Deregulation of the Histone Lysine-Specific Demethylase 1 Is Involved in Human Hepatocellular Carcinoma

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Samples
2.2. Immunohistochemistry
2.3. Evaluation of Immunohistochemistry
2.4. Cell Lines
2.5. Plasmid DNA Constructs
2.6. Lentiviral Transduction
2.7. Generation of LSD1 Knockout Cell Lines Using the CRISPR/Cas9 Gene Editing System
2.8. Western Blotting Assays
2.9. Colony Formation Assays
2.10. Cell Proliferation Assays
2.11. Cell Cycle Assays
2.12. RNA-Seq
2.13. Statistical Analysis
3. Results
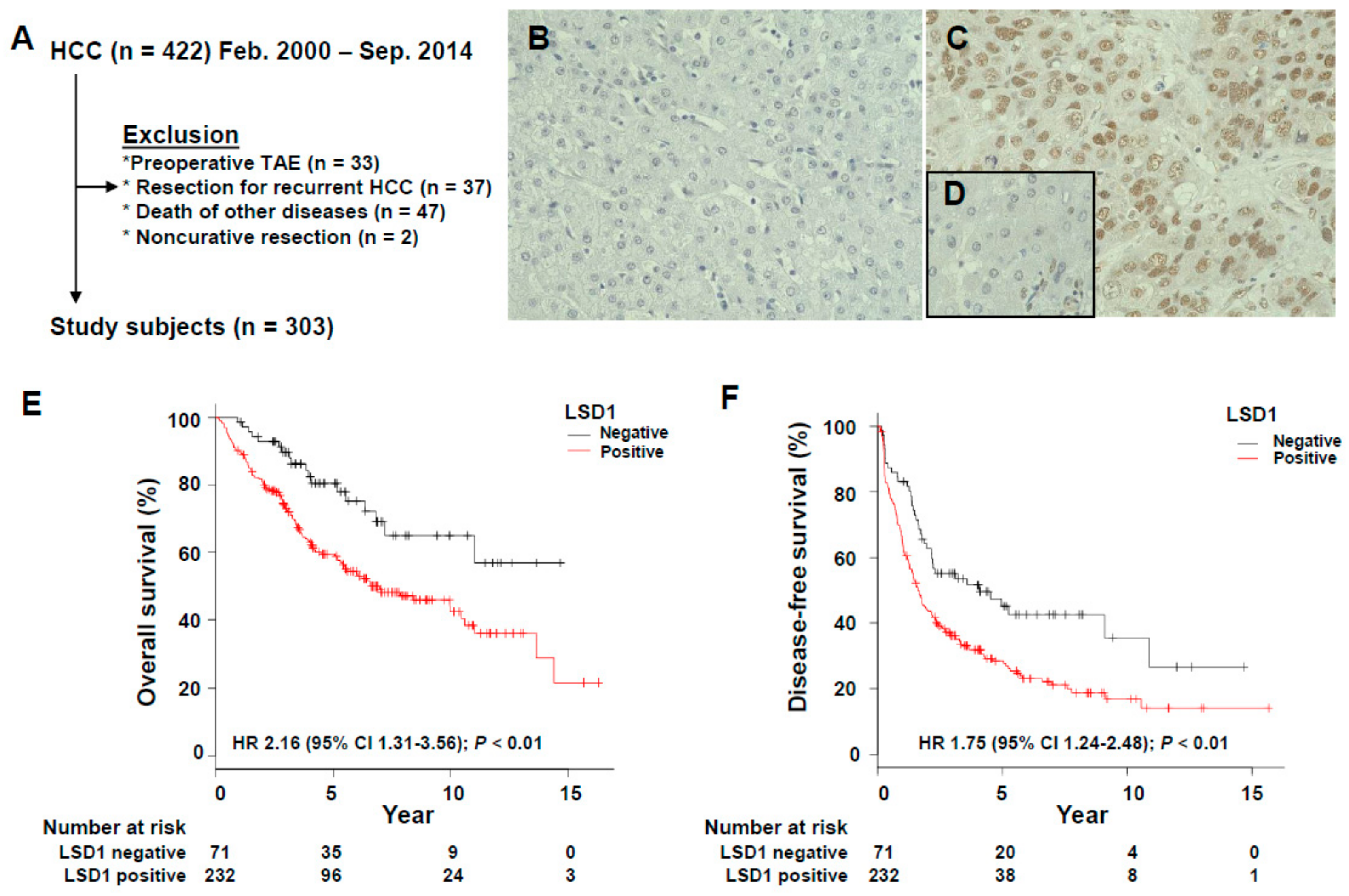
3.1. Association of LSD1 Expression with the Clinicopathological Features of HCC
3.2. Prognostic Significance of LSD1 Expression in HCC
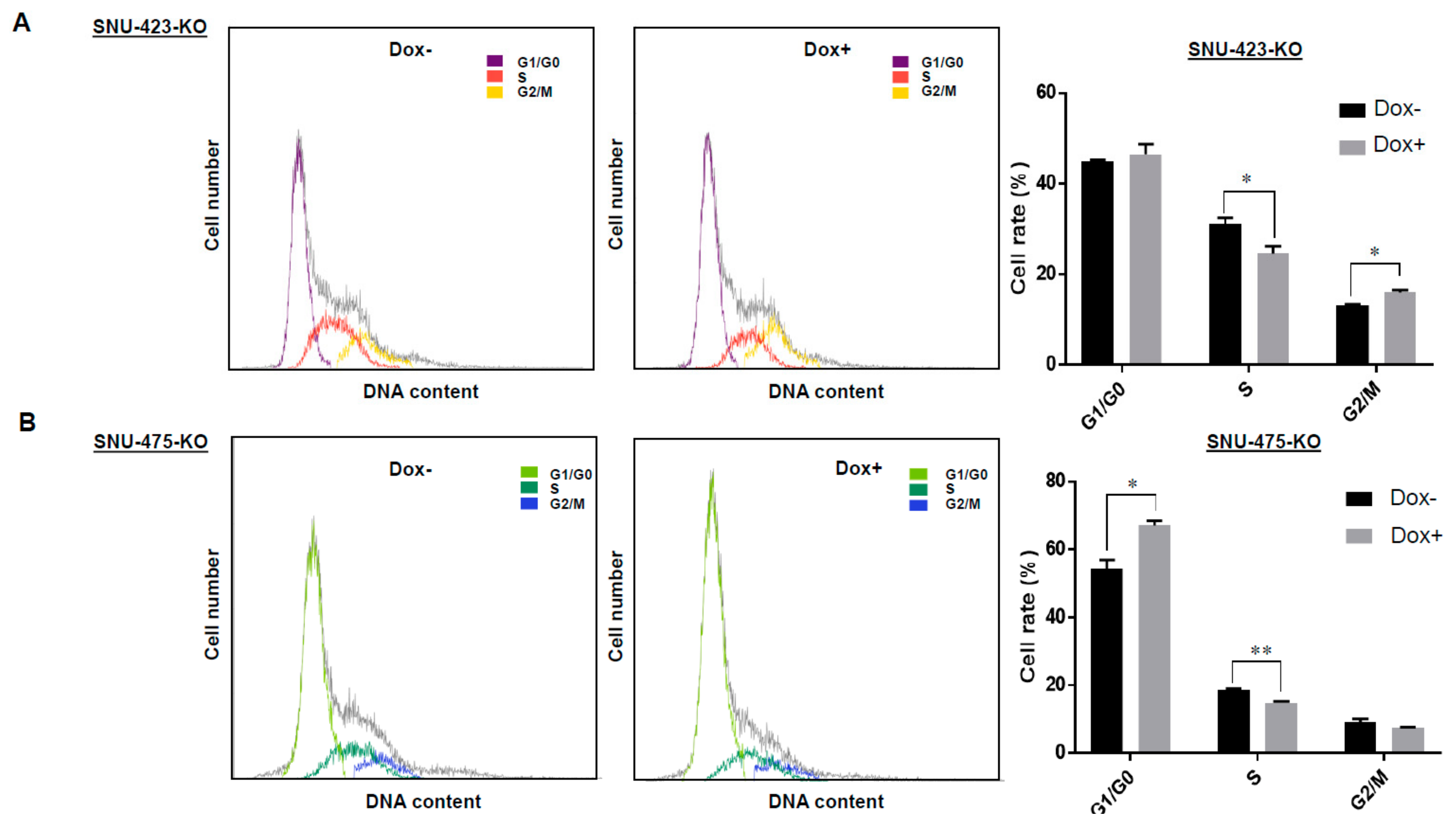
3.3. Establishment of Dox-Inducible LSD1 Knockout HCC Cells Using the CRISPR/Cas9 System
3.4. Effects of LSD1 on Cell Growth and Cell Cycle in HCC Cells
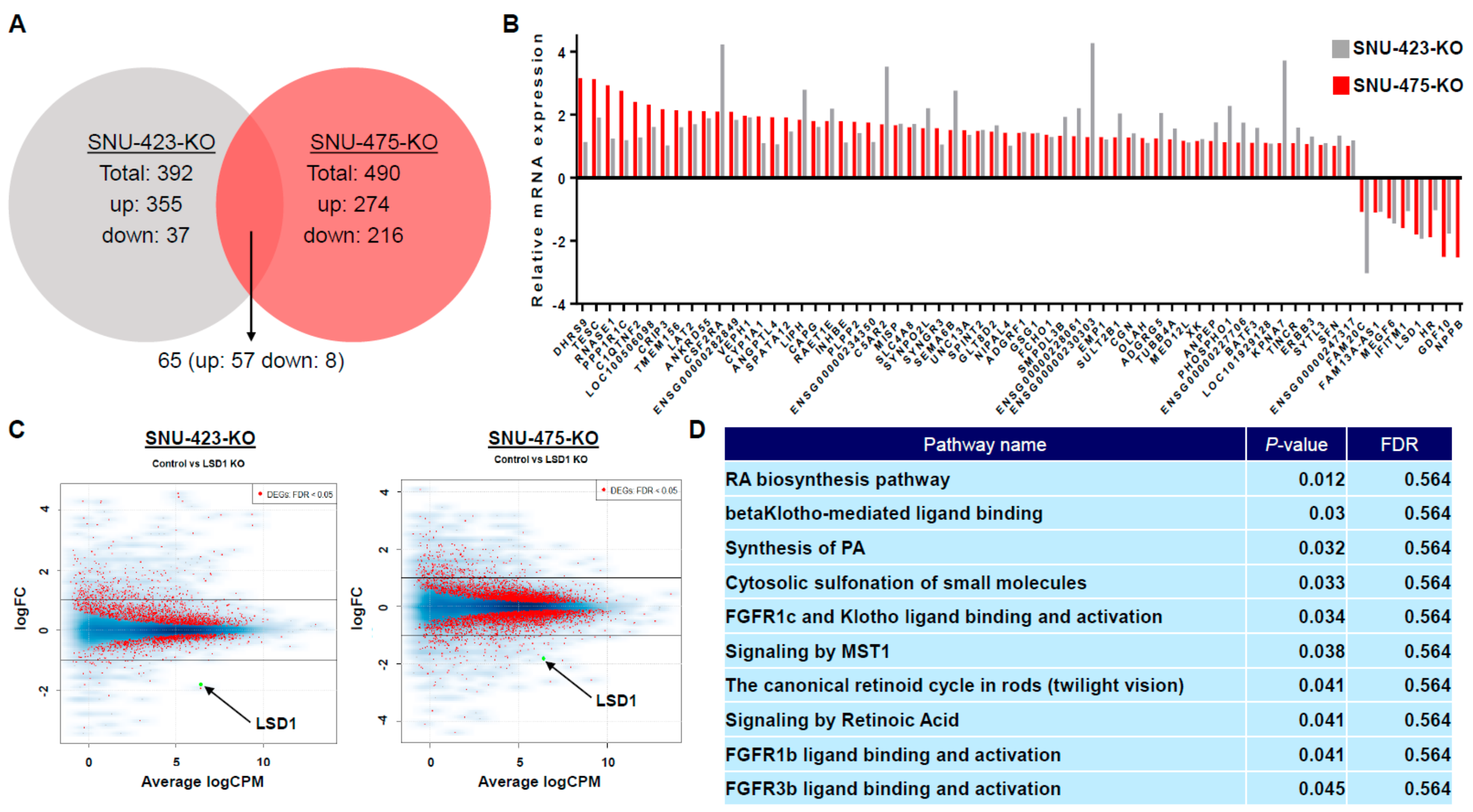
3.5. Transcriptome Analysis of LSD1 Knockout in HCC Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Gao, W.; Kondo, Y.; Shen, L.; Shimizu, Y.; Sano, T.; Yamao, K.; Natsume, A.; Goto, Y.; Ito, M.; Murakami, H.; et al. Variable DNA methylation patterns associated with progression of disease in hepatocellular carcinomas. Carcinogenesis 2008, 29, 1901–1910. [Google Scholar] [CrossRef]
- Herath, N.I.; Leggett, B.A.; MacDonald, G.A. Review of genetic and epigenetic alterations in hepatocarcinogenesis. J. Gastroenterol. Hepatol. 2006, 21, 15–21. [Google Scholar] [CrossRef]
- Nishida, N.; Goel, A. Genetic and epigenetic signatures in human hepatocellular carcinoma: A systematic review. Curr. Genom. 2011, 12, 130–137. [Google Scholar] [CrossRef]
- Pejin, B.; Jovanovic, K.K.; Mojovic, M.; Savic, A.G. New and highly potent antitumor natural products from marine-derived fungi: Covering the period from 2003 to 2012. Curr. Top. Med. Chem. 2013, 13, 2745–2766. [Google Scholar] [CrossRef]
- Cho, H.S.; Toyokawa, G.; Daigo, Y.; Hayami, S.; Masuda, K.; Ikawa, N.; Yamane, Y.; Maejima, K.; Tsunoda, T.; Field, H.I.; et al. The JmjC domain-containing histone demethylase KDM3A is a positive regulator of the G1/S transition in cancer cells via transcriptional regulation of the HOXA1 gene. Int. J. Cancer 2012, 131, E179–E189. [Google Scholar] [CrossRef]
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004, 6, 731–740. [Google Scholar] [CrossRef]
- Kang, D.; Cho, H.S.; Toyokawa, G.; Kogure, M.; Yamane, Y.; Iwai, Y.; Hayami, S.; Tsunoda, T.; Field, H.I.; Matsuda, K.; et al. The histone methyltransferase Wolf-Hirschhorn syndrome candidate 1-like 1 (WHSC1L1) is involved in human carcinogenesis. Genes Chromosomes Cancer 2013, 52, 126–139. [Google Scholar] [CrossRef]
