Integrated Transcriptome Analysis Reveals Plant Hormones Jasmonic Acid and Salicylic Acid Coordinate Growth and Defense Responses upon Fungal Infection in Poplar

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Phytohormone Treatments
2.2. RNA Extraction and Transcriptome Sequencing
2.3. Quantitative Real-time PCR Assay
2.4. Bioinformatic Analysis
3. Results and Discussion
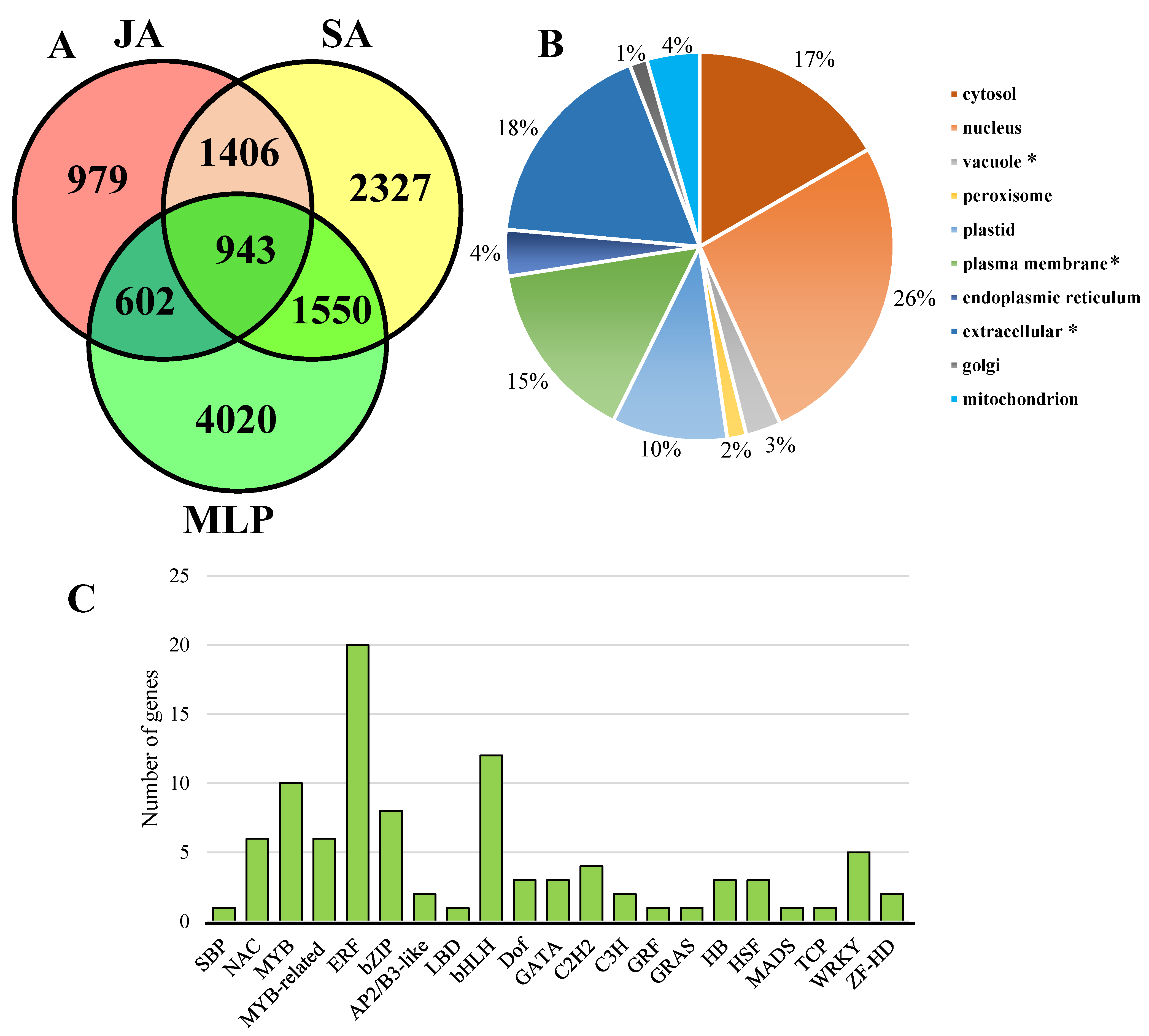
3.1. Identification of Jasmonic Acid-Responsive and Salicylic Acid-Responsive Genes
3.2. Characterization of the Roles of Jasmonic Acid-Responsive and Salicylic Acid-Responsive Genes in the Regulation of Melampsora Defense by Comparative Transcriptome Analysis
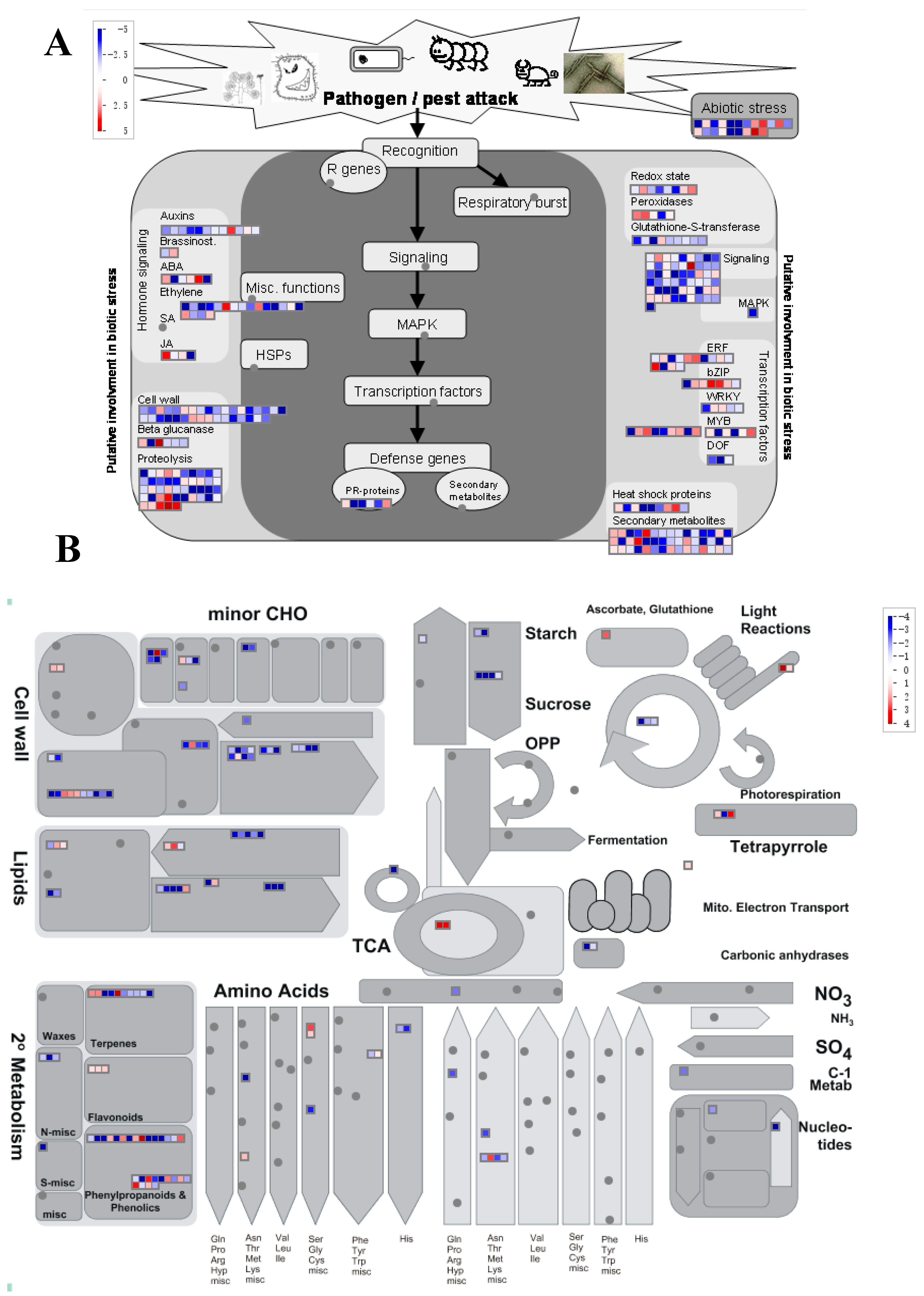
3.3. Jasmonic Acid and Salicylic Acid May Exhibit Different Effects on Plant Fungal Defense
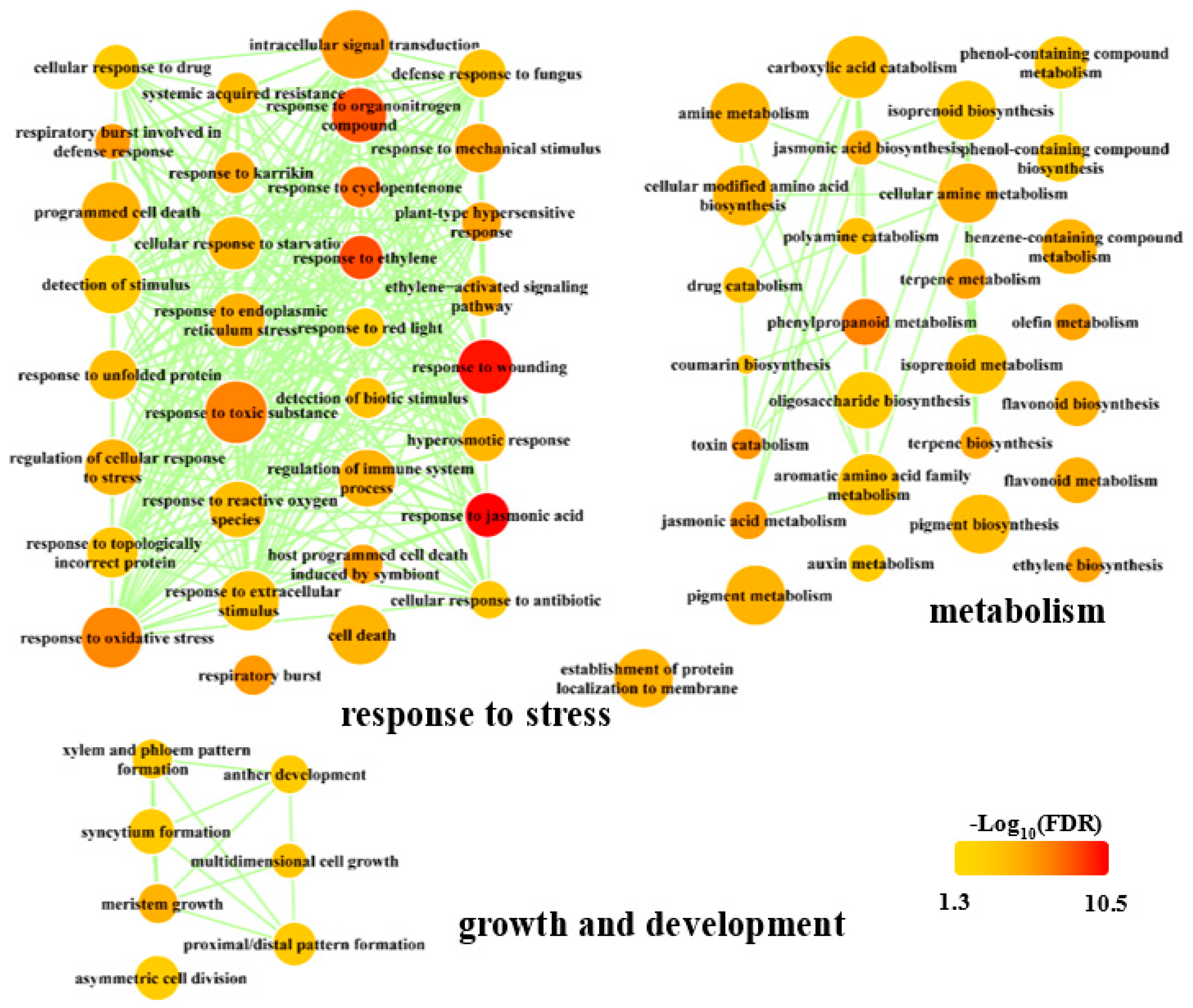
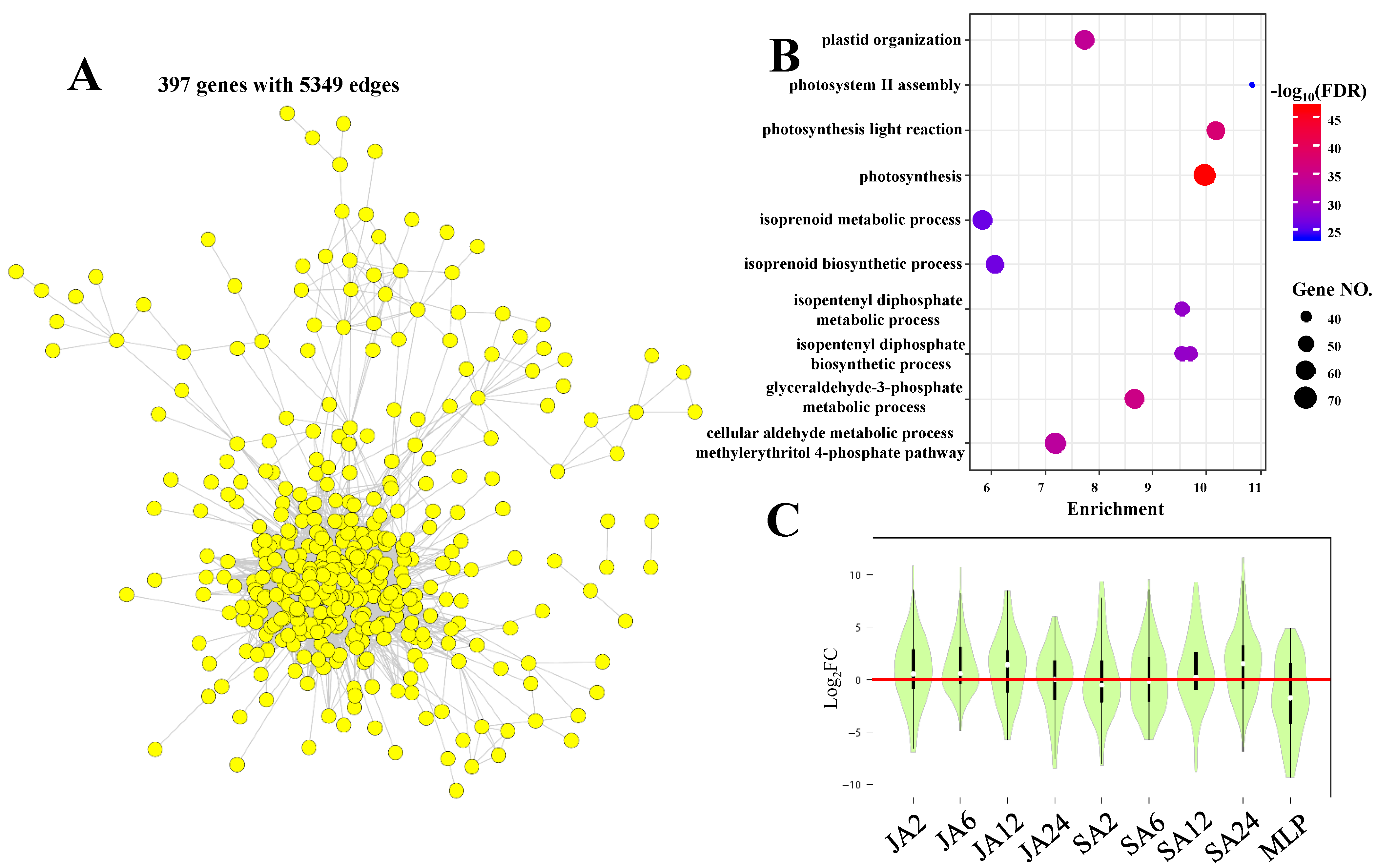
3.4. Common Responsive Genes are Involved in Key Pathways that are Important for Plant Fitness
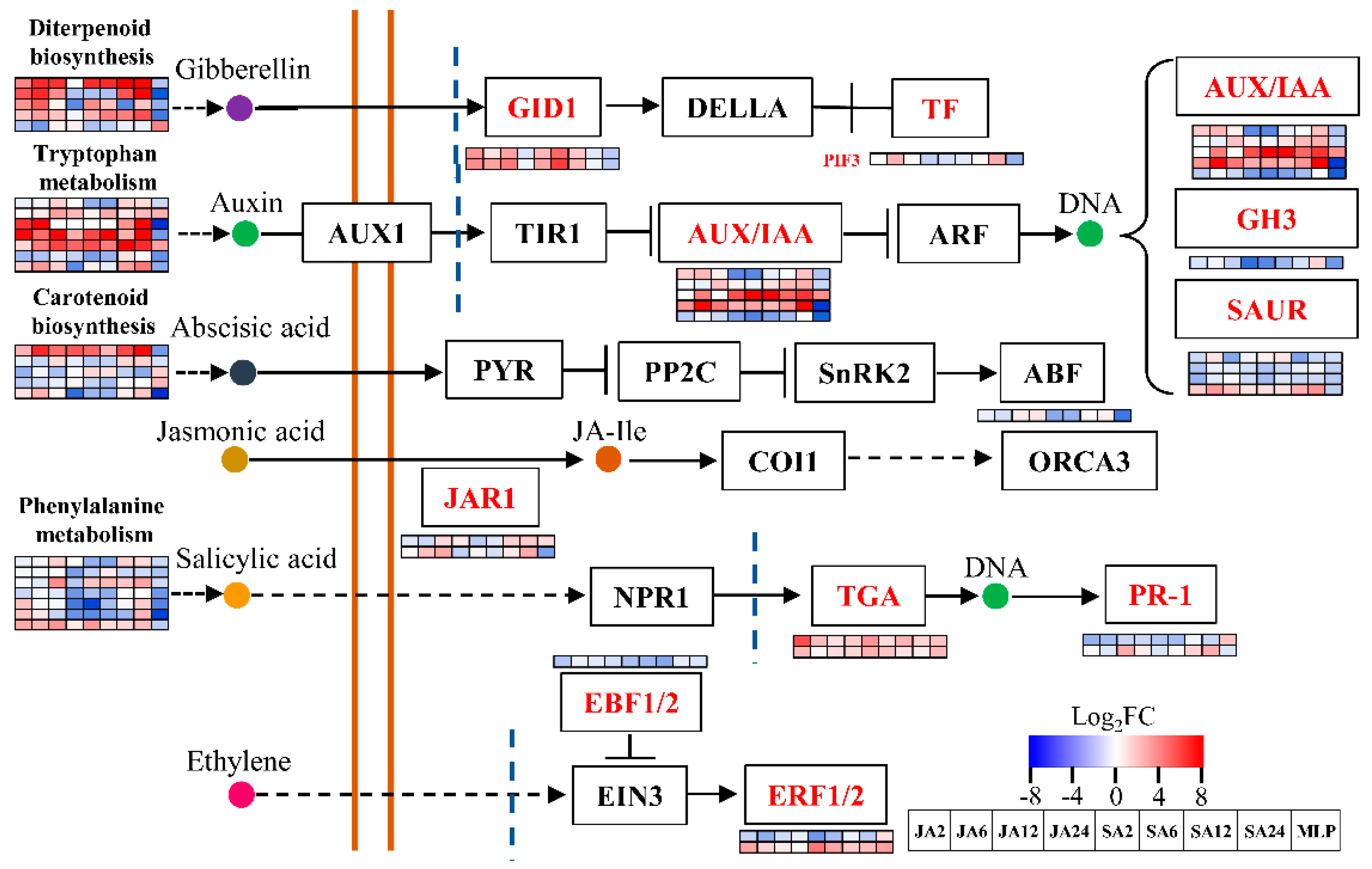
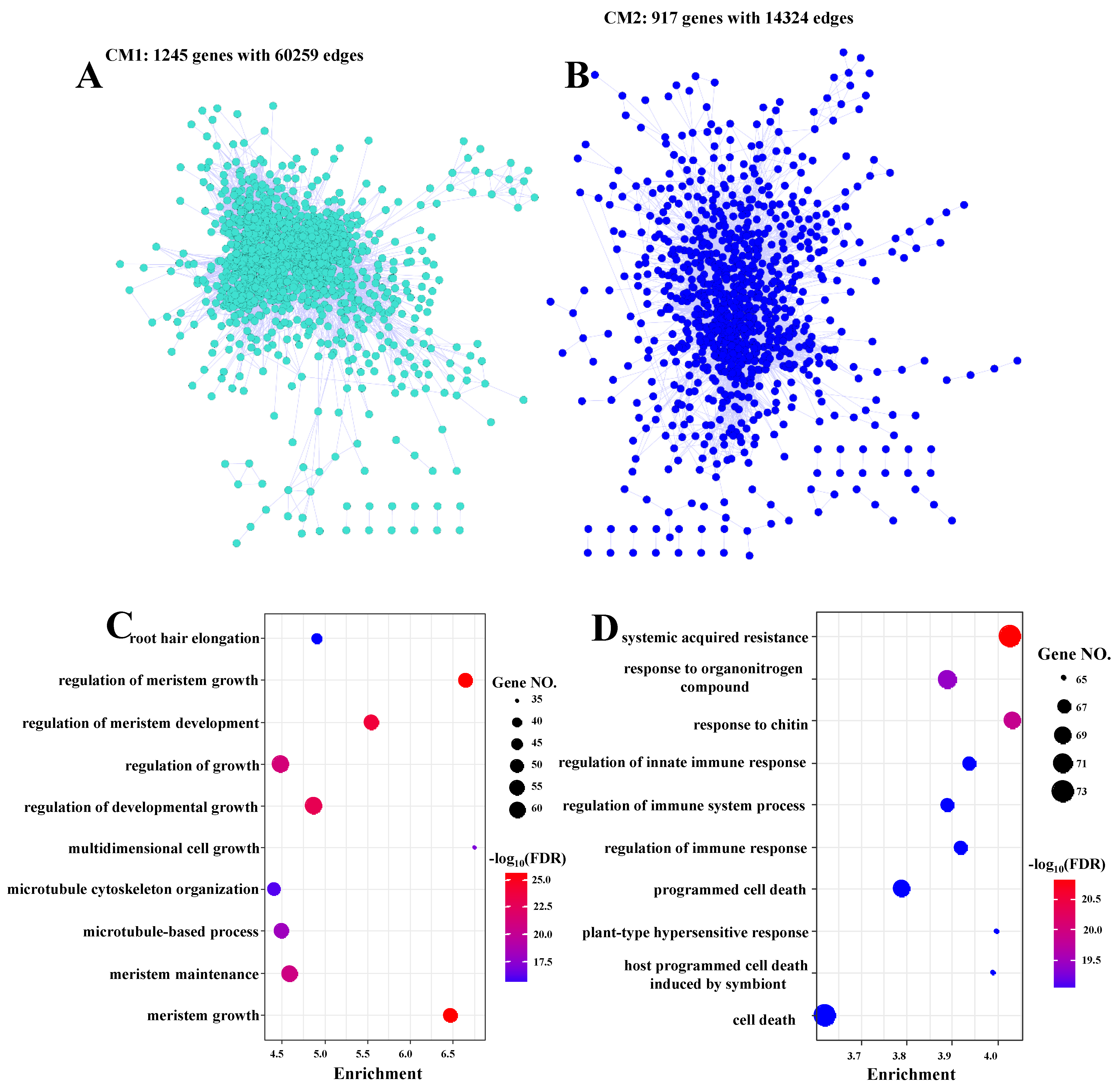
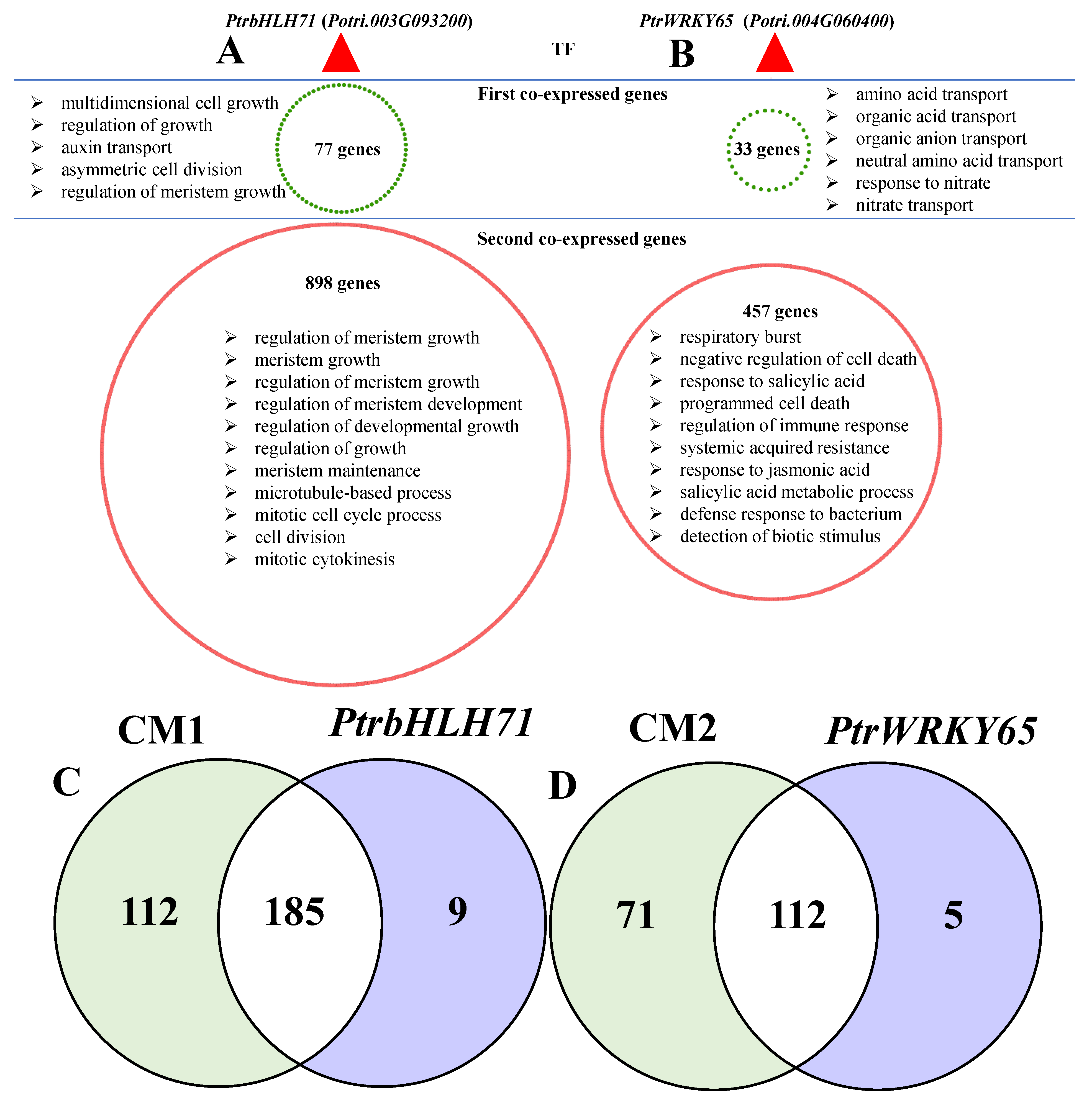
3.5. Co-Expression Networks Confirmed the Possible Roles of Jasmonic Acid and Salicylic Acid in Response to Fungal Defense by Balancing Growth and Biotic Stress Responses
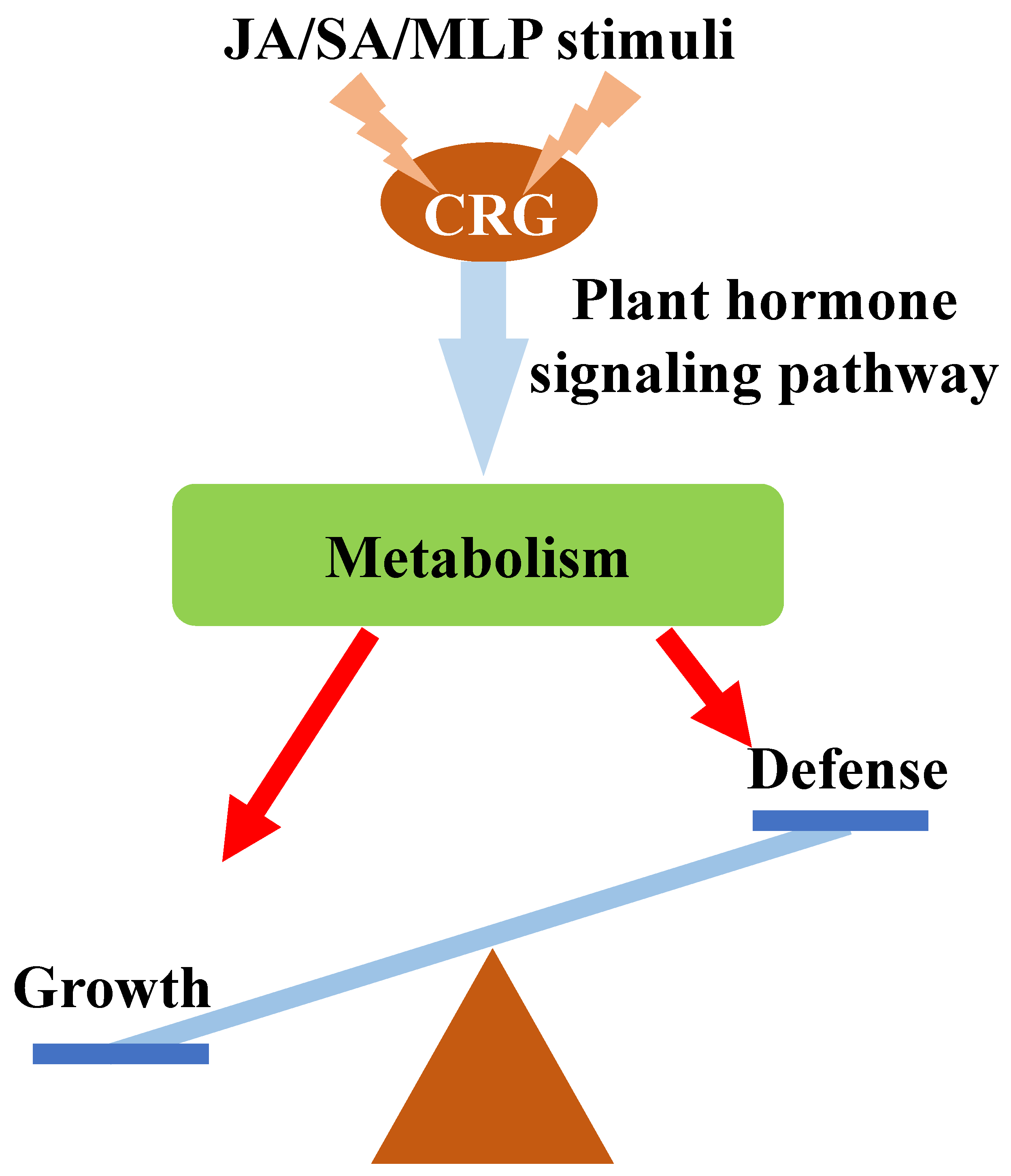
