
Aldosterone: Essential for Life but Damaging to the Vascular Endothelium

Bristol Renal, Dorothy Hodgkin Building, University of Bristol, Whitson Street, Bristol BS1 3NY, UK
*
Author to whom correspondence should be addressed.
Biomolecules 2023, 13(6), 1004; https://doi.org/10.3390/biom13061004
Submission received: 26 May 2023
/
Revised: 12 June 2023
/
Accepted: 15 June 2023
/
Published: 17 June 2023
(This article belongs to the Special Issue Molecular Aspect of Cardiovascular Risk Factors)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The renin angiotensin aldosterone system is a key regulator of blood pressure. Aldosterone is the final effector of this pathway, acting predominantly via mineralocorticoid receptors. Aldosterone facilitates the conservation of sodium and, with it, water and acts as a powerful stimulus for potassium excretion. However, evidence for the pathological impact of excess mineralocorticoid receptor stimulation is increasing. Here, we discussed how in the heart, hyperaldosteronism is associated with fibrosis, cardiac dysfunction, and maladaptive hypertrophy. In the kidney, aldosterone was shown to cause proteinuria and fibrosis and may contribute to the progression of kidney disease. More recently, studies suggested that aldosterone excess damaged endothelial cells. Here, we reviewed how damage to the endothelial glycocalyx may contribute to this process. The endothelial glycocalyx is a heterogenous, negatively charged layer on the luminal surface of cells. Aldosterone exposure alters this layer. The resulting structural changes reduced endothelial reactivity in response to protective shear stress, altered permeability, and increased immune cell trafficking. Finally, we reviewed current therapeutic strategies for limiting endothelial damage and suggested that preventing glycocalyx remodelling in response to aldosterone exposure may provide a novel strategy, free from the serious adverse effect of hyperkalaemia seen in response to mineralocorticoid blockade.
1. Introduction
The development of sodium and potassium gradients between the intra and extra cellular compartments was a key evolutionary step that facilitated life on earth [1]. Without these key electrochemical gradients, events within the cell cycle, such as transcription and translation, would not be possible. To allow terrestrial life, complex systems evolved to conserve salt and water and maintain homeostasis within the intra and extra cellular compartments [1]. This regulation is achieved, in part, by the renin angiotensin aldosterone system (RAAS). All the major components of this system were highly conserved through evolution and were present in ancestral chordates [1]. Aldosterone production is the final step in this regulatory pathway and the most potent stimulator of sodium conservation in mammals. Here, we review how aldosterone is produced and the regulatory steps that control its release and downstream signalling pathways. In addition to the biological role of aldosterone, we discuss identified pathological pathways implicated in human disease, highlighting that endothelial damage may be key. Finally, we highlight how damage to the endothelial glycocalyx may explain key pathological findings before discussing current therapeutic strategies and areas for future research.
2. Aldosterone Production
The enzyme renin is released in response to reduced renal (glomerular) perfusion pressure and in response to reduced sodium concentrations within the tubular lumen or sympathetic nervous system activation. Renin hydrolyses angiotensinogen to angiotensin I which is subsequently activated by angiotensin converting enzyme (ACE) to form angiotensin II. Aldosterone is produced in response to angiotensin II or elevations in extracellular potassium levels. Under physiological conditions, aldosterone synthesis has a circadian rhythm that parallels cortisol in humans [2] and corticosterone in rodents [3].
Aldosterone is produced in the zona glomerulosa of the adrenal cortex. Adenomas in the adrenal cortex or bilateral adrenal hypertrophy can both cause primary hyperaldosteronism resulting in hypertension and hypokalaemia. However, beyond the hypertensive effect, primary hyperaldosteronism increases the risk of cardiovascular events and proteinuria, even when compared to appropriate hypertensive controls [4,5,6,7]. Secondary hyperaldosteronism occurs in a number of common conditions, including idiopathic hypertension [8], obesity [9,10], advanced renal failure [11], obstructive sleep apnoea [8], and sleep disorders including shift work [8]. These patient groups also have an increased risk of developing proteinuria and cardiovascular disease [12,13].
Fluctuations in circulating aldosterone levels are largely the result of variations in de novo synthesis. Steroidogenesis starts with the “early regulatory step” of translocation of cholesterol to the inner mitochondrial membrane [14]. Translocation is facilitated by steroidogenic acute regulatory protein (St AR Protein) and alterations in this step can happen within minutes [14]. The “late regulatory step” in aldosterone production is mediated largely through modulation of expression of the enzyme aldosterone synthase (CYP11β-hydroxylase) (CYP11B2 human) [14]. The direct regulation of this enzyme by circulating factors outside of the RAAS may help explain variations in serum aldosterone levels. Leptin, a hormone produced by adipose tissue, regulates hunger and energy balance, and increases circulating aldosterone levels. Leptin acts, via its own receptor on CYP11β-hydroxylase, modulating a calcium-dependent regulatory mechanism to increase aldosterone production [15]. This finding may help to explain why certain clinical groups have high circulating aldosterone levels even in the absence of ‘traditional’ RAAS stimuli and a diminished response to angiotensin blockade. The discovery of leptin as a direct stimulus for aldosterone production also opens the possibility that there may be other circulating factors capable of stimulating aldosterone release.
The role of extra adrenal aldosterone production remains debated. There is some evidence that cardiac myocytes [16,17], pulmonary vascular endothelial cells (EnC) [18], systemic vascular EnC [19], and cells within the CNS [17] may be able to produce aldosterone in quantities capable of eliciting a local effect. However, recent work has failed to demonstrate significant aldosterone synthesis within the vascular endothelium, and so, its effect within these specialized cells is thought to be limited [20]. Adipocytes surrounding blood vessels may produce a basal level of aldosterone and respond to stimulation with angiotensin II with further increased production [21]. Blocking the effect of locally produced aldosterone within mesenteric arteries from diabetic db/db mice was shown to increase the degree of the vasodilation seen in response to acetylcholine. The authors concluded that in the context of diabetes, adipocyte derived aldosterone may act in an autocrine and paracrine manner to regulate adipocyte differentiation and vascular function, respectively [21]. Similarly, aldosterone may act in a paracrine manner to potentiate localised inflammation. In Wistar rats, local aldosterone production by aldosterone synthase in peripheral sensory neurons activated neuronal mineralocorticoid receptors (MR), contributing to inflammation-induced mechanical hypersensitivity; this effect was attenuated with selective MR blockade [22]. A consensus view on the importance of extra adrenal aldosterone production clinically is yet to be reached and remains an area of ongoing research.
3. Aldosterone Acts on Multiple Tissues and May Act via Multiple Receptors
The classical role of aldosterone is to conserve sodium. This action is achieved within the distal tubules and collecting ducts of the kidney via stimulation of epithelial cytosolic MR. Binding of aldosterone to MR dislodges chaperone proteins from the receptor to form an aldosterone-MR complex. This complex trans-locates, from the cytosol to the nucleus, where it alters target gene transcription. Increased serum and glucocorticoid-induced kinase 1 (SGK-1) leads to altered expression of amiloride-sensitive epithelial sodium channels (ENaC). These channels migrate to the cell membrane and facilitate the conservation of sodium.
The above pathway was well studied, but in recent years, it became apparent that MR expression in other tissues is common. MR are widely expressed in epithelial tissues including the bowel and salivary glands [23], but also in non-epithelial tissues such as vascular smooth muscle [24,25,26,27], skeletal muscle [28], cells of the immune system [29], vascular EnCs [24,30,31,32,33,34], cardiac myocytes [35,36], and adipocytes [37]. Our group confirmed that even within the human glomerulus, EnC and podocytes express MR [38]. The function of these receptors, in cells distant from the renal tubules, is not yet fully understood.
Aldosterone and the much more prevalent glucocorticoids, such as cortisol, have an equal affinity for the MR binding zone. Specificity is achieved in a tissue-specific manner though the expression of 11 –beta-hydroxysteroid dehydrogenase type 2 (11 β-HSD2) [39]. This enzyme, even when present at very low levels, metabolizes glucocorticoids to their metabolically inactive substrates [40]. 11 β-HSD1 in contrast can catalyse this reaction in either direction but generally favours the generation of active cortisol from inactive cortisone. Thus, the balance in activity of these two enzymes within the cytosol dictates the level of MR stimulation by circulating glucocorticoids. In mice, deficiency of 11 β-HSD2 in the vascular endothelium increased inflammatory markers within the endothelium and accelerated atherogenesis in ApoE-/-mice [41]. The MR antagonist eplerenone reversed this effect [41]. Knock out of 11 β-HSD2 in rats renders animals polydipsic, polyuric, and proteinuric with histological changes within the kidneys that are consistent with chronic kidney disease [42]. In human umbilical vein EnC (HUVEC), cortisol was noted to reduce EnC nitric oxide synthase (eNOS) production [43]. This effect was markedly increased following pre-treatment of cells with small interfering RNA (siRNA) targeting 11 β-HSD2, an effect that could be attenuated with spironolactone. These results suggest that knock down of 11 β-HSD2 allowed cortisol to remain active in the cytosol, modulating eNOS via MR [43]. Down regulation of 11 β-HSD2 was also linked with salt sensitive hypertension in human linkage studies and animal models [39,44,45]. Unilateral nephrectomy, in animal models, reduced 11 β-HSD2 expression within the remaining kidney and predisposed animals to a salt responsive hypertension [39,46,47]. In humans, a number of studies demonstrated a prolonged plasma half-life for cortisol in patients with CKD. A study of 95 adult renal patients also demonstrated a direct correlation between the expression of 11 β-HSD2 and creatinine clearance [48]. In humans, we know that liquorice ingestion (which inhibits 11 β-HSD2) or functional polymorphisms in 11 β-HSD2 expression can predispose individuals to a salt sensitive form of hypertension, but the full effect of reduced functioning renal mass on this complex system is yet to be studied [49,50].