- Toyokawa, G.; Cho, H.S.; Iwai, Y.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Shimizu, N.; Tanaka, H.; Tsunoda, T.; et al. The histone demethylase JMJD2B plays an essential role in human carcinogenesis through positive regulation of cyclin-dependent kinase 6. Cancer Prev. Res. 2011, 4, 2051–2061. [Google Scholar] [CrossRef]
- Toyokawa, G.; Cho, H.S.; Masuda, K.; Yamane, Y.; Yoshimatsu, M.; Hayami, S.; Takawa, M.; Iwai, Y.; Daigo, Y.; Tsuchiya, E.; et al. Histone lysine methyltransferase wolf-hirschhorn syndrome candidate 1 is involved in human carcinogenesis through regulation of the wnt pathway. Neoplasia 2011, 13, 887–898. [Google Scholar] [CrossRef]
- Zhou, Z.; Jiang, H.; Tu, K.; Yu, W.; Zhang, J.; Hu, Z.; Zhang, H.; Hao, D.; Huang, P.; Wang, J.; et al. ANKHD1 is required for SMYD3 to promote tumor metastasis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 18. [Google Scholar] [CrossRef]
- Yang, Z.H.; Dang, Y.Q.; Ji, G. Role of epigenetics in transformation of inflammation into colorectal cancer. World J. Gastroenterol. 2019, 25, 2863–2877. [Google Scholar] [CrossRef]
- Baylin, S.B. Resistance, epigenetics and the cancer ecosystem. Nat. Med. 2011, 17, 288–289. [Google Scholar] [CrossRef]
- Jung, H.; Kim, H.S.; Kim, J.Y.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Esteller, M.; Lee, S.H.; Choi, J.K. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 2019, 10, 4278. [Google Scholar] [CrossRef]
- Richon, V.M.; Johnston, D.; Sneeringer, C.J.; Jin, L.; Majer, C.R.; Elliston, K.; Jerva, L.F.; Scott, M.P.; Copeland, R.A. Chemogenetic analysis of human protein methyltransferases. Chem. Biol. Drug Des. 2011, 78, 199–210. [Google Scholar] [CrossRef]
- Cloos, P.A.; Christensen, J.; Agger, K.; Helin, K. Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008, 22, 1115–1140. [Google Scholar] [CrossRef]
- Hamamoto, R.; Nakamura, Y. Dysregulation of protein methyltransferases in human cancer: An emerging target class for anticancer therapy. Cancer Sci. 2016, 107, 377–384. [Google Scholar] [CrossRef]
- Hamamoto, R.; Saloura, V.; Nakamura, Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat. Rev. Cancer 2015, 15, 110–124. [Google Scholar] [CrossRef]
- Hayami, S.; Yoshimatsu, M.; Veerakumarasivam, A.; Unoki, M.; Iwai, Y.; Tsunoda, T.; Field, H.I.; Kelly, J.D.; Neal, D.E.; Yamaue, H.; et al. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: Involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol. Cancer 2010, 9, 59. [Google Scholar] [CrossRef]
- Yoshimatsu, M.; Toyokawa, G.; Hayami, S.; Unoki, M.; Tsunoda, T.; Field, H.I.; Kelly, J.D.; Neal, D.E.; Maehara, Y.; Ponder, B.A.; et al. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int. J. Cancer 2011, 128, 562–573. [Google Scholar] [CrossRef]