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dempsey, D.A.; Vlot, A.C.; Wildermuth, M.C.; Klessig, D.F. Salicylic Acid biosynthesis and metabolism. Arabidopsis Book 2011, 9, e0156. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, C.M.; Van der Does, D.; Zamioudis, C.; Leon-Reyes, A.; Van Wees, S.C. Hormonal modulation of plant immunity. Annu. Rev. Cell Dev. Biol. 2012, 28, 489–521. [Google Scholar] [CrossRef] [PubMed]
- Druege, U.; Franken, P.; Hajirezaei, M.R. Plant Hormone Homeostasis, Signaling, and Function during Adventitious Root Formation in Cuttings. Front. Plant Sci. 2016, 7, 381. [Google Scholar] [CrossRef] [PubMed]
- Campos-Rivero, G.; Osorio-Montalvo, P.; Sanchez-Borges, R.; Us-Camas, R.; Duarte-Ake, F.; De-la-Pena, C. Plant hormone signaling in flowering: An epigenetic point of view. J. Plant Physiol. 2017, 214, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Atamian, H.S.; Harmer, S.L. Circadian regulation of hormone signaling and plant physiology. Plant Mol. Biol. 2016, 91, 691–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chini, A.; Fonseca, S.; Fernandez, G.; Adie, B.; Chico, J.M.; Lorenzo, O.; Garcia-Casado, G.; Lopez-Vidriero, I.; Lozano, F.M.; Ponce, M.R.; et al. The JAZ family of repressors is the missing link in jasmonate signalling. Nature 