Aldosterone does not act solely via modulation of nuclear transcription of target genes (Figure 1). ‘Nonclassical’ receptors including GPER were also demonstrated to be aldosterone-responsive [51]. The first detectable response to aldosterone occurs within minutes of exposure. This non-classical response result in aldosterone activating enzymes within the cytosol and extra nuclear compartments within minutes to activate protein kinases [52]. Activated kinases subsequently phosphorylate membrane channels and secondary enzymatic targets to modulate sodium and potassium channel activity or availability and pro-inflammatory pathways [51,53]. Aldosterone also stimulates receptors located at the vascular EnC surface [54]. The isolation of MR in the cell membrane fractions from Wistar rats and human embryonic kidney cells (HEK 293) adds further weight to the argument that cell surface MR exists and may be functional [55,56]. In rats, the function of surface receptors was studied using pegylated aldosterone. Pegylated aldosterone is incapable of crossing cell membranes but still activates ERK 1/2 phosphorylation by binding to receptors on H9c2 cells surface. In Sprague Dawley rats, activation of these surface receptors did not increase infarct size (in contrast to non-pegylated aldosterone). Spironolactone did not prevent ERK1/2 phosphorylation but the G-protein associated oestrogen receptor (GPER or GPR 30) inhibitor G36 did prevent phosphorylation, suggesting that aldosterone may activate GPER on the cell surface [57]. Despite being labelled as an oestrogen receptor, GPER is sensitive to aldosterone at very low doses [51]. This field remains controversial; however, due to a lack of evidence of competitive aldosterone binding; thus, not all conditions to define GPER as an aldosterone receptor were met [58]. GPER on a breast cancer-derived EnC line, for example, suggested that aldosterone does not bind to GPER; however, it does induce a direct interaction between MR and GPER and between GPER and epidermal growth factor receptor (EGFR). In this study, the presence and activation of both MR and GPER was required for proliferation and migration of breast cancer cells to occur [59]. Whilst these findings need to be validated in other cell lines, they open the possibility that the net effect of aldosterone exposure on different tissues may be dependent on the variable expression of both MR and GPER. However, although the status of GPER as an aldosterone receptor remains uncertain, several groups found that GPER seems to be involved in aldosterone signalling. A recent work proposed a novel autocrine-paracrine GPER-mediated mechanism of aldosterone synthesis in human adrenocortical cell lines (HAC15). Incubation of HAC15 with aldosterone increased expression of aldosterone synthase (CYP11B2), an effect unchanged by addition of the MR antagonist canrenone. In contrast, antagonism of GPER by G36 abolished this increase in CYP11B2 expression, suggesting that aldosterone acts via GPER to enhance CYP11B2 gene expression [60]. Rat aorta EnC were used to study the effects of GPER stimulation due to their unique lack of MR but consistent GPER expression [61]. In this preparation, GPER stimulation mediated pro-apoptotic and anti-proliferative effects as well as vasodilatation [61]. The vasodilator effect seen was in contrast to the action of aldosterone via MR, where, generally, vasoconstriction is considered the predominant effect resulting from impaired NO production [62]. Similarly, seemingly opposing effects of GPER versus MR activation were recently reported in rat cardiomyocytes. Aldosterone acting via MR-induced hypertrophy of myocytes. GPER activation in the presence of G1 (a specific GPER agonist) and aldosterone halted this MR-mediated hypertrophy [63].
The complex interaction between 11 β-HSD1/2 and recent discoveries of aldosterone’s rapid actions, cell surface receptor, and potential action via GPER all add additional layers of complexity to the process of aldosterone signalling in the vascular endothelium. They also present exciting new targets through which the pathological effects of aldosterone on the vascular endothelium may be modulated.
4. The Vascular Endothelium
EnC form the innermost cell layer of the vascular system. These specialist cells adapted to function optimally within different areas of the vascular tree where they contribute to the regulation of fluid filtration, vessel tone, coagulation, neutrophil recruitment, and hormone trafficking [64].
In the last few decades, researchers showed that the EnC membrane is not the innermost structure of blood vessels. Lying on the luminal aspect of the EnC is an anionic biopolymer layer called the glycocalyx. This complex structure consists of components anchored to the cell surface such as proteoglycans and sialoproteins, along with elements adsorbed from the circulation including albumin [65]. The anionic charge is largely due to the expression of the glycosaminoglycans heparan sulphate (HS) and chondroitin sulphate(CS). The integrity of the overall structure seems to be dependent on both fixed and adsorbed components. Many of the specialist functions performed by the endothelium are dependent on a ‘healthy’ glycocalyx [65]. There is growing evidence that this innermost layer can be affected negatively by MR stimulation [66].
5. Aldosterone Disrupts Flow-Mediated Dilatation
Flow-mediated dilatation (FMD) is defined as the EnC-dependent process facilitating the relaxation of a vessel in response to shear stress. FMD occurs as EnC respond to the movement of fluid over their luminal surface by producing NO. EnC dysfunction, detected by altered FMD, is an important independent risk factor for cardiovascular disease and remains clinically significant once adjustments are made for underlying common pathologies ,including diabetes [67,68], smoking [69,70], and hypertension [71].
Patients with high circulating levels of aldosterone have impaired EnC-dependent FMD [72]. In addition, elevated levels of biomarkers of EnC dysfunction (von Willebrand Factor, ICAM-1, ox-LDL) were seen in hyperaldosteronism [6]. Patients with a relative excess of aldosterone, compared to renin levels, also have impaired NO-mediated vasodilatation [73]. Recent work found FMD was significant reduced in patients with primary hyperaldosteronism as compared to those with essential resistant hypertension [74]. Fortunately, some reports suggest EnC dysfunction due to MR stimulation is reversible. Spironolactone increases EnC-dependent flow-mediated dilatation in patients with heart failure, even when used in addition to other medical treatments [75]. However, in a recent randomized controlled trial (RCT), spironolactone failed to improve FMD in early autosomal dominant poly cystic kidney disease (ADPKD) patients. Although the small sample size (n = 60) and the probable absence of significant EnC dysfunction in this cohort (those with significant co-morbidities and/or renal impairment, eGFR < 60, were excluded) may explain the results observed [76].
The eGlx is integral to the process of FMD [77,78]. To date, at least ten candidate mechanosensitive molecules were identified within the eGlx [78]. Conflicting data emerged from enzymatic degradation studies and gene deletion studies, making isolation of the most important mechanosensitive molecules difficult [79]. HS and syndecan-4 remain important candidate molecules. Our recent work in a type 1 diabetic mouse model suggests that eGlx damage occurs through matrix metalloproteinase (MMP)-induced syndecan-4 shedding. Treatment of mice with an MMP inhibitor restored eGlx, reduced albuminuria and glomerular permeability, thus presenting MMP inhibition as an attractive, novel therapy in those with diabetic kidney disease [80].
6. Aldosterone Increases Permeability to Macromolecules
The vascular endothelium plays a key role in regulating the movement of macromolecules from the blood into the tissues. Aldosterone, at a dose of 10−9 mol/L, increased the EnC permeability to proteins <70 Kda within 60 min both in cultured HUVECs and ex vivo arteries [81]. In contrast, the permeability of HUVECs was found to be unaffected following 3 days exposure to aldosterone [82]. It seems likely that the duration of aldosterone exposure is important. Whilst the study of systemic EnC permeability is complex, the impact of aldosterone and MR inhibition on the renal glomerulus was studied by several groups, including our own. Protein largely enters the urine in significant quantities following increased glomerular protein permeability [65,83]. The glomerular ‘sieve’ is composed of multiple layers, all of which are vital to the integrity of the glomerular filtration barrier (Figure 2).
The EnC were once thought to have a minimal impact on the pathogenesis of proteinuria. However, more recent research highlighted the importance of the EnC, and the glycocalyx they produce, in regulating glomerular protein passage [65,85]. Both primary and secondary hyperaldosteronism in humans are linked to proteinuria [6,12,13,86,87]. An MR blockade was shown to be effective in reducing urinary protein leak [87]. Reducing urinary protein loss through the glomerular filtration barrier reduces patients’ risk of progressive kidney failure and is a key strategy for managing chronic kidney disease [88]. The reduction in proteinuria in response to MR blockade is independent of blood pressure reduction and occurs even when MR blockade is used in addition to other RAAS blocking agents [38,87,89,90,91]. Further evidence for this blood pressure independent effect was gathered by normalizing blood pressure (with agents not targeting RAAS components), which failed to prevent renal fibrosis and proved ineffective at reducing urinary protein loss [92,93]. We showed that spironolactone, even when used in addition to ACE inhibition, reduced the levels of circulating glycocalyx-degrading enzymes in patients with diabetes. In addition, our study highlighted that MR inhibition in a diabetic rat model preserved the glycocalyx and reduced albuminuria, again by reducing glycocalyx degrading enzyme activity and preserving the EGlx [84]. Podocytes also express MR and are damaged by elevated aldosterone [94]. However, podocyte-specific MR knock out mice still become proteinuric and systemic aldosterone blockade in these animals remains effective in reducing glomerular injury, suggesting other cell types are important in the pathogenesis of proteinuria in response to MR stimulation [95].
It is possible that crosstalk between cells of the renal glomerulus in response to aldosterone stimulation contributes to the development of proteinuria. Aldosterone acts via MR to increase endothelin-1 (ET-1) expression in Sprague Dawley rat kidney tissue [96] Endothelin-1 increases heparanase release from podocytes and this enzyme is known to cleave heparan sulphate, a major component of the glycocalyx, loss of which is associated with proteinuria [65,97]. Conversely, Atrasentan, an endothelin A receptor antagonist, increased eGlx coverage and reduced albuminuria in diabetic mice [98]. The RCT (SONAR), showed that atrasentan reduced CKD progression in type 2 diabetics; however, there was a greater incidence of heart failure in the treated group (despite careful selection in a pre-randomisation enrichment period designed to minimise this risk). Whilst this effect was not statistically significant between groups, the trend suggests that widespread use of endothelin receptor antagonists may be limited by the risk of heart failure associated with these medications [99].
The increased permeability of the systemic endothelium in response to aldosterone, and the associated glycocalyx remodelling, hint towards the importance of the glycocalyx in the pathogenesis of proteinuria in response to aldosterone excess. The possibility of glomerular eGlx protection as a potential pharmacological target in proteinuric renal disease will rely on ongoing research to elucidate the steps in this complex process.
7. Aldosterone Promotes Inflammation
The ability of white blood cells to egress from the vascular compartment to a site of tissue injury or infection is an essential part of the innate immune system (Figure 3).
However, when the regulation of this process becomes defective, unwanted inflammation and fibrosis may occur. Aldosterone exposure rapidly increases the adhesion of polymorphonuclear leukocytes to the HUVEC surface [100]. These effects were maximal within one hour of aldosterone exposure. Increased protein expression of vascular cell adhesion molecule (VCAM)-1, e-selectin, and intercellular adhesion molecule-1 (ICAM-1) were thought to be responsible for the effect [100]. In ApoE knockout mice, who received aldosterone and an atherogenic diet, ICAM-1 expression increased, as did atherosclerotic plaque size; this was not seen in ApoE/I-CAM double knockouts [101]. MR deletion from endothelial cells prevented adverse cardiac remodelling in mice in response to the MR agonist deoxycorticosterone acetate (DOCA) in the presence of high salt [102]. This was associated with a reduced expression of the VCAM-1 and ICAM-1 in MR-deficient endothelial cells and less macrophage infiltration of the heart [33]. In human coronary EnC, ICAM-1-dependent leucocyte adhesion was inhibited in MR-knockdown cells and by spironolactone [103]. Taken together, these implicate aldosterone-induced ICAM-1 expression in the progression of atherosclerosis, an effect that appears inhibited by MR blockade.