- Cho, H.S.; Hayami, S.; Toyokawa, G.; Maejima, K.; Yamane, Y.; Suzuki, T.; Dohmae, N.; Kogure, M.; Kang, D.; Neal, D.E.; et al. RB1 methylation by SMYD2 enhances cell cycle progression through an increase of RB1 phosphorylation. Neoplasia 2012, 14, 476–486. [Google Scholar] [CrossRef]
- Cho, H.S.; Kelly, J.D.; Hayami, S.; Toyokawa, G.; Takawa, M.; Yoshimatsu, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Ponder, B.A.; et al. Enhanced expression of EHMT2 is involved in the proliferation of cancer cells through negative regulation of SIAH1. Neoplasia 2011, 13, 676–684. [Google Scholar] [CrossRef]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Hayami, S.; Kelly, J.D.; Cho, H.S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.; et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef]
- Kahl, P.; Gullotti, L.; Heukamp, L.C.; Wolf, S.; Friedrichs, N.; Vorreuther, R.; Solleder, G.; Bastian, P.J.; Ellinger, J.; Metzger, E.; et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006, 66, 11341–11347. [Google Scholar] [CrossRef]
- Schulte, J.H.; Lim, S.; Schramm, A.; Friedrichs, N.; Koster, J.; Versteeg, R.; Ora, I.; Pajtler, K.; Klein-Hitpass, L.; Kuhfittig-Kulle, S.; et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: Implications for therapy. Cancer Res. 2009, 69, 2065–2071. [Google Scholar] [CrossRef]
- Romo-Morales, A.; Aladowicz, E.; Blagg, J.; Gatz, S.A.; Shipley, J.M. Catalytic inhibition of KDM1A in Ewing sarcoma is insufficient as a therapeutic strategy. Pediatr. Blood Cancer 2019, 66, e27888. [Google Scholar] [CrossRef]
- Shigekawa, Y.; Hayami, S.; Ueno, M.; Miyamoto, A.; Suzaki, N.; Kawai, M.; Hirono, S.; Okada, K.I.; Hamamoto, R.; Yamaue, H. Overexpression of KDM5B/JARID1B is associated with poor prognosis in hepatocellular carcinoma. Oncotarget 2018, 9, 34320–34335. [Google Scholar] [CrossRef]
- Kogure, M.; Takawa, M.; Cho, H.S.; Toyokawa, G.; Hayashi, K.; Tsunoda, T.; Kobayashi, T.; Daigo, Y.; Sugiyama, M.; Atomi, Y.; et al. Deregulation of the histone demethylase JMJD2A is involved in human carcinogenesis through regulation of the G(1)/S transition. Cancer Lett. 2013, 336, 76–84. [Google Scholar] [CrossRef]
- Kogure, M.; Takawa, M.; Saloura, V.; Sone, K.; Piao, L.; Ueda, K.; Ibrahim, R.; Tsunoda, T.; Sugiyama, M.; Atomi, Y.; et al. The oncogenic polycomb histone methyltransferase EZH2 methylates lysine 120 on histone H2B and competes ubiquitination. Neoplasia 2013, 15, 1251–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takawa, M.; Cho, H.S.; Hayami, S.; Toyokawa, G.; Kogure, M.; Yamane, Y.; Iwai, Y.; Maejima, K.; Ueda, K.; Masuda, A.; et al. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 2012, 72, 3217–3227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takawa, M.; Masuda, K.; Kunizaki, M.; Daigo, Y.; Takagi, K.; Iwai, Y.; Cho, H.S.; Toyokawa, G.; Yamane, Y.; Maejima, K.; et al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 2011, 102, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Saloura, V.; Cho, H.S.; Kyiotani, K.; Alachkar, H.; Zuo, Z.; Nakakido, M.; Tsunoda, T.; Seiwert, T.; Lingen, M.; Licht, J.; et al. WHSC1 promotes oncogenesis through regulation of NIMA-related-kinase-7 in squamous cell carcinoma of the head and neck. Mol. Cancer Res. 2015, 13, 293–304. [Google Scholar] [CrossRef] [Green Version]
- Toyokawa, G.; Masuda, K.; Daigo, Y.; Cho, H.S.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Chino, M.; Field, H.I.; et al. Minichromosome Maintenance Protein 7 is a potential therapeutic target in human cancer and a novel prognostic marker of non-small cell lung cancer. Mol. Cancer 2011, 10, 65. [Google Scholar] [CrossRef] [Green Version]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Alzayady, K.; Stewart, R.; Ye, P.; Yang, S.; Li, W.; Shi, Y. Histone demethylase LSD1 regulates neural stem cell proliferation. Mol. Cell. Biol. 2010, 30, 1997–2005. [Google Scholar] [CrossRef] [Green Version]
- Ancelin, K.; Syx, L.; Borensztein, M.; Ranisavljevic, N.; Vassilev, I.; Briseno-Roa, L.; Liu, T.; Metzger, E.; Servant, N.; Barillot, E.; et al. Maternal LSD1/KDM1A is an essential regulator of chromatin and transcription landscapes during zygotic genome activation. Elife 2016, 5, e08851. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.S.; Suzuki, T.; Dohmae, N.; Hayami, S.; Unoki, M.; Yoshimatsu, M.; Toyokawa, G.; Takawa, M.; Chen, T.; Kurash, J.K.; et al. Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells. Cancer Res. 2011, 71, 1–6. [Google Scholar] [CrossRef]
- Wang, M.; Liu, X.; Jiang, G.; Chen, H.; Guo, J.; Weng, X. Relationship between LSD1 expression and E-cadherin expression in prostate cancer. Int. Urol. Nephrol. 2015, 47, 485–490. [Google Scholar] [CrossRef]
- Althoff, K.; Beckers, A.; Odersky, A.; Mestdagh, P.; Koster, J.; Bray, I.M.; Bryan, K.; Vandesompele, J.; Speleman, F.; Stallings, R.L.; et al. MiR-137 functions as a tumor suppressor in neuroblastoma by downregulating KDM1A. Int. J. Cancer 2013, 133, 1064–1073. [Google Scholar] [CrossRef]
- Lv, T.; Yuan, D.; Miao, X.; Lv, Y.; Zhan, P.; Shen, X.; Song, Y. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS ONE 2012, 7, e35065. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.K.; Yu, H.F.; Wang, D.R.; Dong, P.; Chen, L.; Wu, W.G.; Ding, W.J.; Liu, Y.B. Overexpression of lysine specific demethylase 1 predicts worse prognosis in primary hepatocellular carcinoma patients. World J. Gastroenterol. 2012, 18, 6651–6656. [Google Scholar] [CrossRef]