2007, 448, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Stolz, S.; Chetelat, A.; Reymond, P.; Pagni, M.; Dubugnon, L.; Farmer, E.E. A downstream mediator in the growth repression limb of the jasmonate pathway. Plant Cell 2007, 19, 2470–2483. [Google Scholar] [CrossRef]
- Thines, B.; Katsir, L.; Melotto, M.; Niu, Y.; Mandaokar, A.; Liu, G.; Nomura, K.; He, S.Y.; Howe, G.A.; Browse, J. JAZ repressor proteins are targets of the SCF(COI1) complex during jasmonate signalling. Nature 2007, 448, 661–665. [Google Scholar] [CrossRef]
- Dempsey, D.A.; Klessig, D.F. How does the multifaceted plant hormone salicylic acid combat disease in plants and are similar mechanisms utilized in humans? BMC Biol. 2017, 15, 23. [Google Scholar] [CrossRef]
- Vlot, A.C.; Dempsey, D.A.; Klessig, D.F. Salicylic Acid, a multifaceted hormone to combat disease. Annu. Rev. Phytopathol. 2009, 47, 177–206. [Google Scholar] [CrossRef]
- Van der Does, D.; Leon-Reyes, A.; Koornneef, A.; Van Verk, M.C.; Rodenburg, N.; Pauwels, L.; Goossens, A.; Korbes, A.P.; Memelink, J.; Ritsema, T.; et al. Salicylic acid suppresses jasmonic acid signaling downstream of SCFCOI1-JAZ by targeting GCC promoter motifs via transcription factor ORA59. Plant Cell 2013, 25, 744–761. [Google Scholar] [CrossRef] [PubMed]
