Thermal Inactivation of a Cold-Active Esterase PMGL3 Isolated from the Permafrost Metagenomic Library

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Cloning and Mutant Construction
2.2. Protein Purification
2.3. Esterase Assay
2.4. Circular Dichroism Spectra
2.5. Size-exclusion Chromatography
2.6. Fluorescence Measurements
2.7. Bioinformatic Analysis
3. Results
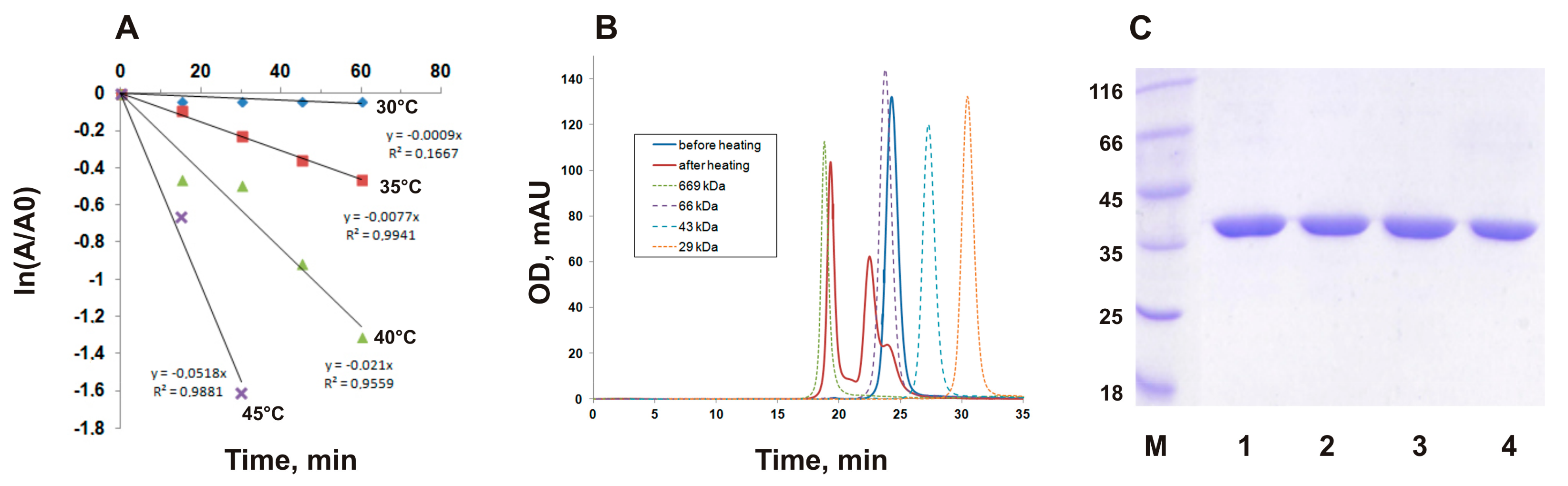
3.1. Thermal Inactivation of PMGL3 is Accompanied by Oligomerization
3.2. Disulfide Bond Formation does not Contribute to Oligomerization of PMGL3
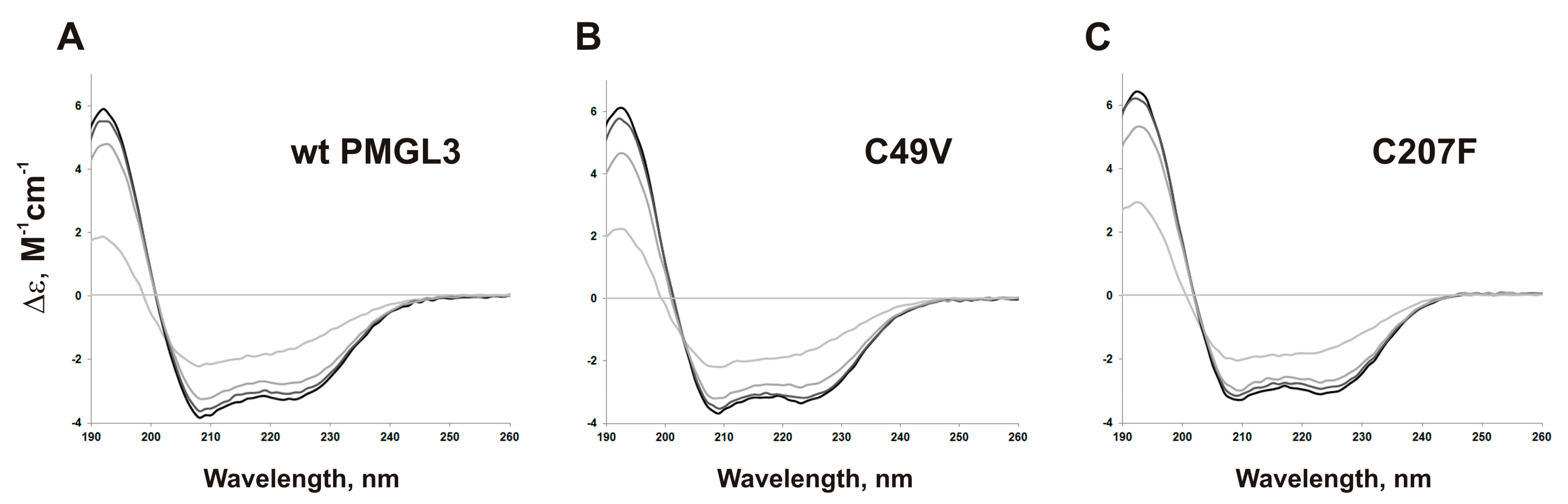
3.3. Construction and Properties of the Mutant Proteins C49V and C207F
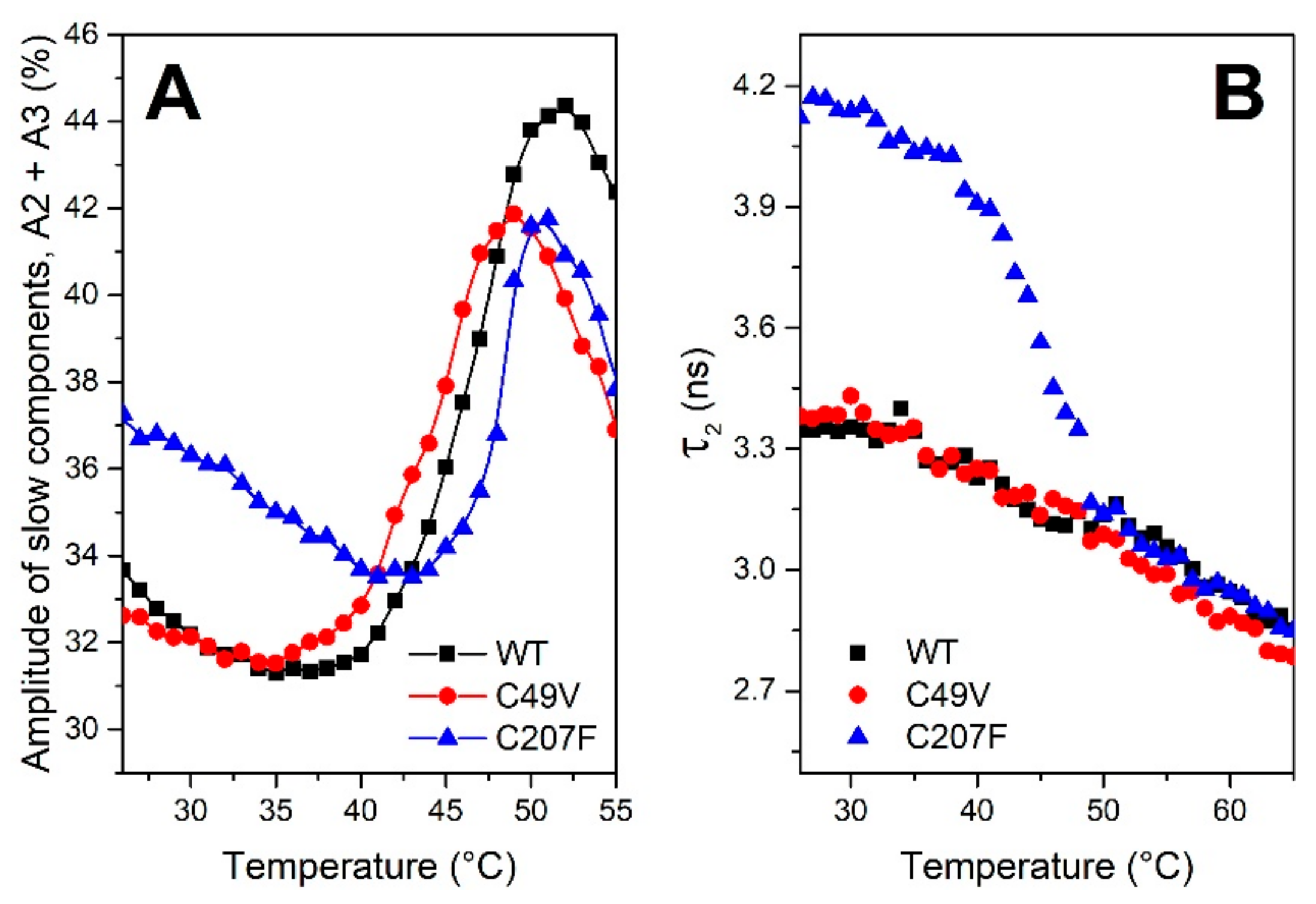
3.4. bis-ANS Binding Studies of PMGL3 and Mutant Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Margesin, R.; Feller, G.; Gerday, C.; Russell, N.J. Cold-adapted microorganisms: Adaptation strategies and biotechnological potential. Encycl. Environ. Microbiol. 2002. [Google Scholar] [CrossRef]
- D’Amico, S.; Claverie, P.; Collins, T.; Georlette, D.; Gratia, E.; Hoyoux, A.; Meuwis, M.-A.; Feller, G.; Gerday, C. Molecular basis of cold adaptation. Philos. Trans. R. Soc. London 2002, 357, 917–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavicchioli, R.; Siddiqui, K.S.; Andrews, D.; Sowers, K.R. Low-temperature extremophiles and their applications. Curr. Opin. Biotechnol. 2002, 13, 253–261. [Google Scholar] [CrossRef]
- Littlechild, J.A. Enzymes from extreme environments and their industrial applications. Front. Bioeng. Biotechnol. 2015, 3, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barroca, M.; Santos, G.; Gerday, C.; Collins, T. Biotechnological Aspects of Cold-Active Enzymes. In Psychrophiles: From Biodiversity to Biotechnology; Margesin, R., Ed.; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 461–475. [Google Scholar] [CrossRef]
- Santiago, M.; Ramírez-Sarmiento, C.A.; Zamora, R.A.; Parra, L.P. Discovery, Molecular Mechanisms, and Industrial Applications of Cold-Active Enzymes. Front. Microbiol. 2016, 7, 1408. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Martínez-Martínez, M.; Bargiela, R.; Streit, W.R.; Golyshina, O.V.; Golyshin, P.N. Estimating the success of enzyme bioprospecting through metagenomics: Current status and future trends. Microb. Biotechnol. 2016, 9, 22–34. [Google Scholar] [CrossRef]
- Siddiqui, K.S.; Cavicchioli, R. Cold-adapted enzymes. Annu. Rev. Biochem. 2006, 75, 403–433. [Google Scholar] [CrossRef] [Green Version]