- Wissmann, M.; Yin, N.; Muller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Gunther, T.; Buettner, R.; et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef]
- Piao, L.; Suzuki, T.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. SUV39H2 methylates and stabilizes LSD1 by inhibiting polyubiquitination in human cancer cells. Oncotarget 2015, 6, 16939–16950. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zhao, Y.; Wang, L.; Bohrer, L.R.; Pan, Y.; Wang, L.; Huang, H. LSD1 promotes S-phase entry and tumorigenesis via chromatin co-occupation with E2F1 and selective H3K9 demethylation. Oncogene 2018, 37, 534–543. [Google Scholar] [CrossRef]
- Shiota, G.; Kanki, K. Retinoids and their target genes in liver functions and diseases. J. Gastroenterol. Hepatol. 2013, 28, 33–37. [Google Scholar] [CrossRef]
- Cortes, E.; Lachowski, D.; Rice, A.; Chronopoulos, A.; Robinson, B.; Thorpe, S.; Lee, D.A.; Possamai, L.A.; Wang, H.; Pinato, D.J.; et al. Retinoic acid receptor-beta is downregulated in hepatocellular carcinoma and cirrhosis and its expression inhibits myosin-driven activation and durotaxis in hepatic stellate cells. Hepatology 2019, 69, 785–802. [Google Scholar] [CrossRef]
- Shimizu, M.; Imai, K.; Takai, K.; Moriwaki, H. Role of acyclic retinoid in the chemoprevention of hepatocellular carcinoma: Basic aspects, clinical applications, and future prospects. Curr. Cancer Drug Targets 2012, 12, 1119–1128. [Google Scholar]
- Xu, G.; Xiao, Y.; Hu, J.; Xing, L.; Zhao, O.; Wu, Y. The combined effect of retinoic acid and LSD1 siRNA inhibition on cell death in the human neuroblastoma cell line SH-SY5Y. Cell. Physiol. Biochem. 2013, 31, 854–862. [Google Scholar] [CrossRef] [Green Version]
- Smitheman, K.N.; Severson, T.M.; Rajapurkar, S.R.; McCabe, M.T.; Karpinich, N.; Foley, J.; Pappalardi, M.B.; Hughes, A.; Halsey, W.; Thomas, E.; et al. Lysine specific demethylase 1 inactivation enhances differentiation and promotes cytotoxic response when combined with all-trans retinoic acid in acute myeloid leukemia across subtypes. Haematologica 2019, 104, 1156–1167. [Google Scholar] [CrossRef] [Green Version]
- Dai, S.; Zhou, Z.; Chen, Z.; Xu, G.; Chen, Y. Fibroblast growth factor receptors (FGFRs): Structures and small molecule inhibitors. Cells 2019, 8, 614. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Chen, H.; Patterson, A.V.; Smaill, J.B.; Ding, K. Fibroblast growth factor receptor 4 (FGFR4) selective inhibitors as hepatocellular carcinoma therapy: Advances and prospects. J. Med. Chem. 2019, 62, 2905–2915. [Google Scholar] [CrossRef]