- Lemarie, S.; Robert-Seilaniantz, A.; Lariagon, C.; Lemoine, J.; Marnet, N.; Jubault, M.; Manzanares-Dauleux, M.J.; Gravot, A. Both the Jasmonic Acid and the Salicylic Acid Pathways Contribute to Resistance to the Biotrophic Clubroot Agent Plasmodiophora brassicae in Arabidopsis. Plant Cell Physiol. 2015, 56, 2158–2168. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, R.; Heise, A.M.; Persicke, M.; Muller, C. Interactions between the jasmonic and salicylic acid pathway modulate the plant metabolome and affect herbivores of different feeding types. Plant Cell Environ. 2014, 37, 1574–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libault, M.; Farmer, A.; Joshi, T.; Takahashi, K.; Langley, R.J.; Franklin, L.D.; He, J.; Xu, D.; May, G.; Stacey, G. An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant J. 2010, 63, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Kusunoki, K.; Nakano, Y.; Tanaka, K.; Sakata, Y.; Koyama, H.; Kobayashi, Y. Transcriptomic variation among six Arabidopsis thaliana accessions identified several novel genes controlling aluminium tolerance. Plant Cell Environ. 2017, 40, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Righetti, K.; Vu, J.L.; Pelletier, S.; Vu, B.L.; Glaab, E.; Lalanne, D.; Pasha, A.; Patel, R.V.; Provart, N.J.; Verdier, J.; et al. Inference of Longevity-Related Genes from a Robust Coexpression Network of Seed Maturation Identifies Regulators Linking Seed Storability to Biotic Defense-Related Pathways. Plant Cell 2015, 27, 2692–2708. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef]
- Miranda, M.; Ralph, S.G.; Mellway, R.; White, R.; Heath, M.C.; Bohlmann, J.; Constabel, C.P. The transcriptional response of hybrid poplar (Populus trichocarpa × P. deltoides) to infection by Melampsora medusae leaf rust involves induction of flavonoid pathway genes leading to the accumulation of proanthocyanidins. Mol. Plant Microbe Interact. 2007, 20, 816–831. [Google Scholar] [CrossRef]
- Rinaldi, C.; Kohler, A.; Frey, P.; Duchaussoy, F.; Ningre, N.; Couloux, A.; Wincker, P.; Le Thiec, D.; Fluch, S.; Martin, F.; et al. Transcript profiling of poplar leaves upon infection with compatible and incompatible strains of the foliar rust Melampsora larici-populina. Plant Physiol. 2007, 144, 347–366. [Google Scholar] [CrossRef]
- Liang, H.; Staton, M.; Xu, Y.; Xu, T.; Leboldus, J. Comparative expression analysis of resistant and susceptible Populus clones inoculated with Septoria musiva. Plant Sci. 2014, 223, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yao, Y.; Zhang, L.; Xu, M.; Jiang, J.; Dou, T.; Lin, W.; Zhao, G.; Huang, M.; Zhou, Y. A Comprehensive Analysis of the Transcriptomes of Marssonina brunnea and Infected Poplar Leaves to Capture Vital Events in Host-Pathogen Interactions. PLoS ONE 2015, 10, e0134246. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Cao, P.; Xia, W.; Fang, L.; Yu, H. Identification and characterization of long non-coding RNAs in response to early infection by Melampsora larici-populina using genome-wide high-throughput RNA sequencing. Tree Genet. Genomes 2017, 13, 34. [Google Scholar] [CrossRef]
- Xia, W.; Yu, H.; Cao, P.; Luo, J.; Wang, N. Identification of TIFY Family Genes and Analysis of Their Expression Profiles in Response to Phytohormone Treatments and Melampsora larici-populina Infection in Poplar. Front. Plant Sci. 2017, 8, 493. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Chan, C.K. Analysis of RNA-Seq Data Using TopHat and Cufflinks. Methods Mol. Biol. 2016, 1374, 339–361. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.M.; Castleden, I.R.; Tanz, S.K.; Aryamanesh, N.; Millar, A.H. SUBA4: The interactive data analysis centre for Arabidopsis subcellular protein locations. Nucleic Acids Res. 2016, 45, D1064–D1074. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R.; Kolde, M.R. Package ‘Pheatmap’ Version1.0.10. 2018. Available online: https://cran.r-project.org/web/packages/pheatmap (accessed on 30 May 2018).