- Feller, G.; Gerday, C. Psychrophilic enzymes: Hot topics in cold adaptation. Nat. Rev. Microbiol. 2003, 1, 200–208. [Google Scholar] [CrossRef]
- Feller, G. Psychrophilic enzymes: From folding to function and biotechnology. Scientifica 2013, 2013, 512840. [Google Scholar] [CrossRef]
- Elleuche, S.; Schröder, C.; Sahm, K.; Antranikian, G. Extremozymes—biocatalysts with unique properties from extremophilic microorganisms. Curr. Opin. Biotechnol. 2014, 29, 116–123. [Google Scholar] [CrossRef]
- Kovacic, F.; Mandrysch, A.; Poojari, C.; Strodel, B.; Jaeger, K.-E. Structural features determining thermal adaptation of esterases. Protein Eng. 2016, 29, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornscheuer, U.T.; Kourist, R. Evolving Enzymes for Biocatalysis. In Consequences of Microbial Interactions with Hydrocarbons, Oils, and Lipids: Production of Fuels and Chemicals; Lee, S.Y., Ed.; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 271–287. [Google Scholar] [CrossRef]
- Kourist, R.; Brundiek, H.; Bornscheuer, U.T. Protein engineering and discovery of lipases. Eur. J. Lipid Sci. Technol. 2010, 112, 64–74. [Google Scholar] [CrossRef]
- Tutino, M.L.; Parrilli, E.; Santi, C.; Giuliani, M.; Marino, G.; Pascale, D. Cold-adapted esterases and lipases: A biodiversity still under-exploited. Curr. Chem. Biol. 2010, 4, 74–83. [Google Scholar] [CrossRef]
- Romano, D.; Bonomi, F.; Mattos, M.C.; Fonseca, T.D.; Oliveira, M.D.F.; Molinari, F. Esterases as stereoselective biocatalysts. Biotechnol. Adv. 2015, 33, 547–565. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B.; Ramteke, P.W.; Thomas, G.; Shrivastava, N. Standard review cold-active microbial lipases: A versatile tool for industrial applications. Biotechnol. Mol. Biol. Rev. 2007, 2, 39–48. [Google Scholar]
- Ollis, D.L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken, S.M.; Harel, M.; Remington, S.J.; Silman, I.; Schrag, J. The α/β hydrolase fold. Protein Eng. 1992, 5, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Casas-Godoy, L.; Gasteazoro, F.; Duquesne, S.; Bordes, F.; Marty, A.; Sandoval, G. Lipases: An Overview. Methods Mol. Biol. 2018, 1835, 3–38. [Google Scholar] [CrossRef]
- Kim, T.D. Bacterial hormone-sensitive lipases (bHSLs): Emerging enzymes for biotechnological applications. J. Microbiol. Biotechnol. 2017, 27, 1907–1915. [Google Scholar] [CrossRef] [Green Version]