It is important to note that the expression of cell adhesion receptors is also regulated by shear stress [104]. As highlighted previously, the glycocalyx is intimately involved in this process and so, damage to the glycocalyx structure, which limits the EnC protective response to shear stress, may also result in the cell surface becoming more adhesive [104] (Figure 3). These data, when combined with the observation by Oberleithner (that the glycocalyx becomes thin and loses heparan sulphate from its structure in response to aldosterone and salt exposure) [66,105], suggest that the transit of white blood cells from the circulation may increase in response to aldosterone. This may be due to increased adhesion molecule expression, loss of shear sensitivity, and increased adhesion molecule exposure due to reduced expression of other glycocalyx components such as HS.
8. Aldosterone Promotes Atherosclerosis
Elevated aldosterone levels, even within the ‘normal range’, are strong predictors of cardiovascular disease risk [106]. This effect remains even when adjusted for baseline atheroma [107]. Atherosclerosis is a systemic vascular inflammatory disease initiated by cardiovascular risk factors that cause EnC damage [108]. Activated EnC recruit leukocytes to the EnC wall, typically in areas of low shear stress [108]. High circulating levels of aldosterone are linked to an increased rate of atherogenesis, particularly in areas of the circulation that experience low shear stress, e.g., vessel bifurcations, etc. In a mouse model with elevated circulating aldosterone at levels insufficient to cause BP alteration, aldosterone increased recruitment of activated monocytes and T cells to atheroma prone areas, promoting atheroma formation [24]. A recent work in a mouse model of low shear stress (via partial carotid artery ligation) proposed that AMPK inactivation increases Na+ -H+ exchanger (NHE)1 activity and downstream hyaluronidase (HYAL2) 2-mediated glycocalyx degradation. Activation of AMPK with ampkinone reduced HYAL2 activity and halted glycocalyx loss; blocked ICAM-1 and VCAM-1 expression and reduced macrophage recruitment [109]. As discussed previously, aldosterone is implicated in ICAM-1 expression; however, the exact contribution to this signalling pathway and associated glycocalyx loss remains unknown.
9. Pharmacological Manipulation of the Aldosterone—MR System
Although we are only now beginning to understand how aldosterone excess results in tissue damage, the clinical use of MR blocking drugs was widespread since their introduction in 1953. The benefits of MR inhibition in humans, beyond BP control, was established following two landmark studies. The first study was performed by Pitt et al. and was designed to review the effects of spironolactone administration on the morbidity and mortality of patients with heart failure [110]. The second study, again by Pitt et al., looked specifically at patients with left ventricular dysfunction after a myocardial infarction [111]. Both studies reported significantly improved clinical outcomes in patients receiving spironolactone. Since then, the discovery of multiple pathological pathways influenced by MR and the clinical benefits associated with MR inhibition have driven further drug discoveries targeting the RAAS system.
9.1. Angiotensin Inhibition
Blockade of the RAAS at any level was initially thought to inhibit aldosterone secretion to acceptable levels. Both angiotensin-converting enzyme inhibitor (ACEi) and angiotensin receptor blocker (ARB) medication reduce serum aldosterone levels initially [112,113]. However, the phenomenon of aldosterone escape or breakthrough is now recognized clinically. The incidence of aldosterone escape depends on the definition used. The most widely adopted definition is “an increase in plasma aldosterone levels during long-term RAAS blockade, not compared to pre-treatment levels but to aldosterone levels after 2 months treatment” [113].
Using this definition, researchers in Denmark found 41% of type 1 diabetic patients on long term ARBs suffered from aldosterone escape [113]. Sato et al. described a similar incidence of aldosterone escape in hypertensive patients and type 2 diabetic patients [114,115]. Thus, even in the presence of maximal ACEi or ARB therapy, aldosterone may not be fully suppressed. Their use clinically is also limited by their recognized side effects. These include hyperkalaemia, an increased predisposition to acute kidney injury (AKI) [116], hypotension, teratogenicity, and cough.
9.2. Mineralocorticoid Antagonism
The MR antagonist Spironolactone was originally developed as a potassium sparing diuretic for the treatment of oedema. Its usefulness in primary hyperaldosteronism was subsequently discovered. More recently, as the benefits of MR blockade in heart disease became more widely recognized [87,110,111], the use of spironolactone in the UK greatly increased [117]. Spironolactone is readily absorbed with a bioavailability of 80–90% [118]. It has a half-life of only 1.4 h due to metabolism within the liver; however, the major metabolite (7 α-thiomethylspironolactone) is also pharmacologically active and accounts for 80% of the effects of the parent compound. Because of these effects, spironolactone can be administered once daily but it takes several days to reach steady state and several weeks to reach its maximal hypotensive effect [118]. Spironolactone has a relative binding efficacy for MR of 0.11 (Aldosterone = 1) but is not selective for MR blockade [118]. The non-specific binding of spironolactone accounts for the sexual side effects including gynaecomastia, mastodynia, impotence, and menstrual irregularities. These effects occur because of spironolactone’s effect on progesterone receptors (PR) and androgen receptors (AR). Spironolactone’s use is also limited by the predictable retention of potassium that results from aldosterone blockade. This becomes an issue in patients with impaired potassium regulation. For this reason, spironolactone must be used with extreme caution in patients with chronic kidney disease (CKD). The BARACK-D trial should give us valuable information regarding the safety of spironolactone in patients with CKD and its results are eagerly awaited now that recruitment has ended [119].
Eplerenone, a second-generation MR antagonist, became available for clinical use in 2002. Compared to spironolactone, eplerenone is a much more selective antagonist for MR [120]. However, the relative affinity of eplerenone for MR is lower (0.005) and with a plasma half-life of only 4–6 h (with no active metabolites) necessitating twice daily dosing. Eplerenone has a much more limited side effect profile due to its reduced affinity for progesterone receptors (140-fold lower) and androgen receptors (1200-fold lower) relative to spironolactone [120].
A third generation of novel non-steroidal MR antagonists (finerenone, esaxerenone and apararenone) were recently developed. Clinically, finerenone was now evaluated in two landmark clinical randomised control trials (FIDELIO-DKD and FIGARO-DKD) [121,122]. Experiments using radiolabelled drug suggest that finerenone may achieve a more even organ distribution concentration in contrast to spironolactone where renal accumulation was prominent [123]. The ARTS study reported a lower mean elevation in serum potassium in chronic kidney disease 3 (CKD3), chronic heart failure (CHF) patients on finerenone, as compared to those taking spironolactone [124]. The improved side effect profile was hypothetically linked to the shorter plasma half-life; however, hyperkalaemia remains a predictable effect of all MR antagonists, including finerenone and must be considered during clinical use [125].
9.3. Aldosterone Synthase Inhibitors
Blocking the MR receptor leads to further stimulation of the RAAS via potassium accumulation and blood pressure modulation. This, in turn, promotes higher levels of circulating aldosterone, which may eventually necessitate dose increases to maintain the treatment effect. At higher doses, cross reactivity of the MR antagonists with AR and PR became an increasing issue and the resulting side effects more common. Thus, drugs that inhibit aldosterone production/secretion are an attractive therapeutic option. Several agents were in development, including LCI 699 (osilodrostat). However, there are concerns about the non-selectivity of the 11-hydroxylation step [120]. Targeting this enzyme introduces the possibility of interference with the cortisol synthesis pathway, raising concerns about inducing an Addisonian crisis at times of high stress [120]. A phase III study involving 137 patients reported adrenal insufficiency in 28%, hypo-cortisolism in 51%, and adverse events related to adrenal hormone precursors in 42% of the participants [126]. The search continues for more specific molecules to block aldosterone synthesis with new models being established to facilitate this search [127,128]. Sakakibara et al. reported a synthesis of selective aldosterone synthase (CYP11B2) inhibitors with high potency and selectivity over 11β-hydroxylase (CYP11B1) that reduced aldosterone production in cynomolgus monkeys [129]. Sparks et al. reported the development of a selective aldosterone synthase (CYP11B2) inhibitor that reduced aldosterone production in cynomolgus monkeys without the accumulation of steroid precursors (11-doxycortisol, 11-deoxycorticosterone) seen with LCI 699 [130]. Most recently, a phase II trial of Baxdrostat demonstrated a promising human safety profile when used in patients with resistant hypertension [131]. This 12-week placebo-controlled trial confirmed a significant (additional 11 mmHg reduction in systolic BP) compared to control participants [131]. A longer trial including ambulatory BP is now needed to see how trial can be translated to long-term clinical practice.
10. Conclusions
The eGlx forms a vital, but fragile, component of the vascular system. Inappropriate levels of salt and aldosterone result in damage to the endothelium, leading to functional impairment of the eGlx. Pharmaceutical companies are investing large amounts of capital to find drugs capable of preventing the negative clinical impact of aldosterone excess. Newer agents will have improved side effect profiles and improved specificity for MR. However, the failure to look for new targets within this signalling cascade left us focused on MR, which has a physiological, highly conserved role in the regulation of total body sodium and potassium. Targeting MR will inevitably result in elevated serum potassium levels and may preclude the use of these agents in patients with kidney disease. Focusing on how the negative effects of aldosterone develop may reveal safer pharmacological targets in the battle to prevent EnC damage.
Author Contributions
Conceptualization, M.C., S.C.S. and M.J.B. data curation, M.C., L.J.S., S.C.S. and M.J.B.; writing—original draft preparation, M.C., L.J.S., S.C.S. and M.J.B.; writing—review and editing, M.C., L.J.S., S.C.S. and M.J.B.; visualization, M.C., L.J.S., S.C.S. and M.J.B.; funding acquisition S.C.S. and M.J.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the MRC MR/M018237/1/MRC_/Medical Research Council/United Kingdom (M Butler) MR/T031921/1/MRC_/Medical Research Council/United Kingdom (M Butler), British Heart Foundation FS/CRTF/22/24361 (L Skinner), British Heart Foundation PG/20/10187 (M Crompton).