- Adler, D. Vioplot: Violin Plot. R Package Version 0.2. 2005. Available online: http://CRAN.R-project.org/package=vioplot (accessed on 6 March 2018).
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant graphics for data analysis. J. Stat. Softw. 2010, 35, 65–88. [Google Scholar]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; Rokhsar, D.S. Phytozome: A comparative platform for green plant genomics. Nucleic acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Grennan, A.K. Ethylene Response Factors in Jasmonate Signaling and Defense Response. Plant Physiol. 2008, 146, 1457–1458. [Google Scholar] [CrossRef] [Green Version]
- Gutterson, N.; Reuber, T.L. Regulation of disease resistance pathways by AP2/ERF transcription factors. Curr. Opin. Plant Biol. 2004, 7, 465–471. [Google Scholar] [CrossRef]
- McGrath, K.C.; Dombrecht, B.; Manners, J.M.; Schenk, P.M.; Edgar, C.I.; Maclean, D.J.; Scheible, W.-R.; Udvardi, M.K.; Kazan, K. Repressor-and activator-type ethylene response factors functioning in jasmonate signaling and disease resistance identified via a genome-wide screen of Arabidopsis transcription factor gene expression. Plant Physiol. 2005, 139, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.L.; Kazan, K.; McGrath, K.C.; Maclean, D.J.; Manners, J.M. A role for the GCC-box in jasmonate-mediated activation of the PDF1. 2 gene of Arabidopsis. Plant Physiol. 2003, 132, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Oñate-Sánchez, L.; Anderson, J.P.; Young, J.; Singh, K.B. AtERF14, a Member of the ERF Family of Transcription Factors, Plays a Nonredundant Role in Plant Defense. Plant Physiol. 2007, 143, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Pré, M.; Atallah, M.; Champion, A.; De Vos, M.; Pieterse, C.M.J.; Memelink, J. The AP2/ERF Domain Transcription Factor ORA59 Integrates Jasmonic Acid and Ethylene Signals in Plant Defense. Plant Physiol. 2008, 147, 1347–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Xu, J.; He, Y.; Yang, K.-Y.; Mordorski, B.; Liu, Y.; Zhang, S. Phosphorylation of an ERF Transcription Factor by Arabidopsis MPK3/MPK6 Regulates Plant Defense Gene Induction and Fungal Resistance. Plant Cell Online 2013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yao, W.; Dong, N.; Liang, H.; Liu, H.; Huang, R. A novel ERF transcription activator in wheat and its induction kinetics after pathogen and hormone treatments. J. Exp. Bot. 2007, 58, 2993–3003. [Google Scholar] [CrossRef] [Green Version]
- Dong, N.; Liu, X.; Lu, Y.; Du, L.; Xu, H.; Liu, H.; Xin, Z.; Zhang, Z. Overexpression of TaPIEP1, a pathogen-induced ERF gene of wheat, confers host-enhanced resistance to fungal pathogen Bipolaris sorokiniana. Funct. Integr. Genom. 2010, 10, 215–226. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, M.; Li, L.; Xu, Z.; Chen, X.; Guo, J.; Ma, Y. Overexpression of the soybean GmERF3 gene, an AP2/ERF type transcription factor for increased tolerances to salt, drought, and diseases in transgenic tobacco. J. Exp. Bot. 2009, 60, 3781–3796. [Google Scholar] [CrossRef]
- Liu, D.; Chen, X.; Liu, J.; Ye, J.; Guo, Z. The rice ERF transcription factor OsERF922 negatively regulates resistance to Magnaporthe oryzae and salt tolerance. J. Exp. Bot. 2012, 63, 3899–3911. [Google Scholar] [CrossRef]
- Jisha, V.; Dampanaboina, L.; Vadassery, J.; Mithöfer, A.; Kappara, S.; Ramanan, R. Overexpression of an AP2/ERF type transcription factor OsEREBP1 confers biotic and abiotic stress tolerance in rice. PLoS ONE 2015, 10, e0127831. [Google Scholar] [CrossRef]
- Lu, J.; Ju, H.; Zhou, G.; Zhu, C.; Erb, M.; Wang, X.; Wang, P.; Lou, Y. An EAR-motif-containing ERF transcription factor affects herbivore-induced signaling, defense and resistance in rice. Plant J. 2011, 68, 583–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Qi, T.; Fan, M.; Zhang, X.; Gao, H.; Huang, H.; Wu, D.; Guo, H.; Xie, D. The bHLH subgroup IIId factors negatively regulate jasmonate-mediated plant defense and development. PLoS Genet. 2013, 9, e1003653. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Duan, Y.; Yin, J.; Ye, S.; Zhu, J.; Zhang, F.; Lu, W.; Fan, D.; Luo, K. Genome-wide identification and characterization of the Populus WRKY transcription factor family and analysis of their expression in response to biotic and abiotic stresses. J. Exp. Bot. 2014, 65, 6629–6644. [Google Scholar] [CrossRef] [PubMed]
- Levee, V.; Major, I.; Levasseur, C.; Tremblay, L.; MacKay, J.; Seguin, A. Expression profiling and functional analysis of Populus WRKY23 reveals a regulatory role in defense. New Phytol. 2009, 184, 48–70. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Jiang, Y.; Ye, S.; Karim, A.; Ling, Z.; He, Y.; Yang, S.; Luo, K. PtrWRKY73, a salicylic acid-inducible poplar WRKY transcription factor, is involved in disease resistance in Arabidopsis thaliana. Plant Cell Rep. 2015, 34, 831–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, F.; Melotto, M.; Yao, J.; He, S.Y. Jasmonate signaling and manipulation by pathogens and insects. J. Exp. Bot. 2017, 68, 1371–1385. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-J.; Luo, J. The PIN-FORMED Auxin Efflux Carriers in Plants. Int. J. Mol. Sci. 2018, 19, 2759. [Google Scholar] [CrossRef]