- Lin, Z.Z.; Hsu, C.; Jeng, Y.M.; Hu, F.C.; Pan, H.W.; Wu, Y.M.; Hsu, H.C.; Cheng, A.L. Klotho-beta and fibroblast growth factor 19 expression correlates with early recurrence of resectable hepatocellular carcinoma. Liver Int. 2019, 39, 1682–1691. [Google Scholar] [CrossRef]
- Repana, D.; Ross, P. Targeting FGF19/FGFR4 pathway: A novel therapeutic strategy for hepatocellular carcinoma. Diseases 2015, 3, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.B.; Park, Y.L.; Song, Y.A.; Kim, K.Y.; Lee, G.H.; Cho, D.H.; Myung, D.S.; Park, K.J.; Lee, W.S.; Chung, I.J.; et al. Small interfering RNA-directed targeting of RON alters invasive and oncogenic phenotypes of human hepatocellular carcinoma cells. Oncol. Rep. 2011, 26, 1581–1586. [Google Scholar]
- Matsushita, J.; Okamura, K.; Nakabayashi, K.; Suzuki, T.; Horibe, Y.; Kawai, T.; Sakurai, T.; Yamashita, S.; Higami, Y.; Ichihara, G.; et al. The DNA methylation profile of liver tumors in C3H mice and identification of differentially methylated regions involved in the regulation of tumorigenic genes. BMC Cancer 2018, 18, 317. [Google Scholar] [CrossRef]
- Sehrawat, A.; Gao, L.; Wang, Y.; Bankhead, A., 3rd; McWeeney, S.K.; King, C.J.; Schwartzman, J.; Urrutia, J.; Bisson, W.H.; Coleman, D.J.; et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc. Natl. Acad. Sci. USA 2018, 115, E4179–E4188. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | LSD1-Positive (n = 232) | LSD1-Negative (n = 71) | p-Value |
|---|---|---|---|
| Patient characteristics | |||
| Sex, Female | 66 (28) | 10 (14) | 0.018 |
| Male | 166 (72) | 61 (86) | |
| Age | 69 (62–75) | 72 (66–76) | 0.11 |
| Alcohol abuse | 80 (34) | 38 (54) | 0.0027 |
| Smoking | 90 (39) | 34 (48) | 0.094 |
| Hepatitis status | |||
| HBV Ag positive | 42 (18) | 8 (11) | 0.39 |
| HCV Ab positive | 139 (60) | 32 (45) | 0.038 |
| No infection | 52 (22) | 27 (38) | 0.013 |
| Preoperative laboratory tests | |||
| Albumin, g/dL | 4.2 (3.8–4.4) | 4.1 (3.8–4.4) | 0.78 |
| Total bilirubin, mg/dL | 0.8 (0.6–1) | 0.7 (0.6–1) | 0.16 |
| Prothrombin time, % | 87.0 (80.1–96.3) | 87.8 (78.3–97.9) | 0.63 |
| ICG R15, % | 13.0 (9.0–19.0) | 12.1 (9.0–16.5) | 0.32 |
| AST | 45 (30.3–64.8) | 37 (25.5–62.5) | 0.054 |
| ALT | 39 (27.0–64.8) | 36 (21.5–55.5) | 0.34 |
| Platelet count, ×104/μL | 14.1 (10.2–19.0) | 16.6 (13.2–19.6) | 0.012 |
| Child Pugh grade | 1 | ||
| A | 216 (93) | 67 (94) | |
| B | 16 (7) | 4 (6) | |
| C | - | - | |
| AFP, ng/mL | 33.8 (6.8–377.1) | 7.1 (4.1–29.9) | <0.001 |
| PIVKA-II, mAU/mL | 221 (43.0–2333.0) | 117 (32.5–875.0) | 0.054 |
| CEA, ng/mL | 2.1 (1.2–3.0) | 1.8 (0.9–2.8) | 0.1 |