- Arpigny, J.; Jaeger, K. Bacterial lipolytic enzymes: Classification and properties. Biochem. J. 1999, 343, 177–183. [Google Scholar] [CrossRef]
- Petrovskaya, L.E.; Novototskaya-Vlasova, K.A.; Gapizov, S.S.; Spirina, E.V.; Durdenko, E.V.; Rivkina, E.M. New member of the hormone-sensitive lipase family from the permafrost microbial community. Bioengineered 2017, 8, 420–423. [Google Scholar] [CrossRef] [Green Version]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, M.J. The oxidative environment and protein damage. Biochim. Biophys. Acta 2005, 1703, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Maksimov, E.; Moldenhauer, M.; Shirshin, E.; Parshina, E.; Sluchanko, N.; Klementiev, K.; Tsoraev, G.; Tavraz, N.; Willoweit, M.; Schmitt, F.-J. A comparative study of three signaling forms of the orange carotenoid protein. Photosynth. Res. 2016, 130, 389–401. [Google Scholar] [CrossRef]
- Li, P.Y.; Chen, X.L.; Ji, P.; Li, C.Y.; Wang, P.; Zhang, Y.; Xie, B.B.; Qin, Q.L.; Su, H.N.; Zhou, B.C.; et al. Interdomain hydrophobic interactions modulate the thermostability of microbial esterases from the hormone-sensitive lipase family. J. Biol. Chem. 2015, 290, 11188–11198. [Google Scholar] [CrossRef] [Green Version]
- Dou, S.; Kong, X.D.; Ma, B.D.; Chen, Q.; Zhang, J.; Zhou, J.; Xu, J.H. Crystal structures of Pseudomonas putida esterase reveal the functional role of residues 187 and 287 in substrate binding and chiral recognition. Biochem. Biophys. Res. Commun. 2014, 446, 1145–1150. [Google Scholar] [CrossRef]
- De Simone, G.; Galdiero, S.; Manco, G.; Lang, D.; Rossi, M.; Pedone, C. A snapshot of a transition state analogue of a novel thermophilic esterase belonging to the subfamily of mammalian hormone-sensitive lipase. J. Mol. Biol. 2000, 303, 761–771. [Google Scholar] [CrossRef]
- Hawe, A.; Sutter, M.; Jiskoot, W. Extrinsic Fluorescent Dyes as Tools for Protein Characterization. Pharm. Res. 2008, 25, 1487–1499. [Google Scholar] [CrossRef] [Green Version]
- Joseph, B.; Ramteke, P.W.; Thomas, G. Cold active microbial lipases: Some hot issues and recent developments. Biotechnol. Adv. 2008, 26, 457–470. [Google Scholar] [CrossRef]
- Zale, S.E.; Klibanov, A.M. On the role of reversible denaturation (unfolding) in the irreversible thermal inactivation of enzymes. Biotechnol. Bioeng. 1983, 25, 2221–2230. [Google Scholar] [CrossRef]