- Luo, J.; Zhou, J.-J.; Zhang, J.-Z. Aux/IAA Gene Family in Plants: Molecular Structure, Regulation, and Function. Int. J. Mol. Sci. 2018, 19, 259. [Google Scholar] [CrossRef]
- Dong, X. SA, JA, ethylene, and disease resistance in plants. Curr. Opin. Plant biol. 1998, 1, 316–323. [Google Scholar] [CrossRef]
- Liao, D.; Wang, S.; Cui, M.; Liu, J.; Chen, A.; Xu, G. Phytohormones Regulate the Development of Arbuscular Mycorrhizal Symbiosis. Int. J. Mol. Sci. 2018, 19, 3146. [Google Scholar] [CrossRef]
- Herrera-Medina, M.J.; Tamayo, M.I.; Vierheilig, H.; Ocampo, J.A.; García-Garrido, J.M. The jasmonic acid signalling pathway restricts the development of the arbuscular mycorrhizal association in tomato. J. Plant Growth Regul. 2008, 27, 221. [Google Scholar] [CrossRef]
- Luo, J.; Shi, W.; Li, H.; Janz, D.; Luo, Z.-B. The conserved salt-responsive genes in the roots of Populus× canescens and Arabidopsis thaliana. Environ. Exp. Bot. 2016, 129, 48–56. [Google Scholar] [CrossRef]
- Luo, J.; Zhou, J.; Li, H.; Shi, W.; Polle, A.; Lu, M.; Sun, X.; Luo, Z.-B. Global poplar root and leaf transcriptomes reveal links between growth and stress responses under nitrogen starvation and excess. Tree Physiol. 2015, 35, 1283–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagard, M.; Launay, A.; Clément, G.; Courtial, J.; Dellagi, A.; Farjad, M.; Krapp, A.; Soulié, M.-C.; Masclaux-Daubresse, C. Nitrogen metabolism meets phytopathology. J. Exp. Bot. 2014, 65, 5643–5656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonini, S.; Bencivenga, S.; Trick, M.; Ostergaard, L. Auxin-Induced Modulation of ETTIN Activity Orchestrates Gene Expression in Arabidopsis. Plant Cell 2017. [Google Scholar] [CrossRef] [PubMed]
- Contento, A.L.; Kim, S.-J.; Bassham, D.C. Transcriptome profiling of the response of Arabidopsis suspension culture cells to Suc starvation. Plant Physiol. 2004, 135, 2330–2347. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, H.; Liu, T.; Polle, A.; Peng, C.; Luo, Z.-B. Nitrogen metabolism of two contrasting poplar species during acclimation to limiting nitrogen availability. J. Exp. Bot. 2013, 64, 4207–4224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, H.; Jiao, Y.; Jia, J.; Wang, X.; Li, H.; Shi, W.; Peng, C.; Polle, A.; Luo, Z.-B. Phosphorus and nitrogen physiology of two contrasting poplar genotypes when exposed to phosphorus and/or nitrogen starvation. Tree Physiol. 2015, 36, 22–38. [Google Scholar] [CrossRef] [Green Version]
- Laule, O.; Fürholz, A.; Chang, H.-S.; Zhu, T.; Wang, X.; Heifetz, P.B.; Gruissem, W.; Lange, M. Crosstalk between cytosolic and plastidial pathways of isoprenoid biosynthesis in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2003, 100, 6866–6871. [Google Scholar] [CrossRef]
- Rohmer, M.; Seemann, M.; Horbach, S.; Bringer-Meyer, S.; Sahm, H. Glyceraldehyde 3-Phosphate and Pyruvate as Precursors of Isoprenic Units in an Alternative Non-mevalonate Pathway for Terpenoid Biosynthesis. J. Am. Chem. Soc. 1996, 118, 2564–2566. [Google Scholar] [CrossRef]
- Chappell, J. Biochemistry and Molecular Biology of the Isoprenoid Biosynthetic Pathway in Plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1995, 46, 521–547. [Google Scholar] [CrossRef]
- Major, I.T.; Nicole, M.C.; Duplessis, S.; Seguin, A. Photosynthetic and respiratory changes in leaves of poplar elicited by rust infection. Photosynth. Res. 2010, 104, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Scholes, J.D.; Rolfe, S.A. Photosynthesis in localised regions of oat leaves infected with crown rust (Puccinia coronata): Quantitative imaging of chlorophyll fluorescence. Planta 1996, 199, 573–582. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, J.; Xia, W.; Cao, P.; Xiao, Z.; Zhang, Y.; Liu, M.; Zhan, C.; Wang, N. Integrated Transcriptome Analysis Reveals Plant Hormones Jasmonic Acid and Salicylic Acid Coordinate Growth and Defense Responses upon Fungal Infection in Poplar. Biomolecules 2019, 9, 12. https://doi.org/10.3390/biom9010012
Luo J, Xia W, Cao P, Xiao Z, Zhang Y, Liu M, Zhan C, Wang N. Integrated Transcriptome Analysis Reveals Plant Hormones Jasmonic Acid and Salicylic Acid Coordinate Growth and Defense Responses upon Fungal Infection in Poplar. Biomolecules. 2019; 9(1):12. https://doi.org/10.3390/biom9010012
Chicago/Turabian StyleLuo, Jie, Wenxiu Xia, Pei Cao, Zheng’ang Xiao, Yan Zhang, Meifeng Liu, Chang Zhan, and Nian Wang. 2019. "Integrated Transcriptome Analysis Reveals Plant Hormones Jasmonic Acid and Salicylic Acid Coordinate Growth and Defense Responses upon Fungal Infection in Poplar" Biomolecules 9, no. 1: 12. https://doi.org/10.3390/biom9010012