| CA 19-9, U/mL | 10.8 (5.8–19.5) | 9.1 (4.2–14.1) | 0.017 |
| Pathological characteristics | |||
| Tumor maximum size, cm | 3.5 (2.5–5.6) | 4.0 (2.5–6.1) | 0.47 |
| Tumor number, multiple | 51 (22) | 12 (17) | 0.4 |
| HCC differentiation | 0.0054 | ||
| Well | 49 (21) | 23 (32) | |
| Moderate | 132 (57) | 43 (61) | |
| Poor | 51 (22) | 5 (7) | |
| Hepatic vein invasion | 43 (19) | 5 (7) | 0.017 |
| Portal vein invasion | 60 (26) | 14 (20) | 0.34 |
| Tumor category | 0.057 | ||
| T1 | 30 (13) | 8 (11) | |
| T2 | 116 (50) | 41 (58) | |
| T3 | 55 (24) | 20 (28) | |
| T4 | 31 (13) | 2 (3) | |
| Fibrosis staging F4 | 119 (51) | 23 (32) | 0.0061 |
| Activity grading A2-3 | 72 (31) | 18 (25) | 0.45 |
| TNM stage (UICC) | 0.051 | ||
| I | 32 (14) | 7 (10) | |
| II | 114 (49) | 42 (59) | |
| III | 54 (23) | 20 (28) | |
| IVA | 31 (13) | 2 (3) | |
| IVB | 1 (0.4) | - | |
| Variables | Univariate Analysis | |||||
|---|---|---|---|---|---|---|
| OS | DFS | |||||
| HR | 95% CI | p-Value | HR | 95% CI | p-Value | |
| Sex (male vs. female) | 0.89 | 0.60–1.33 | 0.57 | 0.93 | 0.68–1.28 | 0.66 |
| Age (≤66 vs. >66) | 1.16 | 0.80–1.66 | 0.43 | 0.93 | 0.70–1.22 | 0.59 |
| HBV infection (yes or no) | 1.09 | 0.68–1.75 | 0.72 | 1.02 | 0.71–1.48 | 0.91 |
| HCV infection (yes or no) | 1.35 | 0.94–1.94 | 0.099 | 1.23 | 0.93–1.62 | 0.14 |
| No infection (yes or no) | 0.66 | 0.43–1.01 | 0.056 | 0.83 | 0.60–1.13 | 0.23 |
| Child Pugh grade (B or C vs. A) | 1.96 | 1.08–3.55 | 0.027 | 1.58 | 0.93–2.67 | 0.09 |
| AFP (≤20 vs. >20 ng/mL) | 2.35 | 1.64–3.89 | <0.001 | 1.68 | 1.28–2.21 | <0.001 |
| PIVKA-II (<40 vs. ≥40 mAU/mL) | 1.42 | 0.93–2.17 | 0.11 | 1.30 | 0.94–1.79 | 0.11 |
| CEA (≤5 vs. >5 ng/mL) | 0.81 | 0.36–1.85 | 0.62 | 0.84 | 0.47–1.50 | 0.55 |
| CA19-9 (≤37 vs. >37 U/mL) | 1.20 | 0.61–2.37 | 0.6 | 0.87 | 0.48–1.56 | 0.64 |
| Tumor size (≤5 vs. >5 cm) | 1.45 | 1.01–2.10 | 0.047 | 1.42 | 1.06–1.91 | 0.018 |
| Tumor number (multiple vs. single) | 2.14 | 1.46–3.14 | <0.001 | 2.39 | 1.75–3.26 | <0.001 |
| Differentiation (mod/por vs. wel) | 1.57 | 1.01–2.43 | 0.043 | 1.48 | 1.06–2.07 | 0.021 |
| Hepatic vein invasion (yes vs. no) | 2.36 | 1.56–3.55 | <0.001 | 2.40 | 1.70–3.37 | <0.001 |
| Portal vein invasion (yes vs. no) | 2.13 | 1.48–3.08 | <0.001 | 1.93 | 1.43–2.60 | <0.001 |
| Tumor category (T3–4 vs. 1–2) | 3.00 | 2.12–4.25 | <0.001 | 2.44 | 1.85–3.22 | <0.001 |
| Fibrosis stage (4 vs. 0–3) | 1.59 | 1.12–2.26 | 0.0099 | 1.61 | 1.23–2.12 | <0.001 |
| Activity stage (2–3 vs. 0–1) | 1.01 | 0.69–1.48 | 0.95 | 1.18 | 0.88–1.54 | 0.27 |
| TNM stage (III–IV vs. I–II) | 2.97 | 2.09–4.22 | <0.001 | 2.57 | 1.95–3.39 | <0.001 |
| LSD1 (positive vs. negative) | 2.16 | 1.31–3.56 | 0.0024 | 1.75 | 1.24–2.48 | 0.0016 |