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 11 β-HSD1 | 11 –beta-hydroxysteroid dehydrogenase type 1 |
| 11 β-HSD2 | 11 –beta-hydroxysteroid dehydrogenase type 2 |
| ACE | angiotensin converting enzyme |
| ACEi | angiotensin-converting enzyme inhibitor |
| ADPKD | autosomal dominant polycystic kidney disease |
| AKI | acute kidney injury |
| AMPK | AMP-activated protein kinase |
| ApoE | Apolipoprotein E |
| AR | androgen receptor |
| ARB | angiotensin receptor blockers |
| BP | blood pressure |
| CHF | congestive heart failure |
| CKD | chronic kidney disease |
| CNS | central nervous system |
| CYP11B1 | cytochrome P450 family 11 subfamily B member 1 |
| CYP11B2 | cytochrome P450 family 11 subfamily B member 2 |
| DOCA | deoxycorticosterone acetate |
| EGFR | epidermal growth factor receptor |
| ENaC | epithelial sodium channels |
| EnC | endothelial cell |
| eGlx | endothelial cell glycocalyx |
| eNOS | endothelial nitric oxide synthase |
| ERK 1/2 | extracellular signal-regulated kinase 1/2 |
| ET-1 | endothelin-1 |
| FMD | flow-mediated dilatation |
| GPER (or GPR 30) | G-protein associated oestrogen receptor |
| HAC | human adrenocortical carcinoma |
| HEK | human embryonic kidney |
| HS | heparan sulphate |
| HUVEC | human umbilical vein endothelial cells |
| HYAL2 | hyaluronidase 2 |
| ICAM-1 | intercellular adhesion molecule-1 |
| MMP | matrix metalloproteinase |
| MR | mineralocorticoid receptor |
| NHE | Na+-H+ exchanger |
| NO | nitric oxide |
| ox-LDL | oxidized low-density lipoprotein |
| PR | progesterone receptor |
| RAAS | renin angiotensin aldosterone system |
| RCT | randomized controlled trial |
| SGK-1 | serum and glucocorticoid- induced kinase 1 |
| siRNA | small interfering RNA |
| St AR Protein | steroidogenic acute regulatory protein |
| VCAM-1 | vascular cell adhesion molecule-1 |
References
- Rossier, B.C.; Baker, M.E.; Studer, R.A. Epithelial sodium transport and its control by aldosterone: The story of our internal environment revisited. Physiol. Rev. 2015, 95, 297–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kem, D.C.; Weinberger, M.H.; Gomez-Sanchez, C.; Kramer, N.J.; Lerman, R.; Furuyama, S.; Nugent, C.A. Circadian rhythm of plasma aldosterone concentration in patients with primary aldosteronism. J. Clin. Investig. 1973, 52, 2272–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Sanchez, C.; Holland, O.B.; Higgins, J.R.; Kem, D.C.; Kaplan, N.M. Circadian rhythms of serum renin activity and serum corticosterone, prolactin, and aldosterone concentrations in the male rat on normal and low-sodium diets. Endocrinology 1976, 99, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Catena, C.; Colussi, G.; Nadalini, E.; Chiuch, A.; Baroselli, S.; Lapenna, R.; Sechi, L.A. Cardiovascular outcomes in patients with primary aldosteronism after treatment. Arch. Intern. Med. 2008, 168, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Milliez, P.; Girerd, X.; Plouin, P.F.; Blacher, J.; Safar, M.E.; Mourad, J.J. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J. Am. Coll. Cardiol. 2005, 45, 1243–1248. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Yin, G.S.; Tang, J.Y.; Ma, D.J.; Ru, J.; Huang, X.H. Endothelial dysfunction in patients with primary aldosteronism: A biomarker of target organ damage. J. Hum. Hypertens. 2014, 28, 711–715. [Google Scholar] [CrossRef]
- Zennaro, M.C.; Boulkroun, S.; Fernandes-Rosa, F.L. Pathogenesis and treatment of primary aldosteronism. Nat. Rev. Endocrinol. 2020, 16, 578–589. [Google Scholar] [CrossRef]
- Shibata, H.; Itoh, H. Mineralocorticoid receptor-associated hypertension and its organ damage: Clinical relevance for resistant hypertension. Am. J. Hypertens. 2012, 25, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Bomback, A.S.; Klemmer, P.J. Renal injury in extreme obesity: The important role of aldosterone. Kidney Int. 2008, 74, 1216, author reply 1216–1217. [Google Scholar] [CrossRef] [Green Version]
- Nagase, M. Activation of the aldosterone/mineralocorticoid receptor system in chronic kidney disease and metabolic syndrome. Clin. Exp. Nephrol. 2010, 14, 303–314. [Google Scholar] [CrossRef]
- Bomback, A.S.; Kshirsagar, A.V.; Ferris, M.E.; Klemmer, P.J. Disordered aldosterone-volume relationship in end-stage kidney disease. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2009, 10, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Adeseun, G.A.; Rosas, S.E. The impact of obstructive sleep apnea on chronic kidney disease. Curr. Hypertens. Rep. 2010, 12, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, R.; Nagasawa, Y.; Iwatani, H.; Shinzawa, M.; Obi, Y.; Teranishi, J.; Ishigami, T.; Yamauchi-Takihara, K.; Nishida, M.; Rakugi, H.; et al. Self-reported sleep duration and prediction of proteinuria: A retrospective cohort study. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2012, 59, 343–355. [Google Scholar] [CrossRef]
- Hattangady, N.G.; Olala, L.O.; Bollag, W.B.; Rainey, W.E. Acute and chronic regulation of aldosterone production. Mol. Cell. Endocrinol. 2012, 350, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Huby, A.C.; Antonova, G.; Groenendyk, J.; Gomez-Sanchez, C.E.; Bollag, W.B.; Filosa, J.A.; Belin de Chantemele, E.J. The Adipocyte-Derived Hormone Leptin is a Direct Regulator of Aldosterone Secretion, Which Promotes Endothelial Dysfunction and Cardiac Fibrosis. Circulation 2015, 132, 2134–2145. [Google Scholar] [CrossRef]
- Fujisaki, M.; Nagoshi, T.; Nishikawa, T.; Date, T.; Yoshimura, M. Rapid induction of aldosterone synthesis in cultured neonatal rat cardiomyocytes under high glucose conditions. BioMed Res. Int. 2013, 2013, 161396. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, S.M.; Connell, J.M.; Davies, E. Non-adrenal synthesis of aldosterone: A reality check. Mol. Cell. Endocrinol. 2012, 350, 163–167. [Google Scholar] [CrossRef]
- Maron, B.A.; Oldham, W.M.; Chan, S.Y.; Vargas, S.O.; Arons, E.; Zhang, Y.Y.; Loscalzo, J.; Leopold, J.A. Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation 2014, 130, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Takeda, Y.; Miyamori, I.; Yoneda, T.; Hatakeyama, H.; Inaba, S.; Furukawa, K.; Mabuchi, H.; Takeda, R. Regulation of aldosterone synthase in human vascular endothelial cells by angiotensin II and adrenocorticotropin. J. Clin. Endocrinol. Metab. 1996, 81, 2797–2800. [Google Scholar] [CrossRef]
- Ahmad, N.; Romero, D.G.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. Do human vascular endothelial cells produce aldosterone? Endocrinology 2004, 145, 3626–3629. [Google Scholar] [CrossRef] [Green Version]
- Briones, A.M.; Nguyen Dinh Cat, A.; Callera, G.E.; Yogi, A.; Burger, D.; He, Y.; Correa, J.W.; Gagnon, A.M.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: Implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension 2012, 59, 1069–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, D.M.; Shaqura, M.; Li, X.; Shakibaei, M.; Beyer, A.; Treskatsch, S.; Schafer, M.; Mousa, S.A. Aldosterone Synthase in Peripheral Sensory Neurons Contributes to Mechanical Hypersensitivity during Local Inflammation in Rats. Anesthesiology 2020, 132, 867–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, K.T. Aldosteronism revisited: Perspectives on less well-recognized actions of aldosterone. J. Lab. Clin. Med. 2003, 142, 71–82. [Google Scholar] [CrossRef] [PubMed]
- McGraw, A.P.; McCurley, A.; Preston, I.R.; Jaffe, I.Z. Mineralocorticoid receptors in vascular disease: Connecting molecular pathways to clinical implications. Curr. Atheroscler. Rep. 2013, 15, 340. [Google Scholar] [CrossRef] [Green Version]
- Galmiche, G.; Pizard, A.; Gueret, A.; El Moghrabi, S.; Ouvrard-Pascaud, A.; Berger, S.; Challande, P.; Jaffe, I.Z.; Labat, C.; Lacolley, P.; et al. Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone-salt to induce vascular stiffness. Hypertension 2014, 63, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Koenig, J.B.; Jaffe, I.Z. Direct role for smooth muscle cell mineralocorticoid receptors in vascular remodeling: Novel mechanisms and clinical implications. Curr. Hypertens. Rep. 2014, 16, 427. [Google Scholar] [CrossRef] [Green Version]
- Tarjus, A.; Belozertseva, E.; Louis, H.; El Moghrabi, S.; Labat, C.; Lacolley, P.; Jaisser, F.; Galmiche, G. Role of smooth muscle cell mineralocorticoid receptor in vascular tone. Pflug. Arch. Eur. J. Physiol. 2015, 467, 1643–1650. [Google Scholar] [CrossRef]
- Chadwick, J.A.; Hauck, J.S.; Lowe, J.; Shaw, J.J.; Guttridge, D.C.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Rafael-Fortney, J.A. Mineralocorticoid receptors are present in skeletal muscle and represent a potential therapeutic target. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 4544–4554. [Google Scholar] [CrossRef] [Green Version]