- Markossian, K.; Kurganov, B. Protein folding, misfolding, and aggregation. Formation of inclusion bodies and aggresomes. Biochemistry (Moscow) 2004, 69, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, A.V.; Shakhnovich, E.I. Theory of cooperative transitions in protein molecules. II. Phase diagram for a protein molecule in solution. Biopolymers 1989, 28, 1681–1694. [Google Scholar] [CrossRef] [PubMed]
- Palomo, J.M.; Fuentes, M.; Fernández-Lorente, G.; Mateo, C.; Guisan, J.M.; Fernández-Lafuente, R. General Trend of Lipase to Self-Assemble Giving Bimolecular Aggregates Greatly Modifies the Enzyme Functionality. Biomacromolecules 2003, 4, 1–6. [Google Scholar] [CrossRef]
- Jain, R.; Pandey, A.; Pasupuleti, M.; Pande, V. Prolonged Production and Aggregation Complexity of Cold-Active Lipase from Pseudomonas proteolytica (GBPI_Hb61) Isolated from Cold Desert Himalaya. Mol. Biotechnol. 2017, 59, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Arpigny, J.L.; Feller, G.; Gerday, C. Cloning, sequence and structural features of a lipase from the antarctic facultative psychrophile Psychrobacter immobilis B10. Biochim. Biophys. Acta Gene Struct. Expr. 1993, 1171, 331–333. [Google Scholar] [CrossRef]
- Bordes, F.; Tarquis, L.; Nicaud, J.-M.; Marty, A. Isolation of a thermostable variant of Lip2 lipase from Yarrowia lipolytica by directed evolution and deeper insight into the denaturation mechanisms involved. J. Biotechnol. 2011, 156, 117–124. [Google Scholar] [CrossRef]
- Nam, K.-H.; Park, S.-H.; Lee, W.-H.; Hwang, K.-Y. Biochemical and Structural Analysis of Hormone-sensitive Lipase Homolog EstE7: Insight into the Stabilized Dimerization of HSL-Homolog Proteins. Bull. Kor. Chem. Soc. 2010, 31, 2627–2632. [Google Scholar] [CrossRef] [Green Version]
- Volkin, D.B.; Klibanov, A.M. Thermal destruction processes in proteins involving cystine residues. J. Biol. Chem. 1987, 262, 2945–2950. [Google Scholar]
- Slavica, A.; Dib, I.; Nidetzky, B. Single-Site Oxidation, Cysteine 108 to Cysteine Sulfinic Acid, in D-Amino Acid Oxidase from Trigonopsis variabilis and Its Structural and Functional Consequences. Appl. Environ. Microbiol. 2005, 71, 8061–8068. [Google Scholar] [CrossRef] [Green Version]
- Perry, L.J.; Wetzel, R. The role of cysteine oxidation in the thermal inactivation of T4 lysozyme. Protein Eng. 1987, 1, 101–105. [Google Scholar] [CrossRef]
