP94 (gp96)-associated peptides. J. Immunol. 2002, 168, 4282–4286. [Google Scholar] [CrossRef] [Green Version]
- Berwin, B.; Rosser, M.; Brinker, K.; Nicchitta, C. Transfer of GRP94 (Gp96)-associated peptides onto endosomal MHC class I molecules. Traffic 2002, 3, 358–366. [Google Scholar] [CrossRef]
- Binder, R.J.; Han, D.K.; Srivastava, P.K. CD91: A receptor for heat shock protein gp96. Nat. Immunol. 2000, 1, 151–155. [Google Scholar] [CrossRef]
- Song, L.; Kim, D.-s.; Gou, W.; Wang, J.; Wang, P.; Wei, Z.; Liu, B.; Li, Z.; Gou, K.; Wang, H. GRP94 regulates M1 macrophage polarization and insulin resistance. Am. J. Physiol.-Endocrinol. Metab. 2020, 318, E1004–E1013. [Google Scholar] [CrossRef]
- He, B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006, 13, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Dotta, F.; Censini, S.; van Halteren, A.G.; Marselli, L.; Masini, M.; Dionisi, S.; Mosca, F.; Boggi, U.; Muda, A.O.; Del Prato, S. Coxsackie B4 virus infection of β cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc. Natl. Acad. Sci. USA 2007, 104, 5115–5120. [Google Scholar] [CrossRef] [Green Version]
- Mathes, E.; O’dea, E.L.; Hoffmann, A.; Ghosh, G. NF-κB dictates the degradation pathway of IκBα. EMBO J. 2008, 27, 1357–1367. [Google Scholar] [CrossRef] [Green Version]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Heath, V.L.; Hutchings, P.; Fowell, D.J.; Cooke, A.; Mason, D.W. Peptides derived from murine insulin are diabetogenic in both rats and mice, but the disease-inducing epitopes are different: Evidence against a common environmental cross-reactivity in the pathogenicity of type 1 diabetes. Diabetes 1999, 48, 2157–2165. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (Pre-Pro) Insulin Peptides | INS-1E | Control KO | INS-1E + GRP94i | GRP94 KO INS-1E | INS-1E + IL-1β | INS-1E + IFNγ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rep 1 | Rep 2 | Rep 1 | Rep 2 | Rep 1 | Rep 2 | Rep 1 | Rep 2 | Rep 3 | Rep 4 | Rep 1 | Rep 2 | Rep 1 | Rep 2 | |
| VLWEPKPAQAFVK | ✓ | ✓ | ✓ | ✓ | ||||||||||
| ILWEPKPAQAFVK | ✓ | ✓ | ||||||||||||
| ALWMRFLPL | ✓ | ✓ | ||||||||||||
| HLVEALYL | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||
| ALYLVCGERGFF | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||
| ALYLVCGERGFFYTP | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||
| YLVCGERGFF | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||
| FFYTPKS | ✓ | ✓ | ✓ | ✓ | ||||||||||
| VEDPQVPQ | ✓ | ✓ | ✓ | ✓ | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khilji, M.S.; Faridi, P.; Pinheiro-Machado, E.; Hoefner, C.; Dahlby, T.; Aranha, R.; Buus, S.; Nielsen, M.; Klusek, J.; Mandrup-Poulsen, T.; et al. Defective Proinsulin Handling Modulates the MHC I Bound Peptidome and Activates the Inflammasome in β-Cells. Biomedicines 2022, 10, 814. https://doi.org/10.3390/biomedicines10040814
Khilji MS, Faridi P, Pinheiro-Machado E, Hoefner C, Dahlby T, Aranha R, Buus S, Nielsen M, Klusek J, Mandrup-Poulsen T, et al. Defective Proinsulin Handling Modulates the MHC I Bound Peptidome and Activates the Inflammasome in β-Cells. Biomedicines. 2022; 10(4):814. https://doi.org/10.3390/biomedicines10040814
Chicago/Turabian StyleKhilji, Muhammad Saad, Pouya Faridi, Erika Pinheiro-Machado, Carolin Hoefner, Tina Dahlby, Ritchlynn Aranha, Søren Buus, Morten Nielsen, Justyna Klusek, Thomas Mandrup-Poulsen, and et al. 2022. "Defective Proinsulin Handling Modulates the MHC I Bound Peptidome and Activates the Inflammasome in β-Cells" Biomedicines 10, no. 4: 814. https://doi.org/10.3390/biomedicines10040814