- Gilet, A.; Zou, F.; Boumenir, M.; Frippiat, J.P.; Thornton, S.N.; Lacolley, P.; Ropars, A. Aldosterone up-regulates MMP-9 and MMP-9/NGAL expression in human neutrophils through p38, ERK1/2 and PI3K pathways. Exp. Cell. Res. 2015, 331, 152–163. [Google Scholar] [CrossRef]
- Jia, G.; Habibi, J.; DeMarco, V.G.; Martinez-Lemus, L.A.; Ma, L.; Whaley-Connell, A.T.; Aroor, A.R.; Domeier, T.L.; Zhu, Y.; Meininger, G.A.; et al. Endothelial Mineralocorticoid Receptor Deletion Prevents Diet-Induced Cardiac Diastolic Dysfunction in Females. Hypertension 2015, 66, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- Nguyen Dinh Cat, A.; Griol-Charhbili, V.; Loufrani, L.; Labat, C.; Benjamin, L.; Farman, N.; Lacolley, P.; Henrion, D.; Jaisser, F. The endothelial mineralocorticoid receptor regulates vasoconstrictor tone and blood pressure. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 2454–2463. [Google Scholar] [CrossRef] [Green Version]
- Lang, F. Stiff endothelial cell syndrome in vascular inflammation and mineralocorticoid excess. Hypertension 2011, 57, 146–147. [Google Scholar] [CrossRef] [Green Version]
- Rickard, A.J.; Morgan, J.; Chrissobolis, S.; Miller, A.A.; Sobey, C.G.; Young, M.J. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension 2014, 63, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Sekizawa, N.; Yoshimoto, T.; Hayakawa, E.; Suzuki, N.; Sugiyama, T.; Hirata, Y. Transcriptome analysis of aldosterone-regulated genes in human vascular endothelial cell lines stably expressing mineralocorticoid receptor. Mol. Cell. Endocrinol. 2011, 341, 78–88. [Google Scholar] [CrossRef]
- Bienvenu, L.A.; Reichelt, M.E.; Delbridge, L.M.; Young, M.J. Mineralocorticoid receptors and the heart, multiple cell types and multiple mechanisms: A focus on the cardiomyocyte. Clin. Sci. 2013, 125, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Lother, A.; Moser, M.; Bode, C.; Feldman, R.D.; Hein, L. Mineralocorticoids in the heart and vasculature: New insights for old hormones. Annu. Rev. Pharm. Toxicol. 2015, 55, 289–312. [Google Scholar] [CrossRef]
- Armani, A.; Marzolla, V.; Fabbri, A.; Caprio, M. Cellular mechanisms of MR regulation of adipose tissue physiology and pathophysiology. J. Mol. Endocrinol. 2015, 55, R1–R10. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.J.; Ramnath, R.; Kadoya, H.; Desposito, D.; Riquier-Brison, A.; Ferguson, J.K.; Onions, K.L.; Ogier, A.S.; ElHegni, H.; Coward, R.J.; et al. Aldosterone induces albuminuria via matrix metalloproteinase-dependent damage of the endothelial glycocalyx. Kidney Int. 2019, 95, 94–107. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, M.; Andrieu, T.; Neuenschwander, S.; Bruggmann, R.; Mordasini, D.; Frey, F.J.; Vogt, B.; Frey, B.M. Regulation of 11beta-hydroxysteroid dehydrogenase type 2 by microRNA. Hypertension 2014, 64, 860–866. [Google Scholar] [CrossRef] [Green Version]
- Ali, Y.; Kuppusamy, M.; Velarde-Miranda, C.; Gomez-Sanchez, C.M.; Plonczynski, M.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P. 11betaHSD2 Efficacy in Preventing Transcriptional Activation of the Mineralocorticoid Receptor by Corticosterone. J. Endocr. Soc. 2021, 5, bvab146. [Google Scholar] [CrossRef]
- Deuchar, G.A.; McLean, D.; Hadoke, P.W.; Brownstein, D.G.; Webb, D.J.; Mullins, J.J.; Chapman, K.; Seckl, J.R.; Kotelevtsev, Y.V. 11beta-hydroxysteroid dehydrogenase type 2 deficiency accelerates atherogenesis and causes proinflammatory changes in the endothelium in apoe−/− mice. Endocrinology 2011, 152, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Mullins, L.J.; Kenyon, C.J.; Bailey, M.A.; Conway, B.R.; Diaz, M.E.; Mullins, J.J. Mineralocorticoid Excess or Glucocorticoid Insufficiency: Renal and Metabolic Phenotypes in a Rat Hsd11b2 Knockout Model. Hypertension 2015, 66, 667–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Mladinov, D.; Pietrusz, J.L.; Usa, K.; Liang, M. Glucocorticoid response elements and 11 beta-hydroxysteroid dehydrogenases in the regulation of endothelial nitric oxide synthase expression. Cardiovasc. Res. 2009, 81, 140–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotelevtsev, Y.; Brown, R.W.; Fleming, S.; Kenyon, C.; Edwards, C.R.; Seckl, J.R.; Mullins, J.J. Hypertension in mice lacking 11beta-hydroxysteroid dehydrogenase type 2. J. Clin. Investig. 1999, 103, 683–689. [Google Scholar] [CrossRef] [Green Version]
- Lovati, E.; Ferrari, P.; Dick, B.; Jostarndt, K.; Frey, B.M.; Frey, F.J.; Schorr, U.; Sharma, A.M. Molecular basis of human salt sensitivity: The role of the 11beta-hydroxysteroid dehydrogenase type 2. J. Clin. Endocrinol. Metab. 1999, 84, 3745–3749. [Google Scholar] [CrossRef] [Green Version]
- Lauterburg, M.; Escher, G.; Dick, B.; Ackermann, D.; Frey, F.J. Uninephrectomy reduces 11beta-hydroxysteroid dehydrogenase type 1 and type 2 concomitantly with an increase in blood pressure in rats. J. Endocrinol. 2012, 214, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Huesler, C.; Lauterburg, M.; Frey, B.M.; Frey, F.J. Evidence for glucocorticoid-mediated hypertension after uninephrectomy. Physiol. Rep. 2013, 1, e00101. [Google Scholar] [CrossRef]
- Quinkler, M.; Zehnder, D.; Lepenies, J.; Petrelli, M.D.; Moore, J.S.; Hughes, S.V.; Cockwell, P.; Hewison, M.; Stewart, P.M. Expression of renal 11beta-hydroxysteroid dehydrogenase type 2 is decreased in patients with impaired renal function. Eur. J. Endocrinol. 2005, 153, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Melcescu, E.; Phillips, J.; Moll, G.; Subauste, J.S.; Koch, C.A. 11Beta-hydroxylase deficiency and other syndromes of mineralocorticoid excess as a rare cause of endocrine hypertension. Horm. Metab. Res. 2012, 44, 867–878. [Google Scholar] [CrossRef]
- Alikhani-Koupaei, R.; Fouladkou, F.; Fustier, P.; Cenni, B.; Sharma, A.M.; Deter, H.C.; Frey, B.M.; Frey, F.J. Identification of polymorphisms in the human 11beta-hydroxysteroid dehydrogenase type 2 gene promoter: Functional characterization and relevance for salt sensitivity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 3618–3628. [Google Scholar] [CrossRef]
- Williams, J.S. Evolving research in nongenomic actions of aldosterone. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 198–203. [Google Scholar] [CrossRef]
- Dooley, R.; Harvey, B.J.; Thomas, W. Non-genomic actions of aldosterone: From receptors and signals to membrane targets. Mol. Cell. Endocrinol. 2012, 350, 223–234. [Google Scholar] [CrossRef]
- Debonneville, C.; Flores, S.Y.; Kamynina, E.; Plant, P.J.; Tauxe, C.; Thomas, M.A.; Munster, C.; Chraibi, A.; Pratt, J.H.; Horisberger, J.D.; et al. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J. 2001, 20, 7052–7059. [Google Scholar] [CrossRef] [Green Version]
- Wildling, L.; Hinterdorfer, P.; Kusche-Vihrog, K.; Treffner, Y.; Oberleithner, H. Aldosterone receptor sites on plasma membrane of human vascular endothelium detected by a mechanical nanosensor. Pflug. Arch. 2009, 458, 223–230. [Google Scholar] [CrossRef]
- Ricchiuti, V.; Lapointe, N.; Pojoga, L.; Yao, T.; Tran, L.; Williams, G.H.; Adler, G.K. Dietary sodium intake regulates angiotensin II type 1, mineralocorticoid receptor, and associated signaling proteins in heart. J. Endocrinol. 2011, 211, 47–54. [Google Scholar] [CrossRef]
- Grossmann, C.; Husse, B.; Mildenberger, S.; Schreier, B.; Schuman, K.; Gekle, M. Colocalization of mineralocorticoid and EGF receptor at the plasma membrane. Biochim. Biophys. Acta 2010, 1803, 584–590. [Google Scholar] [CrossRef] [Green Version]
- Ashton, A.W.; Le, T.Y.; Gomez-Sanchez, C.E.; Morel-Kopp, M.C.; McWhinney, B.; Hudson, A.; Mihailidou, A.S. Role of Nongenomic Signaling Pathways Activated by Aldosterone during Cardiac Reperfusion Injury. Mol. Endocrinol. 2015, 29, 1144–1155. [Google Scholar] [CrossRef] [Green Version]
- Barton, M.; Meyer, M.R. Nicolaus Copernicus and the rapid vascular responses to aldosterone. Trends Endocrinol. Metab. 2015, 26, 396–398. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; Avino, S.; De Marco, P.; Bussolati, B.; Maggiolini, M.; De Francesco, E.M. GPER is involved in the stimulatory effects of aldosterone in breast cancer cells and breast tumor-derived endothelial cells. Oncotarget 2016, 7, 94–111. [Google Scholar] [CrossRef] [Green Version]
- Caroccia, B.; Seccia, T.M.; Piazza, M.; Prisco, S.; Zanin, S.; Iacobone, M.; Lenzini, L.; Pallafacchina, G.; Domening, O.; Poglitsch, M.; et al. Aldosterone Stimulates Its Biosynthesis Via a Novel GPER-Mediated Mechanism. J. Clin. Endocrinol. Metab. 2019, 104, 6316–6324. [Google Scholar] [CrossRef]