| Variables | Multivariate Analysis | |||||
|---|---|---|---|---|---|---|
| OS | DFS | |||||
| HR | 95% CI | p-Value | HR | 95% CI | p-Value | |
| HCV infection (yes or no) | 1.09 | 0.70–1.69 | 0.70 | 1.06 | 0.76–1.48 | 0.74 |
| Child Pugh grade (B or C vs. A) | 1.53 | 0.71–3.30 | 0.27 | 1.65 | 0.88–3.08 | 0.17 |
| AFP (≤20 vs. >20 ng/mL) | 1.66 | 1.07–2.57 | 0.0025 | 1.28 | 0.91–1.79 | 0.15 |
| PIVKA-II (<40 vs. ≥40 mAU/mL) | 1.178 | 0.71–1.96 | 0.53 | 1.07 | 0.74–1.55 | 0.73 |
| Tumor size (≤5 vs. >5 cm) | 1.08 | 0.65–1.80 | 0.76 | 1.06 | 0.71–1.59 | 0.78 |
| Tumor number (multiple vs. single) | 1.20 | 0.71–2.01 | 0.49 | 1.33 | 0.85–2.07 | 0.21 |
| Differentiation (mod/por vs. wel) | 1.07 | 0.62–1.85 | 0.80 | 0.71 | 0.45–1.13 | 0.15 |
| Hepatic vein invasion (yes vs. no) | 1.02 | 0.57–1.81 | 0.96 | 1.14 | 0.70–1.83 | 0.60 |
| Portal vein invasion (yes vs. no) | 1.18 | 0.72–1.93 | 0.52 | 1.24 | 0.85–1.81 | 0.27 |
| Tumor category (T3–4 vs. 1–2) | 2.26 | 0.37–13.8 | 0.38 | 0.56 | 0.13–2.33 | 0.42 |
| Fibrosis stage (4 vs. 0–3) | 1.23 | 0.65–2.51 | 0.48 | 1.43 | 0.82–2.51 | 0.21 |
| TNM stage (III–IV vs. I–II) | 0.96 | 0.16–5.87 | 0.97 | 3.51 | 0.84–14.7 | 0.086 |
| LSD1 (positive vs. negative) | 1.98 | 1.07–3.64 | 0.029 | 1.74 | 1.15–2.64 | 0.0086 |
| Variables | Multivariate Analysis | |||
|---|---|---|---|---|
| HR | 95% CI | p-Value | ||
| OS | AFP (≤20 vs. >20 ng/mL) | 1.76 | 1.18–2.61 | 0.005 |
| Tumor category (T3–4 vs. 1–2) | 3.01 | 2.05–4.43 | <0.001 | |
| LSD1 (positive vs. negative) | 2.18 | 1.22–3.92 | 0.009 | |
| DFS | Fibrosis stage (4 vs. 0–3) | 1.70 | 1.06–2.70 | 0.0026 |
| TNM stage (III–IV vs. I–II) | 2.22 | 1.52–3.24 | <0.001 | |
| LSD1 (positive vs. negative) | 1.67 | 1.12–2.48 | 0.012 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Bolatkan, A.; Kaneko, S.; Ikawa, N.; Asada, K.; Komatsu, M.; Hayami, S.; Ojima, H.; Abe, N.; Yamaue, H.; et al. Deregulation of the Histone Lysine-Specific Demethylase 1 Is Involved in Human Hepatocellular Carcinoma. Biomolecules 2019, 9, 810. https://doi.org/10.3390/biom9120810
Kim S, Bolatkan A, Kaneko S, Ikawa N, Asada K, Komatsu M, Hayami S, Ojima H, Abe N, Yamaue H, et al. Deregulation of the Histone Lysine-Specific Demethylase 1 Is Involved in Human Hepatocellular Carcinoma. Biomolecules. 2019; 9(12):810. https://doi.org/10.3390/biom9120810
Chicago/Turabian StyleKim, Sangchul, Amina Bolatkan, Syuzo Kaneko, Noriko Ikawa, Ken Asada, Masaaki Komatsu, Shinya Hayami, Hidenori Ojima, Nobutsugu Abe, Hiroki Yamaue, and et al. 2019. "Deregulation of the Histone Lysine-Specific Demethylase 1 Is Involved in Human Hepatocellular Carcinoma" Biomolecules 9, no. 12: 810. https://doi.org/10.3390/biom9120810