- Suemori, A.; Iwakura, M. A systematic and comprehensive combinatorial approach to simultaneously improve the activity, reaction specificity, and thermal stability of p-hydroxybenzoate hydroxylase. J. Biol. Chem. 2007, 282, 19969–19978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Tsai, C.-J.; Nussinov, R. Factors enhancing protein thermostability. Protein Eng. 2000, 13, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominy, B.N.; Minoux, H.; Brooks III, C.L. An electrostatic basis for the stability of thermophilic proteins. Proteins: Struct. Funct. Bioinf. 2004, 57, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Gromiha, M.M.; Pathak, M.C.; Saraboji, K.; Ortlund, E.A.; Gaucher, E.A. Hydrophobic environment is a key factor for the stability of thermophilic proteins. Proteins: Struct. Funct. Bioinf. 2013, 81, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Vogt, G.; Woell, S.; Argos, P. Protein thermal stability, hydrogen bonds, and ion pairs. J. Mol. Biol. 1997, 269, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Mukaiyama, A.; Takano, K. Delineation of the Conformational Thermostability of Hyperthermophilic Proteins Based on Structural and Biophysical Analyses. In Thermostable Proteins; Sen, S., Nilsson, L., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp. 1–20. [Google Scholar]
- Timucin, E.; Sezerman, O.U. The conserved lid tryptophan, W211, potentiates thermostability and thermoactivity in bacterial thermoalkalophilic lipases. PLoS ONE 2013, 8, e85186. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Jutila, A.; Tuominen, E.K.; Patkar, S.A.; Svendsen, A.; Kinnunen, P.K. Impact of the tryptophan residues of Humicola lanuginosa lipase on its thermal stability. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2001, 1547, 329–338. [Google Scholar] [CrossRef]
- Dror, A.; Shemesh, E.; Dayan, N.; Fishman, A. Protein engineering by random mutagenesis and structure-guided consensus of Geobacillus stearothermophilus lipase T6 for enhanced stability in methanol. Appl. Environ. Microbiol. 2014, 80, 1515–1527. [Google Scholar] [CrossRef] [Green Version]
- Santarossa, G.; Lafranconi, P.G.; Alquati, C.; DeGioia, L.; Alberghina, L.; Fantucci, P.; Lotti, M. Mutations in the “lid” region affect chain length specificity and thermostability of a Pseudomonas fragi lipase. FEBS Lett. 2005, 579, 2383–2386. [Google Scholar] [CrossRef] [Green Version]
- Abraham, T.; Abraham, T.; Pil Pack, S.; Je Yoo, Y. Stabilization of Bacillus subtilis Lipase A by increasing the residual packing. Biocatal. Biotransform. 2005, 23, 217–224. [Google Scholar] [CrossRef]