- Gros, R.; Ding, Q.; Liu, B.; Chorazyczewski, J.; Feldman, R.D. Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation. Am. J. Physiol. Cell Physiol. 2013, 304, C532–C540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, K.V.; McCurley, A.T.; Jaffe, I.Z. Direct contribution of vascular mineralocorticoid receptors to blood pressure regulation. Clin. Exp. Pharmacol. Physiol. 2013, 40, 902–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Mattia, R.A.; Mariangelo, J.I.E.; Blanco, P.G.; Jaquenod De Giusti, C.; Portiansky, E.L.; Mundina-Weilenmann, C.; Aiello, E.A.; Orlowski, A. The activation of the G protein-coupled estrogen receptor (GPER) prevents and regresses cardiac hypertrophy. Life Sci. 2020, 242, 117211. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Salmon, A.H.; Ferguson, J.K.; Burford, J.L.; Gevorgyan, H.; Nakano, D.; Harper, S.J.; Bates, D.O.; Peti-Peterdi, J. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J. Am. Soc. Nephrol. 2012, 23, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Oberleithner, H.; Peters, W.; Kusche-Vihrog, K.; Korte, S.; Schillers, H.; Kliche, K.; Oberleithner, K. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflug. Arch. Eur. J. Physiol. 2011, 462, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Mordi, I.; Tzemos, N. Is reversal of endothelial dysfunction still an attractive target in modern cardiology? World J. Cardiol. 2014, 6, 824–835. [Google Scholar] [CrossRef]
- McVeigh, G.E.; Brennan, G.M.; Johnston, G.D.; McDermott, B.J.; McGrath, L.T.; Henry, W.R.; Andrews, J.W.; Hayes, J.R. Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 1992, 35, 771–776. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Adams, M.R.; Clarkson, P.; Robinson, J.; McCredie, R.; Donald, A.; Deanfield, J.E. Passive smoking and impaired endothelium-dependent arterial dilatation in healthy young adults. N. Engl. J. Med. 1996, 334, 150–154. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Sorensen, K.E.; Georgakopoulos, D.; Bull, C.; Thomas, O.; Robinson, J.; Deanfield, J.E. Cigarette smoking is associated with dose-related and potentially reversible impairment of endothelium-dependent dilation in healthy young adults. Circulation 1993, 88, 2149–2155. [Google Scholar] [CrossRef] [Green Version]
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27. [Google Scholar] [CrossRef]
- Nishizaka, M.K.; Zaman, M.A.; Green, S.A.; Renfroe, K.Y.; Calhoun, D.A. Impaired endothelium-dependent flow-mediated vasodilation in hypertensive subjects with hyperaldosteronism. Circulation 2004, 109, 2857–2861. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.J.; Biegelsen, E.S.; Eberhardt, R.T.; Kahn, D.F.; Kingwell, B.A.; Vita, J.A. Low-renin hypertension with relative aldosterone excess is associated with impaired NO-mediated vasodilation. Hypertension 2005, 46, 707–713. [Google Scholar] [CrossRef] [Green Version]
- Demirkiran, A.; Everaars, H.; Elitok, A.; van de Ven, P.M.; Smulders, Y.M.; Dreijerink, K.M.; Tanakol, R.; Ozcan, M. Hypertension with primary aldosteronism is associated with increased carotid intima-media thickness and endothelial dysfunction. J. Clin. Hypertens. 2019, 21, 932–941. [Google Scholar] [CrossRef]
- Abiose, A.K.; Mansoor, G.A.; Barry, M.; Soucier, R.; Nair, C.K.; Hager, D. Effect of spironolactone on endothelial function in patients with congestive heart failure on conventional medical therapy. Am. J. Cardiol. 2004, 93, 1564–1566. [Google Scholar] [CrossRef]
- Nowak, K.L.; Gitomer, B.; Farmer-Bailey, H.; Wang, W.; Malaczewski, M.; Klawitter, J.; You, Z.; George, D.; Patel, N.; Jovanovich, A.; et al. Mineralocorticoid Antagonism and Vascular Function in Early Autosomal Dominant Polycystic Kidney Disease: A Randomized Controlled Trial. Am. J. Kidney Dis. 2019, 74, 213–223. [Google Scholar] [CrossRef]
- Zeng, Y.; Tarbell, J.M. The adaptive remodeling of endothelial glycocalyx in response to fluid shear stress. PLoS ONE 2014, 9, e86249. [Google Scholar] [CrossRef] [Green Version]
- Yen, W.; Cai, B.; Yang, J.; Zhang, L.; Zeng, M.; Tarbell, J.M.; Fu, B.M. Endothelial surface glycocalyx can regulate flow-induced nitric oxide production in microvessels in vivo. PLoS ONE 2015, 10, e0117133. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Simon, S.I.; Curry, F.R. Mechanosensing at the vascular interface. Annu. Rev. Biomed. Eng. 2014, 16, 505–532. [Google Scholar] [CrossRef] [Green Version]
- Ramnath, R.D.; Butler, M.J.; Newman, G.; Desideri, S.; Russell, A.; Lay, A.C.; Neal, C.R.; Qiu, Y.; Fawaz, S.; Onions, K.L.; et al. Blocking matrix metalloproteinase-mediated syndecan-4 shedding restores the endothelial glycocalyx and glomerular filtration barrier function in early diabetic kidney disease. Kidney Int. 2020, 97, 951–965. [Google Scholar] [CrossRef] [Green Version]
- Kirsch, T.; Beese, M.; Wyss, K.; Klinge, U.; Haller, H.; Haubitz, M.; Fiebeler, A. Aldosterone modulates endothelial permeability and endothelial nitric oxide synthase activity by rearrangement of the actin cytoskeleton. Hypertension 2013, 61, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberleithner, H.; Riethmuller, C.; Ludwig, T.; Shahin, V.; Stock, C.; Schwab, A.; Hausberg, M.; Kusche, K.; Schillers, H. Differential action of steroid hormones on human endothelium. J. Cell Sci. 2006, 119, 1926–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, A.H.; Satchell, S.C. Endothelial glycocalyx dysfunction in disease: Albuminuria and increased microvascular permeability. J. Pathol. 2012, 226, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Ferguson, J.K.; Ramnath, R.D.; Onions, K.L.; Ogier, A.S.; Gamez, M.; Down, C.J.; Skinner, L.; Wong, K.H.; Dixon, L.K.; et al. Mineralocorticoid receptor antagonism in diabetes reduces albuminuria by preserving the glomerular endothelial glycocalyx. JCI Insight 2023, 8, e154164. [Google Scholar] [CrossRef]
- Satchell, S. The role of the glomerular endothelium in albumin handling. Nat. Rev. Nephrol. 2013, 9, 717–725. [Google Scholar] [CrossRef]
- Praga, M.; Morales, E. Obesity, proteinuria and progression of renal failure. Curr. Opin. Nephrol. Hypertens. 2006, 15, 481–486. [Google Scholar] [CrossRef]
- Bolignano, D.; Palmer, S.C.; Navaneethan, S.D.; Strippoli, G.F. Aldosterone antagonists for preventing the progression of chronic kidney disease. Cochrane Database Syst. Rev. 2014, 4, CD007004. [Google Scholar] [CrossRef]
- Levin, A.; Stevens, P.E. Summary of KDIGO 2012 CKD Guideline: Behind the scenes, need for guidance, and a framework for moving forward. Kidney Int. 2014, 85, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.K.; Szeto, C.C. Mineralocorticoid receptor antagonist for renal protection. Ren. Fail. 2012, 34, 810–817. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, N.; Kawamura, T.; Okonogi, H.; Ishii, T.; Hosoya, T. The long-term antiproteinuric effect of eplerenone, a selective aldosterone blocker, in patients with non-diabetic chronic kidney disease. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2012, 13, 113–117. [Google Scholar] [CrossRef]
- Sengul, E.; Sahin, T.; Sevin, E.; Yilmaz, A. Effect of spironolactone on urinary protein excretion in patients with chronic kidney disease. Ren. Fail. 2009, 31, 928–932. [Google Scholar] [CrossRef]
- Schreier, B.; Rabe, S.; Schneider, B.; Ruhs, S.; Grossmann, C.; Hauptmann, S.; Blessing, M.; Neumann, J.; Gekle, M. Aldosterone/NaCl-induced renal and cardiac fibrosis is modulated by TGF-beta responsiveness of T cells. Hypertens. Res. 2011, 34, 623–629. [Google Scholar] [CrossRef]
- Piecha, G.; Koleganova, N.; Gross, M.L.; Geldyyev, A.; Adamczak, M.; Ritz, E. Regression of glomerulosclerosis in subtotally nephrectomized rats: Effects of monotherapy with losartan, spironolactone, and their combination. Am. J. Physiol. Ren. Physiol. 2008, 295, F137–F144. [Google Scholar] [CrossRef] [Green Version]
- Su, M.; Dhoopun, A.R.; Yuan, Y.; Huang, S.; Zhu, C.; Ding, G.; Liu, B.; Yang, T.; Zhang, A. Mitochondrial dysfunction is an early event in aldosterone-induced podocyte injury. Am. J. Physiol. Ren. Physiol. 2013, 305, F520–F531. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.L.; Nikolic-Paterson, D.J.; Han, Y.; Ozols, E.; Ma, F.Y.; Young, M.J.; Tesch, G.H. Myeloid Mineralocorticoid Receptor Activation Contributes to Progressive Kidney Disease. J. Am. Soc. Nephrol. 2014, 25, 2231–2240. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.; Brennan, F.E.; Young, M.J.; Fuller, P.J.; Cole, T.J. A direct effect of aldosterone on endothelin-1 gene expression in vivo. Endocrinology 2007, 148, 1511–1517. [Google Scholar] [CrossRef] [Green Version]
- Raina, R.; Chauvin, A.; Chakraborty, R.; Nair, N.; Shah, H.; Krishnappa, V.; Kusumi, K. The Role of Endothelin and Endothelin Antagonists in Chronic Kidney Disease. Kidney Dis. 2020, 6, 22–34. [Google Scholar] [CrossRef]