- D’Amico, S.; Gerday, C.; Feller, G. Temperature Adaptation of Proteins: Engineering Mesophilic-like Activity and Stability in a Cold-adapted α-Amylase. J. Mol. Biol. 2003, 332, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Salwoom, L.; Rahman, R.A.; Zaliha, R.N.; Salleh, A.B.; Convey, P.; Ali, M.; Shukuri, M. New recombinant cold-adapted and organic solvent tolerant lipase from psychrophilic Pseudomonas sp. LSK25, isolated from Signy Island Antarctica. Int. J. Mol. Sci. 2019, 20, 1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.Y.; Zhang, Y.; Xie, B.B.; Zhang, Y.Q.; Hao, J.; Wang, Y.; Wang, P.; Li, C.Y.; Qin, Q.L.; Zhang, X.Y.; et al. Structural and Mechanistic Insights into the Improvement of the Halotolerance of a Marine Microbial Esterase by Increasing Intra- and Interdomain Hydrophobic Interactions. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Yang, G.; Wu, L.; Feng, Y. Role of the NC-loop in catalytic activity and stability in lipase from Fervidobacterium changbaicum. PLoS ONE 2012, 7, e46881. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | t1/2, min (40 °C) | Tm, °C | Km, mM | kcat, min−1 | Vmax, mM/min/mg | kcat/Km, min−1 mM−1 |
|---|---|---|---|---|---|---|
| PMGL3 | 35.7 | 46.1 a/40.1 b | 0.98 | 5490 | 164.7 | 5602 |
| C49V | 27.4 | 43.6 a/36.9 b | 1.29 | 3156 | 94.7 | 2446 |
| C207F | 169.1 | 47.8 a/43.4 b | 0.17 | 3152 | 94.6 | 18,541 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kryukova, M.V.; Petrovskaya, L.E.; Kryukova, E.A.; Lomakina, G.Y.; Yakimov, S.A.; Maksimov, E.G.; Boyko, K.M.; Popov, V.O.; Dolgikh, D.A.; Kirpichnikov, M.P. Thermal Inactivation of a Cold-Active Esterase PMGL3 Isolated from the Permafrost Metagenomic Library. Biomolecules 2019, 9, 880. https://doi.org/10.3390/biom9120880
Kryukova MV, Petrovskaya LE, Kryukova EA, Lomakina GY, Yakimov SA, Maksimov EG, Boyko KM, Popov VO, Dolgikh DA, Kirpichnikov MP. Thermal Inactivation of a Cold-Active Esterase PMGL3 Isolated from the Permafrost Metagenomic Library. Biomolecules. 2019; 9(12):880. https://doi.org/10.3390/biom9120880
Chicago/Turabian StyleKryukova, M.V., L.E. Petrovskaya, E.A. Kryukova, G.Yu. Lomakina, S.A. Yakimov, E.G. Maksimov, K.M. Boyko, V.O. Popov, D.A. Dolgikh, and M.P. Kirpichnikov. 2019. "Thermal Inactivation of a Cold-Active Esterase PMGL3 Isolated from the Permafrost Metagenomic Library" Biomolecules 9, no. 12: 880. https://doi.org/10.3390/biom9120880