- Boels, M.G.; Avramut, M.C.; Koudijs, A.; Dane, M.J.; Lee, D.H.; van der Vlag, J.; Koster, A.J.; van Zonneveld, A.J.; van Faassen, E.; Grone, H.J.; et al. Atrasentan Reduces Albuminuria by Restoring the Glomerular Endothelial Glycocalyx Barrier in Diabetic Nephropathy. Diabetes 2016, 65, 2429–2439. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; Parving, H.H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef]
- Krug, A.W.; Kopprasch, S.; Ziegler, C.G.; Dippong, S.; Catar, R.A.; Bornstein, S.R.; Morawietz, H.; Gekle, M. Aldosterone rapidly induces leukocyte adhesion to endothelial cells: A new link between aldosterone and arteriosclerosis? Hypertension 2007, 50, e156–e157. [Google Scholar] [CrossRef] [Green Version]
- Marzolla, V.; Armani, A.; Mammi, C.; Moss, M.E.; Pagliarini, V.; Pontecorvo, L.; Antelmi, A.; Fabbri, A.; Rosano, G.; Jaffe, I.Z.; et al. Essential role of ICAM-1 in aldosterone-induced atherosclerosis. Int. J. Cardiol. 2017, 232, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lother, A.; Furst, D.; Bergemann, S.; Gilsbach, R.; Grahammer, F.; Huber, T.B.; Hilgendorf, I.; Bode, C.; Moser, M.; Hein, L. Deoxycorticosterone Acetate/Salt-Induced Cardiac but not Renal Injury Is Mediated by Endothelial Mineralocorticoid Receptors Independently From Blood Pressure. Hypertension 2016, 67, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caprio, M.; Newfell, B.G.; la Sala, A.; Baur, W.; Fabbri, A.; Rosano, G.; Mendelsohn, M.E.; Jaffe, I.Z. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ. Res. 2008, 102, 1359–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, M.T.; Duckles, H.; Feng, S.; Hsiao, S.T.; Kim, H.R.; Serbanovic-Canic, J.; Evans, P.C. Mechanoresponsive networks controlling vascular inflammation. Arter. Thromb. Vasc. Biol. 2014, 34, 2199–2205. [Google Scholar] [CrossRef] [Green Version]
- Oberleithner, H.; Riethmuller, C.; Schillers, H.; MacGregor, G.A.; de Wardener, H.E.; Hausberg, M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc. Natl. Acad. Sci. USA 2007, 104, 16281–16286. [Google Scholar] [CrossRef] [Green Version]
- Tomaschitz, A.; Pilz, S.; Ritz, E.; Meinitzer, A.; Boehm, B.O.; Marz, W. Plasma aldosterone levels are associated with increased cardiovascular mortality: The Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Eur. Heart J. 2010, 31, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Ivanes, F.; Susen, S.; Mouquet, F.; Pigny, P.; Cuilleret, F.; Sautiere, K.; Collet, J.P.; Beygui, F.; Hennache, B.; Ennezat, P.V.; et al. Aldosterone, mortality, and acute ischaemic events in coronary artery disease patients outside the setting of acute myocardial infarction or heart failure. Eur. Heart J. 2012, 33, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Wentzel, J.J.; Chatzizisis, Y.S.; Gijsen, F.J.; Giannoglou, G.D.; Feldman, C.L.; Stone, P.H. Endothelial shear stress in the evolution of coronary atherosclerotic plaque and vascular remodelling: Current understanding and remaining questions. Cardiovasc. Res. 2012, 96, 234–243. [Google Scholar] [CrossRef]
- Zhang, J.; Kong, X.; Wang, Z.; Gao, X.; Ge, Z.; Gu, Y.; Ye, P.; Chao, Y.; Zhu, L.; Li, X.; et al. AMP-activated protein kinase regulates glycocalyx impairment and macrophage recruitment in response to low shear stress. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 7202–7212. [Google Scholar] [CrossRef]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M.; et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Haddad, N.; Rajan, J.; Nagaraja, H.N.; Agarwal, A.K.; Hebert, L.A. Usual ACE inhibitor therapy in CKD patients is associated with lower plasma aldosterone levels than usual angiotensin receptor blocker therapy. Kidney Blood Press. Res. 2007, 30, 299–305. [Google Scholar] [CrossRef]
- Schjoedt, K.J. The renin-angiotensin-aldosterone system and its blockade in diabetic nephropathy: Main focus on the role of aldosterone. Dan. Med. Bull. 2011, 58, B4265. [Google Scholar]
- Sato, A.; Saruta, T. Aldosterone escape during angiotensin-converting enzyme inhibitor therapy in essential hypertensive patients with left ventricular hypertrophy. J. Int. Med. Res. 2001, 29, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Hayashi, K.; Naruse, M.; Saruta, T. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension 2003, 41, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Bedford, M.; Farmer, C.K.; Irving, J.; Stevens, P.E. Acute kidney injury: An acceptable risk of treatment with renin-angiotensin system blockade in primary care? Can. J. Kidney Health Dis. 2015, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Struthers, A.D.; Fahey, T.; Watson, A.D.; Macdonald, T.M. Spironolactone use and renal toxicity: Population based longitudinal analysis. BMJ 2010, 340, c1768. [Google Scholar] [CrossRef] [Green Version]
- Sica, D.A. Pharmacokinetics and pharmacodynamics of mineralocorticoid blocking agents and their effects on potassium homeostasis. Heart Fail. Rev. 2005, 10, 23–29. [Google Scholar] [CrossRef]
- Hill, N.R.; Lasserson, D.; Thompson, B.; Perera-Salazar, R.; Wolstenholme, J.; Bower, P.; Blakeman, T.; Fitzmaurice, D.; Little, P.; Feder, G.; et al. Benefits of Aldosterone Receptor Antagonism in Chronic Kidney Disease (BARACK D) trial-a multi-centre, prospective, randomised, open, blinded end-point, 36-month study of 2616 patients within primary care with stage 3b chronic kidney disease to compare the efficacy of spironolactone 25 mg once daily in addition to routine care on mortality and cardiovascular outcomes versus routine care alone: Study protocol for a randomized controlled trial. Trials 2014, 15, 160. [Google Scholar] [CrossRef] [Green Version]
- Deinum, J.; Riksen, N.P.; Lenders, J.W. Pharmacological treatment of aldosterone excess. Pharm. Ther. 2015, 154, 120–133. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Kolkhof, P.; Delbeck, M.; Kretschmer, A.; Steinke, W.; Hartmann, E.; Barfacker, L.; Eitner, F.; Albrecht-Kupper, B.; Schafer, S. Finerenone, a novel selective nonsteroidal mineralocorticoid receptor antagonist protects from rat cardiorenal injury. J. Cardiovasc. Pharmacol. 2014, 64, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Snelder, N.; Heinig, R.; Drenth, H.J.; Joseph, A.; Kolkhof, P.; Lippert, J.; Garmann, D.; Ploeger, B.; Eissing, T. Population Pharmacokinetic and Exposure-Response Analysis of Finerenone: Insights Based on Phase IIb Data and Simulations to Support Dose Selection for Pivotal Trials in Type 2 Diabetes with Chronic Kidney Disease. Clin. Pharm. 2020, 59, 359–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, R.; Joseph, A.; Anker, S.D.; Filippatos, G.; Rossing, P.; Ruilope, L.M.; Pitt, B.; Kolkhof, P.; Scott, C.; Lawatscheck, R.; et al. Hyperkalemia Risk with Finerenone: Results from the FIDELIO-DKD Trial. J. Am. Soc. Nephrol. 2022, 33, 225–237. [Google Scholar] [CrossRef]
- Pivonello, R.; Fleseriu, M.; Newell-Price, J.; Bertagna, X.; Findling, J.; Shimatsu, A.; Gu, F.; Auchus, R.; Leelawattana, R.; Lee, E.J.; et al. Efficacy and safety of osilodrostat in patients with Cushing’s disease (LINC 3): A multicentre phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol. 2020, 8, 748–761. [Google Scholar] [CrossRef]
- Cai, T.Q.; Stribling, S.; Tong, X.; Xu, L.; Wisniewski, T.; Fontenot, J.A.; Struthers, M.; Akinsanya, K.O. Rhesus monkey model for concurrent analyses of in vivo selectivity, pharmacokinetics and pharmacodynamics of aldosterone synthase inhibitors. J. Pharm. Toxicol. Methods 2015, 71, 137–146. [Google Scholar] [CrossRef]
- Papillon, J.P.; Lou, C.; Singh, A.K.; Adams, C.M.; Ksander, G.M.; Beil, M.E.; Chen, W.; Leung-Chu, J.; Fu, F.; Gan, L.; et al. Discovery of N-[5-(6-Chloro-3-cyano-1-methyl-1H-indol-2-yl)-pyridin-3-ylmethyl]-ethanesulfonam ide, a Cortisol-Sparing CYP11B2 Inhibitor that Lowers Aldosterone in Human Subjects. J. Med. Chem. 2015, 58, 9382–9394. [Google Scholar] [CrossRef]
- Sakakibara, R.; Sasaki, W.; Onda, Y.; Yamaguchi, M.; Ushirogochi, H.; Hiraga, Y.; Sato, K.; Nishio, M.; Egi, Y.; Takedomi, K.; et al. Discovery of Novel Pyrazole-Based Selective Aldosterone Synthase (CYP11B2) Inhibitors: A New Template to Coordinate the Heme-Iron Motif of CYP11B2. J. Med. Chem. 2018, 61, 5594–5608. [Google Scholar] [CrossRef]
- Sparks, S.M.; Danger, D.P.; Hoekstra, W.J.; Leesnitzer, T.; Schotzinger, R.J.; Yates, C.M.; Becherer, J.D. Development of Highly Selective Pyrimidine-Based Aldosterone Synthase (CYP11B2) Inhibitors. ACS Med. Chem. Lett. 2019, 10, 1056–1060. [Google Scholar] [CrossRef]
- Freeman, M.W.; Halvorsen, Y.D.; Marshall, W.; Pater, M.; Isaacsohn, J.; Pearce, C.; Murphy, B.; Alp, N.; Srivastava, A.; Bhatt, D.L.; et al. Phase 2 Trial of Baxdrostat for Treatment-Resistant Hypertension. N. Engl. J. Med. 2023, 388, 395–405. [Google Scholar] [CrossRef]
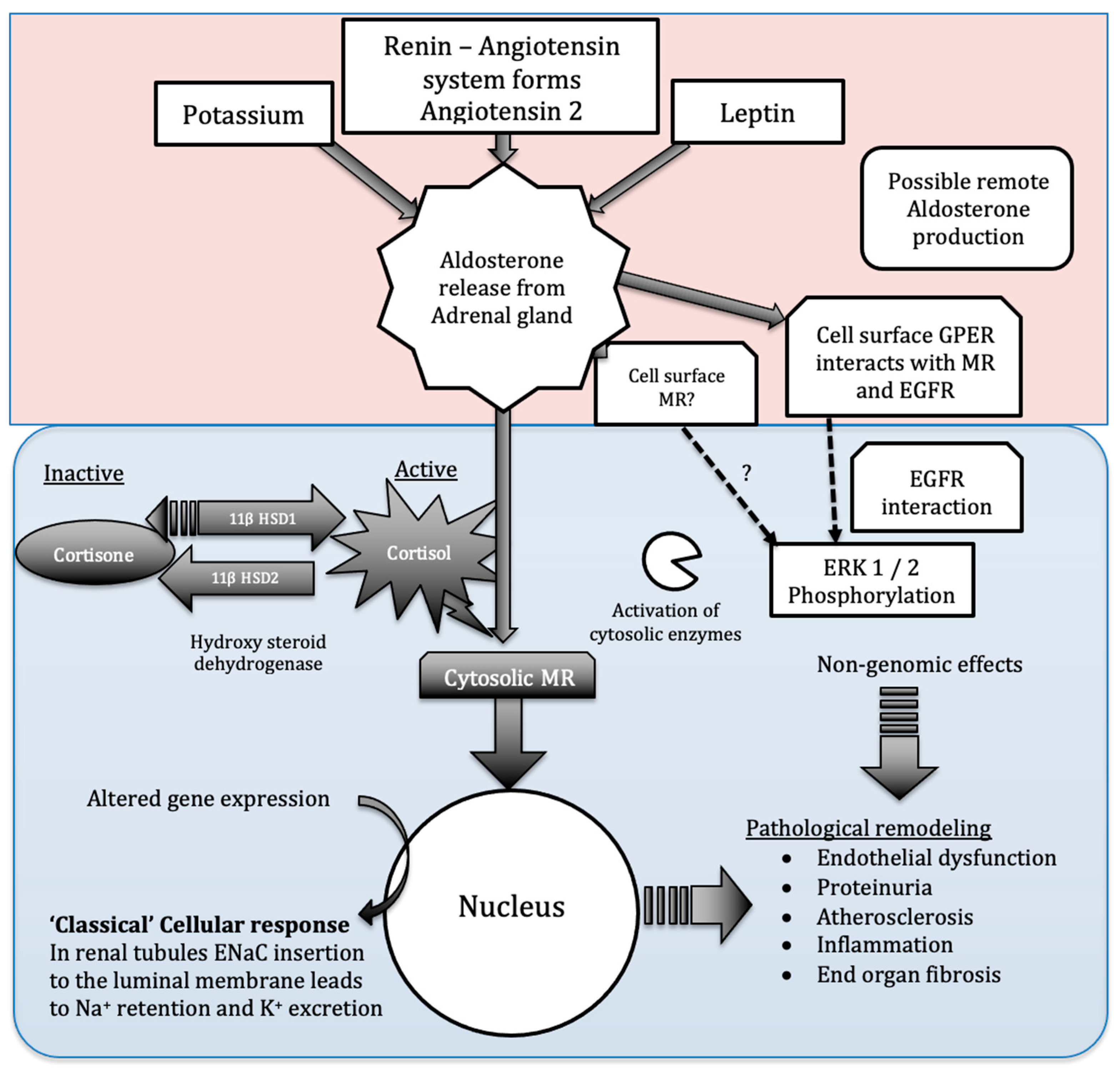
Figure 1.
Multiple pathways coordinate and regulate aldosterone signalling. Multiple stimuli result in aldosterone production within the adrenal glands, the role of localized aldosterone production remains unknown. Circulating aldosterone my act on cell surface MR receptors and GPER in addition to the cytosolic MR receptor. The ‘classical’ aldosterone signalling pathway (via cytosolic MR) is protected from cortisol activation by hydroxysteroid dehydrogenase 2 activity. These complex pathways all represent potential future therapeutic targets.
Figure 1.
Multiple pathways coordinate and regulate aldosterone signalling. Multiple stimuli result in aldosterone production within the adrenal glands, the role of localized aldosterone production remains unknown. Circulating aldosterone my act on cell surface MR receptors and GPER in addition to the cytosolic MR receptor. The ‘classical’ aldosterone signalling pathway (via cytosolic MR) is protected from cortisol activation by hydroxysteroid dehydrogenase 2 activity. These complex pathways all represent potential future therapeutic targets.

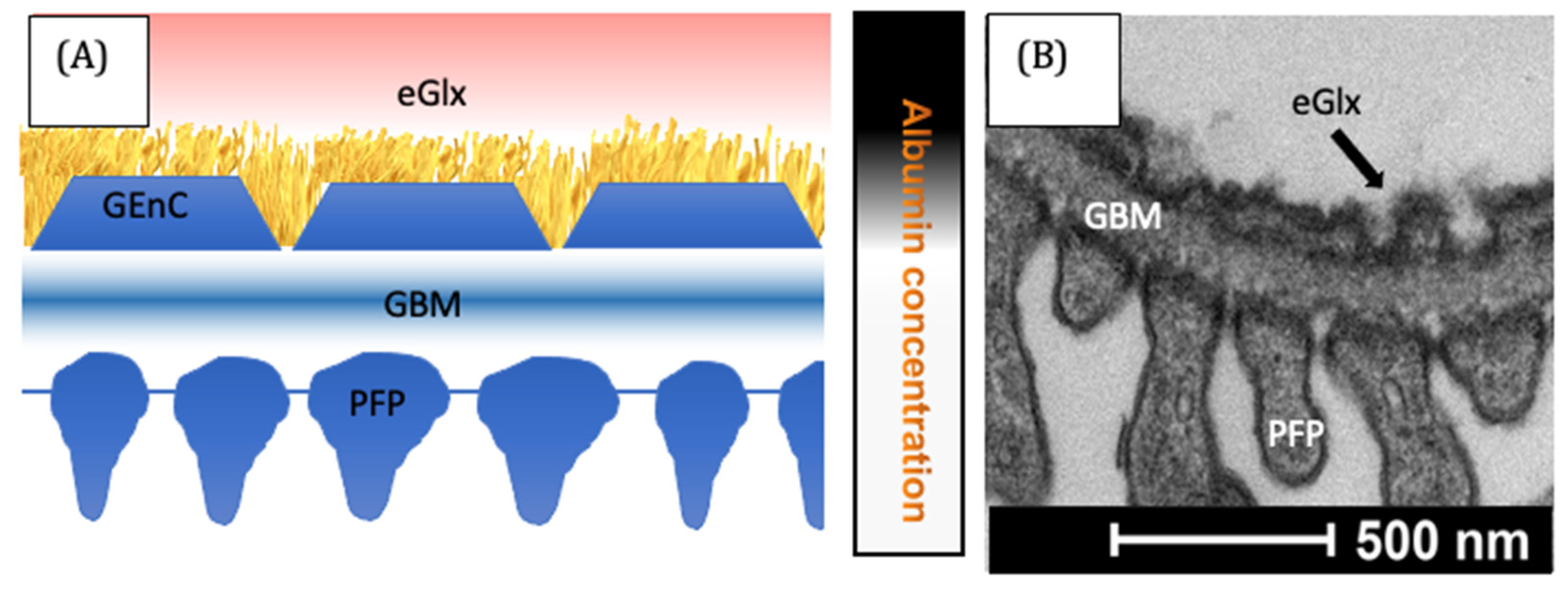
Figure 2.
The glomerular filtration barrier. In the representative diagram, (A) the filtrate passes the layers of the filter. Albumin is largely excluded, as indicated by the local albumin concentration on the right. Electron microscopy (B) demonstrates the functional arrangement of cells within the glomerulus [84]. GBM = glomerular basement membrane, GEnC = glomerular endothelial cells, PFP = podocyte foot process.
Figure 2.
The glomerular filtration barrier. In the representative diagram, (A) the filtrate passes the layers of the filter. Albumin is largely excluded, as indicated by the local albumin concentration on the right. Electron microscopy (B) demonstrates the functional arrangement of cells within the glomerulus [84]. GBM = glomerular basement membrane, GEnC = glomerular endothelial cells, PFP = podocyte foot process.

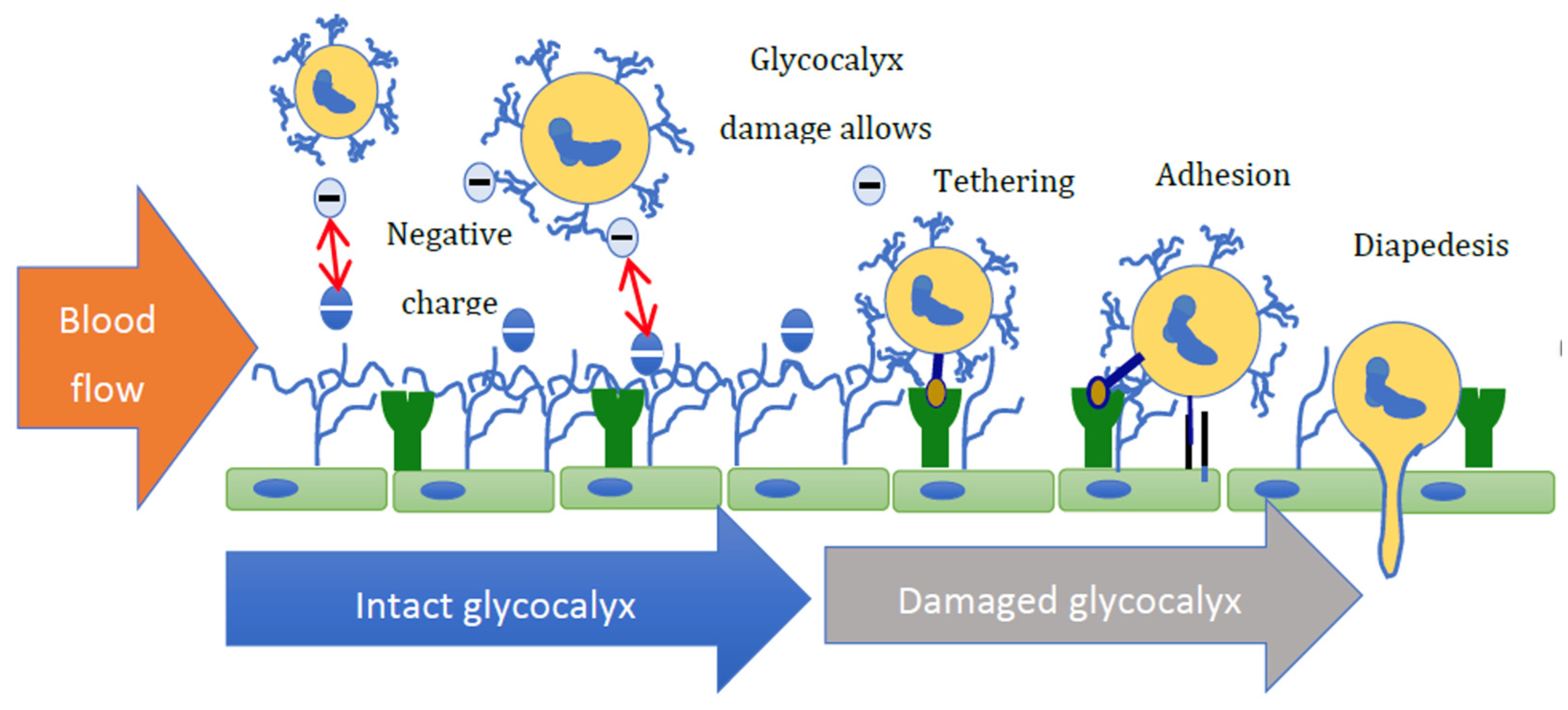
Figure 3.
A suggested mechanism by which aldosterone may affect immune cell transit. In health, repulsion between the negatively charged glycocalyx of white blood cells and the vessel wall limits their interaction. Given the difference in height, the glycocalyx (∼500 nm) shields selectins (∼40 nm). Glycocalyx damage reduces the negative surface charge and allows for electrostatic attraction of white blood cells to the endothelium, which then mediate receptor–ligand interactions, resulting in tethering, rolling, adhesion, and diapedesis.
Figure 3.
A suggested mechanism by which aldosterone may affect immune cell transit. In health, repulsion between the negatively charged glycocalyx of white blood cells and the vessel wall limits their interaction. Given the difference in height, the glycocalyx (∼500 nm) shields selectins (∼40 nm). Glycocalyx damage reduces the negative surface charge and allows for electrostatic attraction of white blood cells to the endothelium, which then mediate receptor–ligand interactions, resulting in tethering, rolling, adhesion, and diapedesis.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Crompton, M.; Skinner, L.J.; Satchell, S.C.; Butler, M.J. Aldosterone: Essential for Life but Damaging to the Vascular Endothelium. Biomolecules 2023, 13, 1004. https://doi.org/10.3390/biom13061004
AMA Style
Crompton M, Skinner LJ, Satchell SC, Butler MJ. Aldosterone: Essential for Life but Damaging to the Vascular Endothelium. Biomolecules. 2023; 13(6):1004. https://doi.org/10.3390/biom13061004
Chicago/Turabian StyleCrompton, Michael, Laura J. Skinner, Simon C. Satchell, and Matthew J. Butler. 2023. "Aldosterone: Essential for Life but Damaging to the Vascular Endothelium" Biomolecules 13, no. 6: 1004. https://doi.org/10.3390/biom13061004
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.